




Abstract
Transcriptional silencing by CpG island hypermethylation has become a critical component in the initiation and progression of lung cancer. The ability of pharmacologic agents to reverse promoter hypermethylation also makes it an attractive target to pursue for prevention of lung cancer. Animal models, together with studies in humans have fostered significant advances in elucidating the role of gene-specific methylation in cancer initiation and progression, the modulation of promoter methylation by carcinogen exposure and the ability to block tumor development by preventing epigenetically mediated gene silencing. These advances represent the beginning of efforts to develop a progression model for lung cancer that should aid efforts for early detection and gene targeting for therapy, and the development of preventive interventions that reverse epigenetic-mediated gene silencing.
Introduction
The past two decades of research have seen a tremendous growth in defining gene targets and pathways disrupted during the development of lung cancer. Some of the earliest findings identified activation of the K-ras oncogene and inactivation of the p53 tumor suppressor gene by point mutation as predominant alterations in lung cancer etiology ( 1 , 2 ). In spite of identifying dysfunction within these two important genes/pathways, many lung tumors were found to arise independently of alteration within these genes. It became apparent through genome studies of loss of heterozygosity that many DNA regions exist that could contain candidate tumor suppressor genes. One region on the short arm of chromosome 9p was found to contain the CDKN2A ( p16 ) gene ( 3 , 4 ). Mutations within the p16 coding sequence were commonly found in melanoma; however, low rates of mutation in other solid tumors such as lung suggested an alternative mechanism for the loss of function ( 4 ). p16 was the first tumor suppressor gene found inactivated in lung cancer predominantly through aberrant hypermethylation of its 5′ promoter region ( 5 ). The p16 gene is inactivated by methylation at prevalences up to 60–70% in adenocarcinomas and squamous cell carcinoma (SCC) of the lung, respectively ( 5 – 9 ). The targeting of p16 for inactivation is likely due to the critical function of this gene in the cell, to inhibit cyclin dependent kinases that bind cyclin D1 and phosphorylate the retinoblastoma gene ( 10 , 11 ).
Since the identification of aberrant promoter methylation as an inactivating mechanism for the p16 gene, epigenetically mediated silencing of transcription has been associated with inactivation of >30 genes in lung cancer ( 12 , 13 ). Transcriptional silencing by CpG island hypermethylation affects genes involved in all aspects of cellular function such as cell-cycle control, differentiation and signalling, apoptosis and invasion ( 13 – 15 ). The commonality of gene promoter methylation in the etiology of lung cancer has led our group to investigate the role methylation plays in cancer initiation and whether exposure to defined carcinogens are associated with specific gene inactivations via methylation. The ability of pharmacologic agents to reverse promoter hypermethylation also makes it an attractive target to pursue for prevention of lung cancer ( 16 ). The following sections discuss the current findings on these research issues and how animal models have aided these studies.
Methylation as an initiating event in lung cancer
The fact that many genes controlling cell regulation are inactivated in lung cancer by promoter methylation suggests that a subset of these genes would likely be involved in initiation and others in progression. A progression model similar to that developed for colon cancer is lacking for lung cancer, due to the difficulty in obtaining premalignant lesions from high-risk people without synchronous invasive tumors. To overcome this obstacle we have studied precursor lesions to adenocarcinoma and SCC in an animal model of lung carcinogenesis and in human subjects, respectively. Our initial studies were focused on the p16 gene ( 8 ). In the rat, 94% of adenocarcinomas induced by the tobacco-specific carcinogen 4-methylnitrosamino-1-(3-pyridyl)-1-butanone (NNK) were hypermethylated at the p16 promoter. This methylation change was also detected frequently in precursor lesions to the tumors—adenomas and hyperplastic lesions. The timing for inactivation of p16 by methylation in SCC was recapitulated in human SCCs where the p16 gene was coordinately methylated in 75% of carcinoma in situ lesions adjacent to SCCs containing this change ( 8 ; Table I ). Moreover, in premalignant lesions obtained from persons without SCC, the frequency of this event increased during disease progression from basal cell hyperplasia (17%) to squamous cell metaplasia (24%) to carcinoma in situ (50%). Further studies substantiating that inactivation of p16 may be a very early event in lung cancer examined bronchial epithelial cells obtained through bronchoscopy from cancer-free smokers ( 17 ). Methylation of p16 was detected in 25 of 137 biopsies (18%) that were classified histologically as normal, metaplastic or mildly dysplastic ( Table I ). In contrast, no methylation of p16 was seen in biopsies obtained from individuals who had never smoked. We also addressed whether in lung cancer cases an association existed between the methylation status of p16 in the bronchial epithelium and the diagnosed tumor. Matched tumor tissue and bronchial epithelium (2–3 bronchial biopsies per case) obtained from lung lobes that did not contain the primary tumor were available from 18 cases of non-small cell lung cancer ( 17 ). An absolute concordance of being either methylated in the tumor and at least one bronchial epithelium site or unmethylated in the tumor and the bronchial epithelium was seen in 17 of 18 cases. The strong association seen between p16 methylation in the bronchial epithelium and corresponding primary tumor substantiates the fact that inactivation of this gene, although not transforming by itself, is likely permissive for the acquisition of additional genetic and epigenetic changes leading to lung cancer.
