





Abstract
Overstimulation of the orthosympathetic system leads to cardiovascular cell and tissue damage through prolonged activation of β-1-2 adrenergic receptors (BARs). The more recent identification of the third isotype of BAR (B3AR) in cardiac myocytes and endothelial cells with a distinctive coupling and effect on cardiac function and remodelling introduced a new facet to this paradigm. In particular, B3AR is up-regulated in cardiac disease and less prone to homologous desensitization, which may reinforce its influence on the diseased myocardium. Mice with transgenic cardiac-specific expression of the human B3AR are protected from cardiac hypertrophy and fibrosis in response to neurohormonal stimulation. B3AR has also been implicated in cardiac protection after ischaemia-reperfusion and the benefits of exercise on the heart. Many of these salvage mechanisms are mediated by B3AR coupling to nitric oxide synthase (eNOS and nNOS) and downstream cGMP/protein kinase G signalling. Notably, B3AR exerts antioxidant protective effects on these and other signalling elements, which may subserve its protective properties in the setting of chronic heart failure. Additional vasorelaxing properties and paracrine NO-mediated signalling by B3AR in endothelium, together with systemic metabolic effects on beige/brown fat complete the pleiotropic protective properties of this new therapeutic target.
1. Introduction
The orthosympathetic system is a key regulator of cardiovascular function, but its prolonged overactivation leads to cardiac damage that eventually progresses towards heart failure.1 This is the underlying concept justifying the use of β-adrenergic blockers to prevent or treat heart failure.2 However, it mostly relies on the pharmacology of the classical β-1 and β-2-adrenergic receptors (B1-2AR). Recently, the third isotype of β-adrenoceptors (B3AR) has come into focus after its clear identification in human cardiovascular tissues.3 Its particular properties and coupling in heart and vessels, as well as other tissues (e.g. brown fat) shed new light in the catecholaminergic regulation of function and remodelling of cardiovascular tissues, with the potential for new therapeutic modalities.
2. Recap
The B3AR started its ‘life’ as a metabolic receptor. Its cDNA was cloned from a human cDNA library in 1989.4 It was shown to be expressed in adipocytes, where it mediates the adrenergic β-oxidation of fatty acids. It shares the structure of typical GPCRs, with ∼40–50% amino acid sequence similarity with B1- and 2-AR, albeit with some key differences.5,6 In particular, most dissimilarities are clustered in the third intracytoplasmic loop and C-terminal tail, where the B3AR lacks the consensus sequences for phosphorylation by PKA and BARK (or GRK2 in the cardiac myocytes). The latter is known to mediate the β-arrestin recruitment, then internalization of the receptor which is responsible for its homologous desensitization. As a result, the B3AR is less prone (than B1- or B2-ARs) to the progressive loss of coupling and efficacy in response to prolonged stimulation by catecholamines. This has been verified in the specific context of cardiac myocytes, and experiments using chimeric receptors have demonstrated the restoration of agonist-induced desensitization when the B3AR-specific sequences mentioned above have been replaced by those of B2AR, confirming the importance of these different amino acid sequences for the unique properties of B3AR.6
