





Abstract
Autophagy is essential to maintain tissue homeostasis, particularly in long-lived cells such as cardiomyocytes. Whereas many studies support the importance of autophagy in the mechanisms underlying obesity-related cardiac dysfunction, the role of autophagy in cardiac lipid metabolism remains unclear. In the heart, lipotoxicity is exacerbated by cardiac lipoprotein lipase (LPL), which mediates accumulation of fatty acids to the heart through intravascular triglyceride (TG) hydrolysis.
In both genetic and dietary models of obesity, we observed a substantial increase in cardiac LPL protein levels without any change in messenger ribonucleic acid (mRNA). This was accompanied by a dramatic down-regulation of autophagy in the heart, as revealed by reduced levels of unc-51 like kinase-1 (ULK1) protein. To further explore the relationship between cardiac LPL and autophagy, we generated cardiomyocyte-specific knockout mice for ulk1 (Myh6-cre/ulk1fl/fl), Lpl (Myh6-cre/Lplfl/fl), and mice with a combined deficiency (Myh6-cre/ulk1fl/flLplfl/fl). Similar to genetic and dietary models of obesity, Myh6-cre/ulk1fl/fl mice had a substantial increase in cardiac LPL levels. When these mice were fed a high-fat diet (HFD), they showed elevated cardiac TG levels and deterioration in heart function. However, with combined deletion of LPL and ULK1 in Myh6-cre/ulk1fl/flLplfl/fl mice, HFD feeding did not lead to alterations in levels of TG or diacylglycerol, or in cardiac function. To further elucidate the role of autophagy in cardiac lipid metabolism, we infused a peptide that enhanced autophagy (D-Tat-beclin1). This effectively lowered LPL levels at the coronary lumen by restoring autophagy in the genetic model of obesity. This decrease in cardiac luminal LPL was associated with a reduction in TG levels and recovery of cardiac function.
These results provide clear evidence of the critical role of modulating cardiac LPL activity through autophagy-mediated proteolytic clearance as a potential novel strategy to overcome obesity-related cardiomyopathy.
1. Introduction
The incidence of obesity is fast increasing, reaching epidemic proportions worldwide. Obesity has been associated with an increased risk of stroke, congestive heart failure, myocardial infarction, and cardiovascular death.1,2 Several different mechanisms linking obesity to cardiovascular disease have been postulated, including the roles of cytokines, insulin resistance, lipids, tension, coagulation, fibrinolysis, inflammation, and epigenetics.1,3–7 Under stress conditions, cardiac muscles are required to maintain self-contraction and pumping ability,8,9 which can be achieved by removal of oxidized, aggregated, and over-phosphorylated proteins and maladaptive organelles.10 These self-preserving processes are collectively referred to as autophagy.10–12 However, the specific role of autophagy in obesity-related cardiac dysfunction remains unclear.
During autophagy, a preautophagosome engulfs cytosolic components and forms an autophagosome, which subsequently fuses with a lysosome, leading to the proteolytic degradation of internal components.13 To date, more than 36 autophagy (Atg)-related proteins have been identified.14 Among these, Atg1 shows strong homology with Caenorhabditis elegans uncoordinated-51 (UNC-51), which has a mammalian homolog known as unc-51 like kinase-1 (ULK1).15,16 Interestingly, ULK1 plays a crucial role in mediating lipid accumulation.17 In the heart, lipotoxicity may cause contractile dysfunction18,19; and lipoprotein lipase (LPL) is one of the important enzymes in cardiac lipid accumulation and cardiomyopathy, particularly under metabolically stressed conditions.20–23
LPL is a member of the lipase family that hydrolyzes triglyceride (TG) in very low-density lipoproteins or chylomicrons at the vascular endothelium.24 The released fatty acids are used to fuel muscles that have high energy demand.25 Despite this essential role of LPL at the coronary luminal surface, endothelial cells are reported not to express LPL messenger ribonucleic acid (mRNA).26 However, a recent report has shown that LPL mRNA expression is present in pure heart endothelial cells at a level of 25% of those present in pure cardiomyocytes.27 Nevertheless, in the heart, the majority of this enzyme is produced by cardiomyocytes and is then transferred to the luminal surface of endothelial cells.28 Several potential mechanisms for this process have been proposed. First, lipase maturation factor 1 produces catalytically active LPL following maturation steps.29,30 Subsequently, phosphorylation of heat shock protein 25 (Hsp25) and protein kinase D (PKD) facilitates myocyte LPL translocation onto the vascular lumen by regulation of the fission of vesicles and actin cytoskeleton reorganization.20 Once bound to the luminal surface, LPL activity can persist through association with an essential cofactor, apolipoprotein C2.31 Alternatively, angiopoietin-like 4 can bind to the active LPL dimer, which favors dissociation into completely inactive LPL monomers.32 Previous studies have demonstrated that higher levels of myocardial TG accumulation and cardiac dysfunction are associated with LPL accumulation in obesity.21,33,34
The objective of this study was to determine the mechanisms by which obesity augments cardiac TG and evokes cardiac dysfunction. We evaluated the role of molecules in autophagy-related proteolytic pathways on cardiac function and LPL accumulation in genetic and dietary obese animal models. Moreover, we used a cell-permeable autophagy-inducing peptide to prevent cardiac dysfunction as a potential therapeutic candidate for obesity-associated cardiomyopathy.