Frequency of p16 methylation in bronchial epithelial cells, premalignant lesions and SCCs
| Subjects | Sample type | Methylation frequency (%) |
|---|---|---|
| Never smokers | BECs a | 0/17 (0) |
| Smokers | BECs | 25/137 (18) |
| Hyperplasia | 2/12 (17) | |
| Metaplasia | 4/17 (24) | |
| Carcinoma in situ | 3/6 (50) b | |
| Lung cancer | Carcinoma in situ | 6/12 (50) |
| SCC | 11/18 (61) |
| Subjects | Sample type | Methylation frequency (%) |
|---|---|---|
| Never smokers | BECs a | 0/17 (0) |
| Smokers | BECs | 25/137 (18) |
| Hyperplasia | 2/12 (17) | |
| Metaplasia | 4/17 (24) | |
| Carcinoma in situ | 3/6 (50) b | |
| Lung cancer | Carcinoma in situ | 6/12 (50) |
| SCC | 11/18 (61) |
BEC, bronchial epithelial cells.
P < 0.05 for linear trend.
Frequency of p16 methylation in bronchial epithelial cells, premalignant lesions and SCCs
| Subjects | Sample type | Methylation frequency (%) |
|---|---|---|
| Never smokers | BECs a | 0/17 (0) |
| Smokers | BECs | 25/137 (18) |
| Hyperplasia | 2/12 (17) | |
| Metaplasia | 4/17 (24) | |
| Carcinoma in situ | 3/6 (50) b | |
| Lung cancer | Carcinoma in situ | 6/12 (50) |
| SCC | 11/18 (61) |
| Subjects | Sample type | Methylation frequency (%) |
|---|---|---|
| Never smokers | BECs a | 0/17 (0) |
| Smokers | BECs | 25/137 (18) |
| Hyperplasia | 2/12 (17) | |
| Metaplasia | 4/17 (24) | |
| Carcinoma in situ | 3/6 (50) b | |
| Lung cancer | Carcinoma in situ | 6/12 (50) |
| SCC | 11/18 (61) |
BEC, bronchial epithelial cells.
P < 0.05 for linear trend.
While p16 is clearly one of the earliest genes known to be altered through methylation in lung cancer, other genes are emerging that are likely inactivated during premalignancy. Methylation of the death associated protein kinase ( DAP-K ) and retinoic acid receptor β ( RAR-β ) genes were detected in approximately half of alveolar hyperplasias induced in mouse lungs exposed chronically to NNK ( 18 , 19 ). Supporting the likelihood that DAP-K inactivation occurs at the time when premalignant changes can be observed histologically, only 3% of bronchial biopsies examined for p16 had methylation of DAP-K ( 17 ). The DNA repair gene, O 6 -methylguanine-DNA methyltransferase ( MGMT ), also appears to be inactivated after the p16 gene. Only 3 of 40 (8%) biopsies with histologies including normal, hyperplasia, metaplasia and dysplasia obtained from heavy smokers showed methylation of this gene ( 20 ). In addition, the prevalence for methylation of MGMT increased between stage I adenocarcinoma and stages II–IV ( 20 ). The timing for inactivation of the genes studied to date appears to coincide with their roles in cellular homeostasis. Disruption of cell-cycle control ( p16 ) would need to be one of the earliest events to allow for clonal expansion. Resistance to apoptosis ( DAP-K ) and loss of normal cell differentiation control ( RAR-β ) would further aid in the development of autonomous premalignant clones. The subsequent loss of DNA repair of alkyl adducts ( MGMT ) could lead to acquisition of somatic mutations in genes such as p53 whose inactivation occurs at more advanced stages of premalignancy (e.g. dysplasia, carcinoma in situ ) ( 21 , 22 ). These studies represent only the beginning of efforts to develop a progression model for SCC and adenocarcinoma that should aid efforts for early detection and gene targeting for lung cancer therapy ( 13 , 23 , 24 ).