The expression of transcripts of B3AR in biopsies of human hearts was first reported as early as 1996.7 Then, the development of an antibody with validated specificity for the human B3AR allowed the immunodetection of proteins in human atrial and ventricular cardiac myocytes, as well as endothelial cells of the small coronary resistance vessels.8 With the same reagents, we showed that B3AR expression is increased in human cardiac tissue from heart failure patients, in whom B1-2 AR are known to be decreased and desensitized.9 This first established an unbalanced expression of the three isotypes in cardiac disease. Subsequently, others showed an up-regulation of cardiac B3AR in diabetic hearts in several animal models.10,11 Besides the heart, B3AR are expressed in smooth muscle cells of the urinary bladder, where they mediate relaxation of the detrusor muscle, thereby improving the filling capacity of the bladder. This justified the development of specific B3AR agonists currently used in the clinic for the treatment of overactive bladder disease.12
3. B3AR coupling and signalling in cardiovascular tissues
The coupling of B3AR to intracellular signalling effectors varies between tissues and cell types. In the vasculature, the expression of B3AR itself is different between vascular beds, vessels of different sizes, and species. In some vessels, B3AR are expressed in both vascular smooth muscle cells and endothelial cells, or in endothelium only. Although B3AR activation generally produces vasorelaxation (albeit this may depend on the pre-constricting agent used), the coupling of B3AR to intracellular effectors also varies according to the cell type. In canine pulmonary arteries, the B3AR vasodilating effect is independent from the endothelium but coupled to an increase in cAMP levels,13 akin to the classical cAMP/PKA vasorelaxing effects observed with B2AR in vascular smooth muscle cells. Activation of B3AR in endothelial cells produces an endothelium-dependent relaxation in aortic and resistance vessels from many animal species,14 both through NOS-dependent and -independent mechanisms. In bovine aortic endothelial cells, B3AR promoted eNOS activation through phosphorylation on Ser1179 and de-phosphorylation on the inhibitory phosphoresidues Ser116 and Thr497; B3AR signalling to eNOS was dependent on the enzyme's association to Rac1, which also induced endothelial cell migration in vitro.15 In human coronary resistance vessels, we showed that the B3AR-induced relaxation is mediated by both NO- and EDHF-dependent responses.8 Although the definitive identity of EDHF remains unclear, one proposed mediator is extracellular K+ that would drive hyperpolarization through activation of the Na+-K+-ATPase.16 This is interesting in view of the recent demonstration that B3AR protects the beta1 subunit of NaKATPase from inactivation by oxidative S-glutathionylation, thereby maintaining the pump activity in the face of oxidant stress, as occurring, e.g. in diabetes.17 This would be in line with antioxidant effects of B3AR, as also observed by us in a setting of acute ischaemia.18 Such property would contribute to maintain both NO and EDHF vasodilatory responses in disease, as well as important paracrine protective signalling (e.g. through NO) to the underlying parenchymal cardiac tissue (Figure 1) .
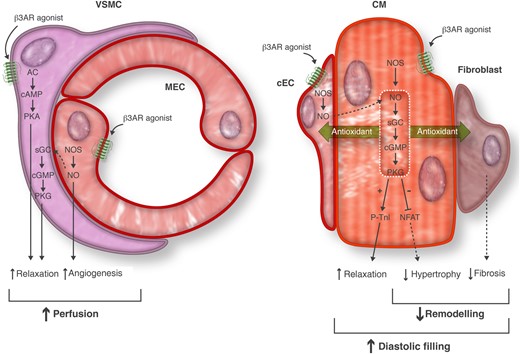
Cell-specific pathways for B3AR signalling. B3AR agonists (or endogenous catecholamines) activate B3AR in cardiac myocytes (CM), cardiac microvascular (capillary) endothelial cells (cEC), macrovascular endothelial cells (MEC), and vascular smooth muscle cells (VSMC) from coronary arteries. In CM and ECs, B3AR activate NOS and downstream soluble guanylyl cyclase (sGC)/cyclic GMP (cGMP)/protein kinase G (PKG) pathway producing autocrine (CM) or paracrine (ECs) relaxation of cardiac and vascular muscle, respectively. In some vessel beds, B3AR also couple to Adenylyl Cyclase (AC)/cyclic AMP (cAMP)/protein kinase A (PKA) to directly relax vessels. In addition to NO-induced pro-angiogenic effects in endothelial cells, these vasorelaxing effects contribute to improved myocardial perfusion. In CM, B3AR decrease hypertrophy, which, together with antioxidant effects in CM and neighbouring cells such as fibroblasts (fib), prevents hypertrophic and fibrotic remodelling, thereby preserving diastolic filling.