There have been numerous studies examining the function of genes inactivated by promoter hypermethylation with respect to impacting malignant transformation. Highlights of the studies for three genes, p16 , DAP-K and RAR-β , will be provided. As mentioned above, p16 plays an important role in cell cycle control by regulating the phosphorylation of the retinoblastoma gene ( 10 , 11 ). A role for p16 in immortalization has been suggested since its loss of function allows cells to escape the immortality checkpoint ‘M0’ ( 25 ). Mouse xenografts or nude mouse studies for mesothelioma cells and head and neck cancer cells, respectively, also showed that reexpression of p16 inhibits tumor formation and diminishes tumor size ( 26 , 27 ). The DAP-K gene is important for γ-interferon, tumor necrosis-α and Fas-induced apoptosis ( 28 , 29 ). Expression of DAP-K has also been shown to suppress oncogenic transformation of primary embryonic fibroblasts by activating p53 in a p19ARF -dependent manner ( 30 ). Thus, DAP-K could play an important role in eliminating premalignant cells during cancer development. Finally, the RAR-β gene is a member of the retinoid family that mediates cell differentiation and proliferation. The antitumor effects of RAR-β are mediated, in part, through inhibition of cell proliferation and metastasis as well as apoptosis ( 31 , 32 ). Thus, the important functions of these genes and others inactivated through promoter hypermethylation supports a causal role for their involvement in the initiation and progression of lung carcinogenesis.
Environmental exposures target genes for promoter hypermethylation
It is well established that specific DNA adducts are associated with specific somatic mutations in oncogenes and tumor suppressor genes. The best example is the K-ras gene where mutation profiles in codons 12 and 61 have been strongly correlated with the promutagenic adducts generated from the metabolism of carcinogens such as NNK, benzo[ a ]pyrene and urethane ( 33 , 34 ). In addition, hot spots for mutation in the p53 gene have also been linked to preferential binding of the diolepoxide adduct generated through activation of benzo[ a ]pyrene ( 35 ). In contrast, there have been no direct studies to determine the role of DNA damage in initiating gene-specific promoter hypermethylation. Our initial approach to this issue was to ask whether carcinogen exposures that damage DNA through different mechanisms such as single-strand versus double-strand breaks would induce lung tumors that arise via inactivation of either different genes or inactivate genes at a higher prevalence. We first examined the methylation of the estrogen receptor α (ER) CpG island in lung tumors from humans, rats and mice ( 36 ). ER methylation was seen at a similar low prevalence (∼20%) in lung tumors induced in humans by cigarette smoke or in rats and mice by NNK. Tumors in unexposed mice or never smokers showed a much higher prevalence for methylation of the ER gene ( Figure 1 ). The most striking differences were seen in rat lung tumors induced by exposure to X-ray or plutonium where the ER gene was methylated in 38 and 82% of tumors, respectively.
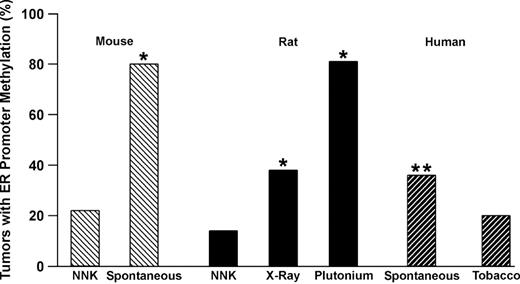
Summary of ER methylation in mouse, rat and human lung tumors. The source of tumors is indicated on the x axis. Spontaneous human tumors are from never smokers. *P < 0.01 compared with NNK; **P < 0.01 compared with tobacco.
A second study examined the prevalence for methylation of a panel of genes in adenocarcinomas from plutonium-exposed workers at MAYAK, the first Russian nuclear enterprise established to manufacture plutonium weapons, compared with non-worker controls ( 37 ). The prevalence for methylation of the MGMT or DAP-K genes did not differ between workers and controls, while a higher prevalence for methylation of the RASSF1A gene was seen in tumors from controls. In marked contrast, the prevalence for methylation of p16 increased significantly in tumors from workers compared with non-worker controls. Stratification of plutonium exposure into tertiles also revealed a striking dose response for methylation of the p16 gene ( Figure 2 ). Workers in the plutonium plant where exposure to internal radiation was highest had a 3.5 times (CI 1.5, 8.5; P = 0.001) greater risk for p16 methylation in their tumors than controls. This increased probability for methylation approximated the 4-fold increase in relative risk for adenocarcinoma in this group of workers exposed to plutonium. In addition, a trend was seen for an increase in the number of genes methylated (>2 genes) with plutonium dose. Together, these studies show that gene-specific promoter methylation could be modulated differentially depending on carcinogen exposure. Furthermore, the targeting of a gene such as p16 through a defined environmental exposure may significantly impact the risk for developing adenocarcinoma.
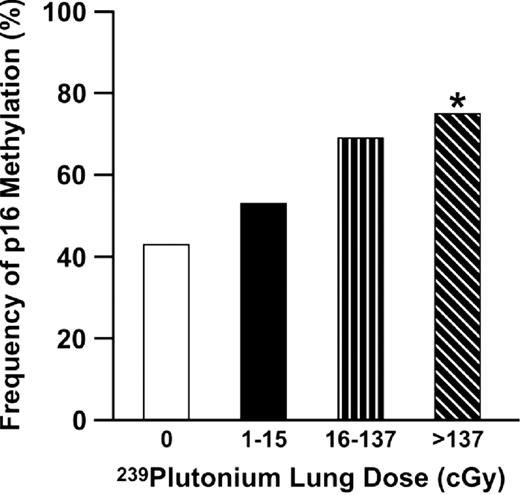
Methylation of the p16 gene increases as a function of plutonium lung dose in adenocarcinomas from MAYAK workers. *P = 0.0008 for linear trend.
A key question generated from these studies is how these different exposures affect the propensity to inactivate a gene through promoter hypermethylation. With respect to plutonium, the α-particles generated deposit large amounts of energy in small volumes along their paths that can lead to double-strand breaks in DNA. A recent study ( 38 ) detected and quantified stable intrachromosomal rearrangements within chromosome 5 in the former workers from the MAYAK facility. The yield of these aberrations was highly correlated with plutonium dose to the bone marrow. The extensive DNA damage suggests that radiation exposure to the lung epithelial cells from workers led to the accumulation of massive amounts of chromosomal damage that possibly overwhelmed DNA repair pathways. Extensive DNA damage could in turn disrupt replication timing because of chromosomal instability or loss of normal transcriptional control and both effects have been proposed as mechanisms through which normally unmethylated regions of DNA become aberrantly methylated ( 39 ).
Other mechanisms to consider include formation of sites of base loss in DNA (abasic AP sites) and other forms of DNA damage that in addition to causing mutations can alter protein/DNA interactions. AP sites occur frequently as spontaneous events in living cells and become elevated following carcinogen exposure ( 40 ). For example, a single AP site within or adjacent to the consensus binding sequences for the transcription factor AP-1 can inhibit transcription factor binding ( 41 ). A link between decreased transcription and promoter methylation was suggested by Song et al . ( 42 ) who evaluated methylation of the glutathione- S transferase P1 gene using stable transfections into prostate cancer cell lines in which the endogenous gene was inactivated through methylation. In those studies, hypermethylation of the transfected gene occurred only after a two-step event that involved ‘seeding’ methylation in combination with reducing transcription through deletion of an Sp1 site. Thus, disruption of normal transcription by DNA damage could be one factor in initiating promoter methylation.
Gene promoter methylation in mouse lung
The preceding sections have already demonstrated how methylation studies in animal models have been used to provide insight into elucidating the timing for gene inactivation during tumor development and the effect of defined exposures on methylation prevalence in tumors. Historically, the mouse has been used as a model for evaluating chemopreventive agents ( 43 ). The identification of carcinogens that induce lung tumors through genetic and epigenetic alterations also seen in human adenocarcinomas would provide improved models to test new chemoprevention strategies. Mutation of the K-ras gene is a common alteration in most murine lung tumors induced by different chemical carcinogens and is found in ∼35% of human adenocarcinomas ( 1 , 33 , 34 ). In contrast, mutation of the p53 gene is rarely seen in mouse lung adenocarcinomas, but occurs in ∼40% of human tumors of this histology type ( 2 ). The mechanism for inactivation of the p16 gene in mouse lung differs by carcinogen exposure. Lung tumors induced by NNK in the A/J mice show diminished expression of the p16 gene consistent with allelic loss, but lack methylation ( 44 ). The p16 gene is deleted in most cell lines derived from NNK-induced tumors ( 44 , 45 ). In contrast, bisulfite sequencing of the p16 promoter in lung tumors induced by methylene chloride or aflatoxin, shows partial-to-complete methylation of CpG sites in 70–80% of tumors ( 46 , 47 ; Table II ). Recently, we have examined methylation of the DAP-K and RAR-β genes in lung tumors induced in various mouse strains by cigarette smoke, NNK, methylene chloride or vinyl carbamate ( 18 , 19 ). Methylation of DAP-K was detected in 40–60% of tumors associated with these exposures ( Table II ). In contrast, most cigarette smoke- and NNK-induced tumors harbored methylated RAR-β alleles, while methylation of this gene was seen in ∼50% of methylene chloride and vinyl carbamate tumors. Thus, these mouse lung tumors largely reflect what is observed in human adenocarcinoma with respect to prevalence for inactivation of the DAP-K and RAR-β genes by promoter hypermethylation ( 48 , 49 ). The utility of this finding may lie in the ability to use these two genes as biomarkers in the evaluation of the effectiveness of agents that affect cytosine methylation and histone deacetylation to cause gene reexpression in lung lesions. The reexpression of genes silenced during premalignancy by promoter hypermethylation has emerged as a new strategy for chemoprevention.