Similar coupling of B3AR to NO production was observed in human cardiac biopsies.19 At the cellular level, although B3AR are clearly expressed in human cardiac myocytes, the expression of the receptor varies from species to species, with low expression in mice, but higher in rats, rabbits, or dogs.20 It is important to remember, though, that expression increases with disease.9 In human ventricular biopsies, the B3AR-induced production of NO was sensitive to pertussis toxin, suggesting B3AR coupling to G-alpha-i proteins, and coupled to NOS-dependent production of cGMP.19 We recently used a transgenic mouse model expressing the human B3AR specifically in cardiac myocytes, at levels driving a functional response similar to that observed in human biopsies. We showed that the receptor co-localized with caveolin3 and eNOS and nNOS in caveolae-enriched membrane fractions, with little change of localization in remodelling (hypertrophic), non-failing hearts. Others have shown that B3AR activates nNOS through phosphorylation on Ser1412 and de-phosphorylation of Ser847 in rat neonatal cardiac myocytes; of note, as in human biopsies, these B3AR-induced post-translational modifications were also sensitive to G-alpha-i/o inhibition.21 By cross-breeding our cardiac-specific B3AR transgenic mice with other transgenics expressing a cGMP-sensitive FRET sensor, we demonstrated coupling of the B3AR to cGMP in cardiac myocytes (which was absent in B3AR KO mice).22 The signal was inhibited by the soluble Guanylyl Cyclase inhibitor, ODQ, pointing to a B3AR/NOS/sGC/cGMP pathway. Whether B3AR can couple to G-alpha-s (and Adenylyl Cyclase/cAMP) in addition to G-alpha-i (as does B2AR in some conditions) is still unresolved, but may depend on the spatial confinement of B3AR together with effectors in specific subcellular compartments. Work is ongoing to identify the coupling to cAMP (and cGMP) using membrane-targeted FRET sensors, as well as the regulation of B3AR-stimulated cyclic nucleotides by different isoforms of PDEs, in normal and remodelling hearts. Downstream cGMP, we found that B3AR activates PKG-dependent signalling that results in inhibition of hypertrophy.22 Among other targets, Titin or troponin-I (TnI) phosphorylation would result in decreased myofilament calcium sensitivity23 and improved diastolic relaxation that would promote LV filling.
The coupling of the cardiac myocyte B3AR to electrical currents may vary in atrial vs. ventricular cells. Most studies have examined the regulation of Ica-L, which is increased by B3AR agonists in human atrial cells ex vivo,24 whereas the response is less prominent in ventricular cells.25 Caution should be used, though in extrapolating data using high concentration of agonists in vitro with imperfect selectivity (and potential off-target effects on B1-2 ARs).
4. From coupling to function…
Early studies of the effects of B3AR on contraction force were performed in isolated cardiac muscle specimens with uncontrolled preloading conditions and using high doses of agonists. Under these circumstances, BRL37344 (up to 1 µM) produced a decrease of contraction force in unloaded human cardiac biopsies.7 A similar effect was reproduced in identical models from several mammalian and even vertebrate species.20,26 A distinctive property of B3AR is to oppose the classical positive inotropic effect of B1-2AR, thereby acting as a countervailing ‘brake’ to prevent adrenergic overactivation.7,9 This has been verified in cardiac preparations by several groups,10,27,28 albeit with some variability depending on the species used (and expression of B3AR, as mentioned above). B3AR may also regulate cardiac muscle relaxation,29 possibly through cGMP/PKG-dependent phosphorylation (see above) from NO within cardiac myocytes or paracrinally released from cardiac endothelium.