Gene promoter methylation in murine lung tumors
| Mouse strain | Exposure | Gene | Methylation frequency (%) |
|---|---|---|---|
| A/J | NNK | p16 | 0/15 (0) a |
| AC3F1 | Aflatoxin | p16 | 51/61 (88) b |
| B6C3F1 | Methylene chloride | p16 | 12/17 (70) b |
| A/J | NNK | DAP-K | 13/25 (52) c |
| B6C3F1 | Cigarette smoke | DAP-K | 9/21 (43) c |
| B6C3F1 | Methylene chloride | DAP-K | 9/18 (50) c |
| C3A | Vinyl chloride | DAP-K | 9/15 (60) c |
| A/J | NNK | RAR-β | 34/40 (85) c |
| B6C3F1 | Cigarette smoke | RAR-β | 18/20 (90) c |
| B6C3F1 | Methylene chloride | RAR-β | 10/18 (56) c,d |
| C3A | Vinyl chloride | RAR-β | 9/15 (60) c,d |
| Mouse strain | Exposure | Gene | Methylation frequency (%) |
|---|---|---|---|
| A/J | NNK | p16 | 0/15 (0) a |
| AC3F1 | Aflatoxin | p16 | 51/61 (88) b |
| B6C3F1 | Methylene chloride | p16 | 12/17 (70) b |
| A/J | NNK | DAP-K | 13/25 (52) c |
| B6C3F1 | Cigarette smoke | DAP-K | 9/21 (43) c |
| B6C3F1 | Methylene chloride | DAP-K | 9/18 (50) c |
| C3A | Vinyl chloride | DAP-K | 9/15 (60) c |
| A/J | NNK | RAR-β | 34/40 (85) c |
| B6C3F1 | Cigarette smoke | RAR-β | 18/20 (90) c |
| B6C3F1 | Methylene chloride | RAR-β | 10/18 (56) c,d |
| C3A | Vinyl chloride | RAR-β | 9/15 (60) c,d |
Methylation assessed by Southern blot.
Methylation assessed by bisulfite sequencing.
Methylation assessed by methylation specific PCR.
P < 0.05 compared with NNK and cigarette smoke.
Gene promoter methylation in murine lung tumors
| Mouse strain | Exposure | Gene | Methylation frequency (%) |
|---|---|---|---|
| A/J | NNK | p16 | 0/15 (0) a |
| AC3F1 | Aflatoxin | p16 | 51/61 (88) b |
| B6C3F1 | Methylene chloride | p16 | 12/17 (70) b |
| A/J | NNK | DAP-K | 13/25 (52) c |
| B6C3F1 | Cigarette smoke | DAP-K | 9/21 (43) c |
| B6C3F1 | Methylene chloride | DAP-K | 9/18 (50) c |
| C3A | Vinyl chloride | DAP-K | 9/15 (60) c |
| A/J | NNK | RAR-β | 34/40 (85) c |
| B6C3F1 | Cigarette smoke | RAR-β | 18/20 (90) c |
| B6C3F1 | Methylene chloride | RAR-β | 10/18 (56) c,d |
| C3A | Vinyl chloride | RAR-β | 9/15 (60) c,d |
| Mouse strain | Exposure | Gene | Methylation frequency (%) |
|---|---|---|---|
| A/J | NNK | p16 | 0/15 (0) a |
| AC3F1 | Aflatoxin | p16 | 51/61 (88) b |
| B6C3F1 | Methylene chloride | p16 | 12/17 (70) b |
| A/J | NNK | DAP-K | 13/25 (52) c |
| B6C3F1 | Cigarette smoke | DAP-K | 9/21 (43) c |
| B6C3F1 | Methylene chloride | DAP-K | 9/18 (50) c |
| C3A | Vinyl chloride | DAP-K | 9/15 (60) c |
| A/J | NNK | RAR-β | 34/40 (85) c |
| B6C3F1 | Cigarette smoke | RAR-β | 18/20 (90) c |
| B6C3F1 | Methylene chloride | RAR-β | 10/18 (56) c,d |
| C3A | Vinyl chloride | RAR-β | 9/15 (60) c,d |
Methylation assessed by Southern blot.
Methylation assessed by bisulfite sequencing.
Methylation assessed by methylation specific PCR.
P < 0.05 compared with NNK and cigarette smoke.
Effect of modulating promoter methylation on cancer
Gene silencing through hypermethylation is mediated by a series of events that include methylation of cytosines within the gene promoter and the establishment of heterochromatin in which the histone tails are modified through effects on acetylation, phosphorylation, methylation and ubiquitylation ( 16 , 50 ). The cytosine DNA-methyltransferase ( DNMT ) genes play a critical role in the establishment of the transcriptionally repressive complex. They function as de novo methylases to affect the methylation status of normally unmethylated CpG sites and to recruit histone deacetylases to chromatin ( 51 – 55 ). Treatment of cells with the demethylating agent 5-aza-2′-deoxyazacytidine (DAC) leads to reexpression of genes silenced by promoter methylation ( 16 ). DAC inhibits DNA methylation by reducing the DNMT enzymatic activity through the formation of a stable complex between the enzyme and the DAC-substituted DNA ( 56 ). One difficulty in using demethylating agents such as DAC in vivo is the ability to achieve pharmacologically active doses without systemic toxicity. A more effective approach for modulating aberrant promoter methylation therapeutically was endorsed through a study by Cameron et al . ( 57 ). These investigators observed that treatment of colorectal tumor cell lines with trichostatin A, an inhibitor of histone deacetylation, did not transcriptionally reactivate genes silenced by promoter hypermethylation. However, when the same cells were exposed to trichostatin A and a low dose of DAC by itself results in minimal gene reexpression, synergistic reexpression was seen with the two-drug combination.
We extended these studies to address in vivo if a short-term treatment with low dose DAC combined with the HDAC inhibitor sodium phenylbutyrate can prevent lung tumor development initiated by NNK in the A/J mouse ( 58 ). The treatment protocol for these studies is shown in Figure 3A . Short-term treatment of mice with DAC alone decreased the incidence of neoplasms by 30% ( Figure 3B ). Importantly, when DAC was combined with sodium phenylbutyrate, lung tumor development was significantly reduced by >50%; no effect was seen with phenylbutyrate alone. These studies demonstrate in a pure prevention model where the treatment occurs after the carcinogen exposure, but before the development of premalignant lesions [seen ∼14 weeks after NNK treatment ( 59 )], inhibition of aberrant promoter hypermethylation can block lung cancer development. The next step in validating this approach to cancer control is to determine the efficacy of inhibitors of methylation and histone deacetylation on the progression of premalignant lesions present in the A/J mouse lung. This scenario closely mimics the situation faced with the chronic, lifetime smoker who has developed field cancerization throughout his/her lungs. An additional challenge to this chemopreventive approach will be the necessity in using demethylating agents other than DAC, since this compound is a potential mutagen and carcinogen ( 60 , 61 ). In this regard, other agents and small molecules that target the DNMTs , such as zebularine and hydralazine, could prove an effective alternative for use in cancer prevention ( 62 , 63 ). In addition, l -selenomethionine, a nutrient demonstrated to reduce by half the incidence of expected lung cancer, may act in part through inhibition of DNMTs ( 64 , 65 ). Finally, the tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNMTs and reactivates genes silenced by methylation in cancer cell lines ( 66 ). Thus, there is a wealth of alternative compounds that affect DNMTs as well as other inhibitors of histone deacetylation such as valproic acid, which should be tested for efficacy in preventing lung tumor progression in the mouse lung model. Reversing methylation of genes such as DAP-K and RAR-β in hyperplasias and adenomas within the mouse lung, will be a valuable biomarker for establishing the efficacy of these different preventive approaches. Ultimately, one would envision the management of premalignant lung disease through approaches used today to control asthma. A cocktail of agents, some that modulate methylation and histone deacetylation and others affecting specific pathways, are delivered directly to the lungs of current or former smokers via an inhaler ( 67 ). Aberrant promoter hypermethylation clearly plays a prominent role in cancer development, but could also prove to be the key target to reverse or prevent malignant transformation in the aerodigestive tract.
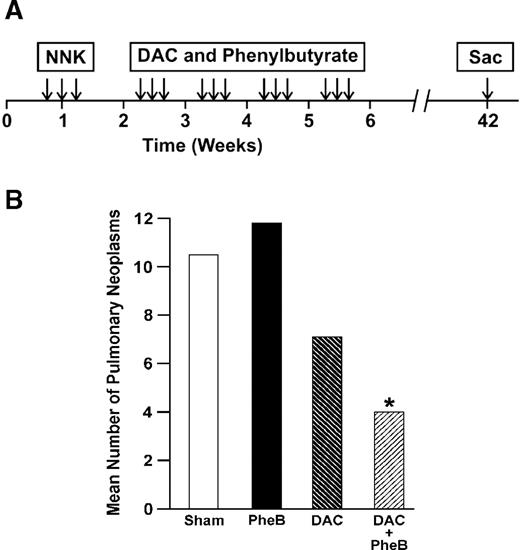
( A ) The timing and duration of treatment of mice with NNK, DAC and phenylbutyrate. This line drawing depicts the order and duration for treatment of A/J mice with NNK followed by the preventive agents DAC and phenylbutyrate. ( B ) Effect of DAC and phenylbutyrate treatment on the mean number of pulmonary neoplasms. The number of pulmonary lesions was decreased significantly ( *P < 0.05) in mice treated with the combination of DAC + sodium phenylbutyrate.