Subsequently, several groups examined the effect of B3AR agonists in vivo in healthy large animals or models of heart failure, with somewhat divergent results. Although some conclude that there is a negative inotropic effect (mainly in HF models),27,30 others do not.31,32 The reasons for discrepancy probably include the type and dose of agonist used (with high doses of unspecific agonists producing opposing positive inotropic effects) and control for reflex orthosympathetic reaction to intense peripheral vasodilatation.31 One study concluded that there is a small negative inotropic effect in healthy sheep, but an improvement of inotropic parameters in sheep with heart failure.32 This was attributed to preservation of Na+-K+-ATPase function in failing cardiac myocytes, that would decrease intracellular Na+ content. Interestingly, reductions in Na+ content and late Na+ current were otherwise proposed to improve relaxation, which would be particularly helpful in heart failure with preserved ejection fraction (HFpEF).33
In addition to the direct regulation of myocyte EC coupling, B3AR may regulate cardiac function through paracrine effects from the coronary endothelium. As B3AR release NO there, one may anticipate effects similar to those of other NO-releasing agonists, e.g. on relaxation.34 B3AR are also expressed in the pulmonary arterial vasculature in some species,13,35 which would contribute to attenuate pulmonary vasoconstriction and reduce pre-capillary pulmonary hypertension.
5. …From function to remodelling
These direct and indirect mechanisms concur to modulate chronic myocardial remodelling. One important aspect is cardiac myocyte hypertrophy. Using the cardiac-specific transgenic model mentioned above, we showed that B3AR attenuate cardiac myocyte hypertrophy in response to continuous or repetitive infusion of isoproterenol or angiotensin II.22 This was evident from a reduction of morphometric indexes (LV/TL ex vivo, echocardiography in vivo) as well as reduced myocyte transverse area (by histological analysis), in parallel with reduced re-expression of the fetal gene program typically accompanying hypertrophy. To precisely analyse the affected signalling in cardiac myocytes, we developed a model of rat neonatal cardiac myocytes with adenoviral expression of the human B3AR (or GFP control) in which we showed a reduction of hypertrophy in response to three different agonists (phenylephrine, endothelin-1, isoproterenol). Using NOS inhibitors, we observed that this anti-hypertrophic effect of B3AR was NO-dependent, as NOS inhibition abrogated the protection in vitro and in vivo. Mechanistically, we showed that one of the potential targets of the B3AR was Nuclear Factor of Activated T-cells (NFAT), the transcriptional activity of which was attenuated upon B3AR expression.22
B3AR expression also greatly reduced myocardial interstitial fibrosis in response to isoproterenol and angiotensin II infusions, in the absence of changes in myocyte death or apoptosis. As the receptor is exclusively expressed in cardiac myocytes in our transgenic model, this suggested a regulation by B3AR of paracrine signalling to neighbouring cells. Indeed, we could model this in vitro with a superfusion assay and observed inhibition of fibroblasts activation upon B3AR expression in cardiac myocytes (Figure 1).
Similar protective effects of B3AR were reported with preferential B3AR agonists (e.g. BRL37344) in mice submitted to transaortic constriction (TAC), with decreased hypertrophy and preserved LV function.36 The implication of NOS (i.e. specifically nNOS) in the protection was further supported from the loss of protection in NOS1-KO mice. More recently, the role of B3AR was examined in the protection afforded by B1AR blockade in a dog model of heart failure due to volume overload (mitral regurgitation).37 The study showed that metoprolol promoted the up-regulation of B3AR expression, its interaction with and activation of nNOS, followed by downstream cGMP production in specific membrane microdomains. This was attributed to protection of soluble guanylyl cyclase from oxidation under B1AR blockade. Therefore, some of the cardioprotective effects of B1AR blockade may well result from enhanced expression and coupling of B3AR to NO/cGMP in the remodelled heart.