Conflict of Interest Statement
The author is a consultant to Oncomethylome. Under a licensing agreement between Lovelace Respiratory Research Institute and Oncomethylome, nested methylation-specific PCR was licensed to Oncomethylome, and the author is entitled to a share of the royalties received by the institute from sales of the licensed technology. The institute, in accordance with its conflict-of-interest policies, is managing the terms of these arrangements.
The author would like to thank all his collaborators who participated in the studies acknowledged in this commentary. Work described from the author's laboratory was supported by ES088801, P30-ES-012072, CA70190, P50CA58184 and DE-FC03-98EH98028.
References
Rodenhuis,S. (
Greenblatt,M.S., Bennett,W.P., Hollstein,M. and Harris,C.C. (
Merlo,A., Gabrielson,E., Askin,F. and Sidransky,D. (
Kamb,A., Gruis,N.A., Weaver-Feldhaus,J., Liu,Q., Harshman,K., Tavtigian,S.V., Stockert,E., Day,R.S.III, Johnson,B.E. and Skolnick,M.H. (
Merlo,A., Herman,J.G., Mao,L., Lee,D.J., Gabrielson,E., Burger,P.C., Baylin,S.B. and Sidransky,D. (
Zöchbauer-Müller,S., Fong,K.M., Virmani,A.K., Geradts,J., Gazdar,A.F. and Minna,J.D. (
Kim,D.-H., Nelson,H.H., Wiencke,J.K., Zheng,S., Christiani,D.C., Waom,J.C., Mark,E.J. and Kelsey,K.T. (
Belinsky,S.A., Nikula,K.J., Palmisano,W.A., Michels,R., Saccomanno,G., Gabrielson,E., Baylin,S.B. and Herman,J.G. (
Divine,K.K., Pulling,L.C., Marron-Terada,P.G., Kang,T., Schwartz,A.G., Bocklage,T.J., Coons,T.A., Gilliland,F.D. and Belinsky,S.A. (
Lukas,J., Parry,D. Aagaard,L., Mann,D.J., Bartkova,J., Strauss,M. Peters,G. and Bartek,J. (
Tsou,J.A., Hagen,J.A., Carpenter,C.L. and Laird-Offringa,I.A. (
Belinsky,S.A. (
Baylin,S.B. and Herman,J.G. (
Jones,P.A. and Laird,P.W. (
Jones,P.A. and Baylin,S.B. (
Belinsky,S.A., Palmisano,W.A., Gilliland,F.D., Crooks,L.A., Divine,K.K., Winters,S.A., Grimes,M.J., Harms,H.J., Tellez,C.S., Smith,T.M., Moots,P.P., Lechner,J.F., Stidley,C.A. and Crowell,R.E. (
Pulling,L.C., Vuillemenot,B.R., Hutt,J.A., Devereux,T.R. and Belinsky,S.A. (
Vuillemenot,B.R., Pulling,L.C., Palmisano,W.A., Hutt,J.A. and Belinsky,S.A. (
Pulling,L.C., Divine,K.K., Klinge,D.M., Gilliland,F.D., Kang,T., Schwartz,A.G., Bocklage,T.J., Gilliland,F.D. and Belinsky,S.A. (
Wistuba,I.I., Behrens,C., Milchgrub,S., Bryant,D., Hung,J., Minna,J.D. and Gazdar,A.F. (
Hirano,T., Franzé, B., Kato,H., Ebihara,Y. and Auer,G. (
Rhee,C.-S., Sen,M., Lu,D., Wu,C., Leoni,L., Rubin,J., Corr,M. and Carson,D.A. (
Lynch,T.J., Bell,D.W., Sordella,R., Gurubhagavatula,S., Okimoto,R.A., Brannigan,B.W., Harris,P.L., Haserlat,S.M., Supko,J.G., Haluska,F.G., Louis,D.N., Christiani,D.C., Settleman,J. and Haber,D.A. (
Wong,D.J., Foster,S.A., Galloway,D.A. and Reid,B.J. (
Frizelle,S.P., Grim,J., Zhou,J., Gupta,P., Curiel,D.T., Geradts,J. and Kratzke,R.A. (
Rocco,J.W., Li,D., Liggett,W.H.J., Duan,L., Saunders,J.K.J., Sidransky,D. and O'Malley,B.W.J. (
Cohen,O., Feinstein,E. and Kimchi,A. (
Cohen,O., Inbal,J., Kissil,J.L., Raveh,T., Berissi,H., Spivak-Kroizaman,T., Feinstein,E. and Kimchi,A. (