These properties may extend beyond hypertrophic remodelling. Indeed, early work had implicated B3AR coupling to eNOS and nNOS in the beneficial effects of exercise on the heart,38 as well as in myocardial protection in the setting of ischaemia/reperfusion in mice.39 The latter was recently replicated in a large (porcine) animal model, in which pre-perfusion of the B3AR agonist, BRL37344 reduced reperfusion damage and improved long-term LV function. Additional cellular work pointed to Akt/NO-mediated prevention of mitochondrial permeability transition pore (mPTP) opening as one underlying mechanism.40 Finally, the B3AR agonist properties of nebivolol conferred superior efficacy over metoprolol in protecting the infarcted mouse heart from adverse remodelling; this was associated with better preserved endothelial dependent vasodilatation, progenitor cells mobilization, and functional recovery.18 Mechanistically, nebivolol exerted a potent inhibition of NADPH oxidase activity and superoxide production that, in endothelial cells, was not sensitive to B1(or-2)AR blockade, but abrogated by full B1-2-3 blockade, pointing to B3AR activation. Interestingly, we had also shown that B3AR agonist properties of nebivolol promote neoangiogenesis,41 which could favour myocardial revascularization.
All these protective effects at the myocardial level are probably reinforced from indirect effects in peripheral cells/tissues, i.e. through coronary vasodilatation via B3AR-induced endothelial dependent relaxation (see above), as well as paracrine release of NO and its effects to improve LV relaxation. Moreover, the antioxidant effects of B3AR signalling may preserve the endothelium of the microvasculature from oxidative activation and the ensuing recruitment of monocytes initiating subendothelial inflammation at the core of sustained endothelial dysfunction. Whether this will prevent chronic development of vascular atherosclerosis or chronic development of diastolic dysfunction initiating HFpEF42 will have to be tested with long-term interventions.
One remote target is now gaining renewed attention, i.e. beige/brown fat. Indeed, although the role of brown fat on thermogenesis and energy expenditure is clearly established in rodent models, the unequivocal identification of residual brown fat in adult humans was only provided recently.43–45 Moreover, the demonstration that specific adipose cell lineages were amenable to ‘browning’ under adrenergic stimulation in humans, in a proportion that may significantly regulate energy expenditure, opened new possibilities for the treatment of human diseases with caloric overload, i.e. obesity and metabolic syndrome.46 Indeed, B3AR are expressed in human beige/brown adipocytes and mediate adrenergic fatty acids β-oxidation there.47 Therefore, systemic administration of B3AR agonists may confer additional cardiovascular protection by improving the metabolic status, i.e. decreased triglyceridemia, decreased obesity, with indirect effects on insulin sensitivity and hypertension. Again, small clinical trials with nebivolol (which has ancillary B3AR agonist properties41 hint at additional metabolic protective effects compared with ‘pure’ B1AR blockers;48Figure 2). As for other β-blockers commonly used post-myocardial infarction or in chronic heart failure, such as metoprolol, bisoprolol, and carvedilol, their affinity and inhibitory properties at native human cardiac B3AR have not been thoroughly characterized; however, the comparison of their pharmacologic properties using recombinant human B3AR in cellular heterologous expression systems showed that metoprolol and bisoprolol have a high affinity for B1AR, as expected, with no binding to B3AR, whereas carvedilol non-selectively binds B1 and B2AR, and has 100-fold lower affinity for B3AR; none of the three molecules exhibited significant agonism or inverse agonism at B3AR.49
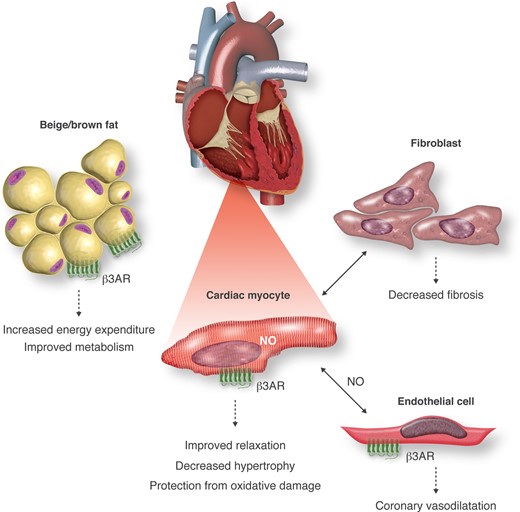
Cardiac protection through autocrine, paracrine, and systemic effects of B3AR signalling. B3AR are expressed in cardiac myocytes, endothelial cells, and adipocytes. B3AR activation in myocytes produces nitric oxide that, together with protection from oxidant stress, contributes to protection from hypertrophy, ischaemia/reperfusion, and oxidative damage. This is reinforced from paracrine NO produced following activation of endothelial B3AR, together with vasorelaxation, angiogenesis, and improved coronary perfusion. Cardiac B3AR activation also modifies paracrine signalling to fibroblasts resulting in decreased fibrosis. Sympathetic activation of B3AR in adipose tissue promotes ‘browning’ of adipocytes with increased energy expenditure (through increased lipolysis and non-shivering thermogenesis) that contributes further cardiovascular benefit from improved metabolic risk factors.