Raveh,T., Droguett,G., Horwitz,M.S., DePinho,R.A. and Kimchi,A. (
Treutin,P.M., Chen,L.I., Buetow,B.S., Zeng,W., Birkebak,T.A., Seewaldt,V.L., Sommer,K.M., Emond,M., Mggio-Price,L. and Swisshelm,K. (
Raffo,P., Emionite,L., Colucci,L., Belmondo,F., Moro,M.G., Bolag,W. and Tom,S. (
You,M., Want,Y., Lineen,A., Stoner,G.D., You,L.A., Maronpot,R.R. and Anderson,M.W. (
Belinsky,S.A., Devereux,T.R., Maronpot,R.R., Stoner,G.D. and Anderson,M.W. (
Denissenko,M.F., Pao,A., Tang,M. and Pfeifer,G.P. (
Issa,J.J., Baylin,S.B. and Belinsky,S.A. (
Belinsky,S.A., Klinge,D.M., Liechty,K.C., March,T.H., Kang,T., Gilliland,F.D., Sotnic,N., Adamova,G., Rusinova,G. and Telnov,V. (
Hande,M.P., Azizova,T.V., Geard,C.R., Burak,L.E., Mitchell,C.R., Khokhryakov,V.F., Vasilenko,E.K. and Brenner,D.J. (
Rountree,M.R., Bachman,K.E., Herman,J.G. and Baylin SB. (
Demple,B., Harrison,L., Wilson,D.M., Bennett,R.A.O., Takagi,T. and Ascione,A.G. (
Parsian,A.J., Funk,M.C., Tao,T.Y. and Hunt,C.R. (
Song,J.Z., Stirzaker,C., Harrison,J., Melki,J.R. and Clark,S.J. (
Malkinson,A.M. (
Belinsky,S.A., Swafford,D.S., Middleton,S.K., Kennedy,C.H. and Tesfaigzi,J. (
Herzog,C.R., Soloff,E.V., McDoniels,A.L., Tyson,F.L., Malkinson,A.M., Haugen-Strano,A., Wiseman,R.W., Anderson,M.W. and You,M. (
Patel,A.C., Anna,C.H., Foley,J.F., Stockton,P.S., Tyson,F.L., Barrett,J.C. and Devereux,T.R. (
Tam,A.S., Devereux,T.R., Patel,A.C., Foley,J.F., Maronpot,R.R. and Massey,T.E. (
Tang,X., Fadlo,R., Khuri, J., Lee,J., Kemp,B.L., Liu,D., Hong,W.K. and Mao,L. (
Virmani,A.K., Rathi,A., Zöchbauer-Müller,S., Sacchi,N., Fukuyama,Y., Bryant,D., Maitra,A., Heda,S., Fong,K.M., Thunnissen,F., Minna,J.D. and Gazdar,A.F. (
Kelly,W.K., O'Connor,O.A. and Marks,P.A. (
Szyf,M., Kaplan,F., Mann,V., Giloh,H., Kedar,E. and Razin,A. (
Robertson,K.D., Uzvolgyi,E., Liang,G., Talmadge,C., Sumegi,J., Gonzales,F.A. and Jones,P.A. (
Okano,M., Bell,D.W., Haber,D.A. and Li,E. (
Roundtree,M.R., Bachman,K.E. and Baylin,S.B. (
Fuks,F., Burgers,W.A., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (
Santi,D.V., Garrett,C.E. and Barr,P.J. (
Cameron,E.E., Bachman,K.E., Myohanen,S., Herman,J.G. and Baylin,S.B. (
Belinsky,S.A., Klinge,D.M., Stidley,C.A., Issa,J.-P., Herman,J.G., March,T.H. and Baylin,S.B. (
Belinsky,S.A., Devereux,T.R., Foley,J.F., Maronpot,R.R. and Anderson,M.W. (
Carr,B.I., Reilly,J.G., Smith,S.S., Winberg,C. and Riggs,A. (
Cunha,K.S., Reguly,M.L., Graf,U. and de Andrade,H.H. (
Cheng,J.C., Weisenberger,D.J., Gonzales,F.A., Liang,G., Xu,G.-L., Hu,Y.-G., Marquez,V.E. and Jones,P.A. (
Segura-Pacheco,B., Trejo-Becerrill,C., Perez-Cardenas,E., Taja-Chayeb,L., Mariscal,I., Chavez,A., Acuña,C., Salazar,A.M., Lizano,M. and Dueñaso-Gonzalez,A. (
Reid,M.E., Duffield-Lillico,A.J., Garland,L., Turnbull,B.W., Clark,L.C. and Marshall,J.R. (
Fiala,E.S., Staretz,M.D., Pandya,G.A., El-Bayoumy,K. and Hamilton,S.R. (
Fang,M.Z., Wang,Y., Ai,N., Hou,Z., Sun,Y., Lu,H., Welsh,W. and Yang,C.S. (

{kind=link}
{kind=link}
{kind=link}