Based on all (pre-)clinical evidence, as summarized above, we designed a Phase IIb, prospective, placebo-controlled randomized trial testing the effect of mirabegron, a new specific B3AR agonist to prevent or reverse the LV hypertrophic remodelling of patients with structural heart disease (Stage B, AHA; measured by cardiac MRI) who are at risk of developing heart failure with preserved ejection fraction (HFpEF); in addition, the effect on LV filling properties (E/E′, by Doppler echocardiography) will be tested as co-primary end point. Secondary end points will include sequential measurements of cardiac fibrosis, exercise tolerance, metabolic parameters, as well as specific biomarkers reflective of myocardial remodelling and function. Mirabegron has been introduced and approved in Europe, USA, and Japan for the treatment of overactive bladder disease, a urological condition characterized by frequent micturition, where the drug was shown to improve bladder filling properties through its myorelaxant properties mediated by activation of B3AR in the detrusor muscle.12 Therefore, our study is based on a principle of ‘drug repurposing’ for a new cardiovascular indication. This investigator-initiated, academic trial involves 12 European partners with nine clinical recruitment sites and is funded by the European Commission under the Horizon 2020 programme. It also contains two substudies that will specifically study the effect of mirabegron on endothelial function (both by digital microtonometry and direct measurement of circulating NO in erythrocytes,50 and on the quantity and activity of beige/brown fat, measured by 18FDG-PET combined with CT scan). The study officially started on 1 May 2015 and is expected to end in the Spring of 2020 (ISRCTN 65055502).
6. Conclusion
After its cDNA cloning in human fat, the B3AR raised enthusiastic hopes for a new cure of obesity, that were prematurely damped from early disappointing results with poorly specific agonists and improper trial designs. The receptor slowly gained recognition in the cardiovascular field after its identification in the human myocardium and description of its uncharacteristic adrenergic properties, i.e. antipathetic to classical B1-2AR. A number of cellular and animal models now support its protective properties at least at early stages of haemodynamic or neurohormonal stresses. Adverse effects on LV function have been observed in some animal models at late stages of heart failure, usually with high doses of agonists, although positive effects on failing hearts have also been observed. Some of this protection involves coupling to NO/cGMP along with antioxidant properties in cardiac myocytes and endothelial cells. In addition to direct protection of cardiac myocytes from hypertrophic remodelling or post-ischaemic damage, B3ARs modulate paracrine signalling from myocytes to surrounding cells (e.g. fibroblasts) and reciprocally, from endothelial cells to vascular and cardiac myocytes. This results in reduction of interstitial fibrosis, increased neoangiogenesis, and coronary vasodilatation, all of which indirectly would favour myocardial perfusion and LV filling, thereby preserving cardiac function. This preclinical evidence was an incentive to launch a clinical trial testing the ‘repurposing’ of mirabegron, a new specific agonist of human B3AR, in patients with structural (hypertrophic) heart disease at risk of developing HFpEF. The trial will include a sub-study on endothelial function, as well as on the (re)activation of beige/brown fat and its possible benefit by reducing metabolic risk factors. Should the results confirm the hypothesis, the B3AR may revive the early enthusiasm for its cardioprotective properties beyond metabolism.
Conflict of interest: none declared.

{kind=link}
{kind=link}