2. Methods
2.1 Mice
B6.V-Lep°b/J, ulk1fl/fl, and Myh6-cre mice were purchased from Jackson Lab. Lplfl/fl mice were provided from UC Davis KOMP Repository. All mice had a C57BL/6 background, and corresponding wild-type (WT) littermate controls were used in experimental protocols. All animal experiments were approved by the Ewha Womans University Animal Care Committee and performed according to National Institutes of Health (NIH) guidelines (Guide for the care and use of laboratory animals).
Ulk1fl/fl mice were mated with Myh6-cre (cre transgene under the control of the cardiac-specific mouse alpha myosin heavy chain promoter) to produce Myh6-cre/ulk1fl/fl. Lplfl/fl mice were mated with Myh6-cre to produce Myh6-cre/Lplfl/fl. Using these mice, we also generated double knockout Myh6-cre/ulk1fl/flLplfl/fl mice. Only male mice aged 6–10 weeks were used in this study, and all were housed in specific pathogen-free conditions.
Mice were genotyped by polymerase chain reaction (PCR) using the following primers: B6.V-Lep°b/J WT allele (5′-TGAGTTTGTCCAAGATGGACC-3′, 5′-GCCATCCAGGCTCTCTGG-3′, and 5′-AATGACCTGGAGAATCTCC-3′, generating 191-bp and 104-bp fragments) or the B6.V-Lep°b/J knockout allele (5′-TGAGTTTGTCCAAGATGGACC-3′, 5′-GCCATCCAGGCTCTCTGG-3′, and 5′-GCAGATGGAGGAGGTCTCA-3′, generating 191-bp and 123-bp fragments); ulk1 WT allele (5′-CAGTTAGGTTCACTGCAGACTTG-3′ and 5′-TTTATCCGTCTTCTGCTATTGG-3′, generating a 291-bp fragment) or the ulk1 floxed allele (5′-CTTGGGTGGAGAGGCTATTC-3′, 5′-AGGTGAGATGACAGGAGATC-3′, generating a 280-bp fragment); Myh6-cre allele (5′-ATGACAGACAGATCCCTCCTATCTCC-3′, 5′-CTCATCACTCGTTGCATCATCGAC-3′, generating a 300-bp fragment); Lpl WT allele (5′-TGCCAGTTCAGCTCTGTGTC-3′ and 5′-ACGCAGAAATAAAGGCAAGC-3′, generating 465-bp or 515-bp fragments). All samples were subjected to the following conditions: 95 °C 2 min (94 °C 20 s, 65 °C 15 s, 68 °C 10 s; 20 cycles), (94 °C 15 s, 50 °C 15 s, 72 °C 10 s; 28 cycles) and 72 °C 3 min.
The 6-week-old male mice were fed either a high-fat diet (HFD; 60% of calories from fat) or a normal-chow diet (ND; 10% of calories from fat) for 28 weeks. To assess the effects of autophagy on cardiac LPL, some mice received D-Tat-scramble or D-Tat-beclin1 (4 μg/min per kg body weight) infusions via a subcutaneously implanted osmotic minipump (Alzet, model 1002) for 3 weeks. Using osmotic pumps, some groups were infused with bafilomycin A1 (10 ng/min per kg body weight) to evaluate light chain 3 (LC3) levels by preventing fusion of autophagosome-lysosome. The mice were anesthetized with 375 mg/kg 2,2,2-tribromoethanol (Avertin, Sigma Chemical Co) by intraperitoneal injection. An osmotic pump containing D-Tat-scramble or D-Tat-beclin1 dissolved in saline solution (0.15 M NaCl) at a concentration calculated to deliver 4 μg/min per kg body weight of drug was inserted into a subcutaneous pocket.
2.2 Plasma measurements
An intraperitoneal glucose tolerance test was performed in 14-h fasted mice by an intraperitoneal injection of glucose (1 g/kg body weight). Following glucose injection, blood samples from the tail vein were collected and blood glucose determined using a glucometer (AccuSoft) and glucose test strips (Accu-Chek; Roche) at varying intervals.20
2.3 Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Heart tissue samples were fixed with formalin for 10 h and washed. Following permeabilization with 0.2% Triton-X 100, deoxyribonucleic acid (DNA) fragmentation staining was performed with a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay kit from abcam Inc. Apoptotic cells were counted using fluorescence microscopy.
2.4 Quantification of myocardial fibrosis
Heart tissue samples were fixed overnight in 4% paraformaldehyde and embedded in paraffin. After serial sectioning of the heart tissue, 5-μm sections were stained with Masson trichrome. Fibrotic (blue-stained) areas within sections were observed using a light microscope. The percentage of fibrotic area was calculated as the summed blue-stained areas divided by total ventricular area.
2.5 Measurement of TG, DAG, ceramide, and fatty acyl-CoA
Cardiac lipids were extracted from the hearts using chloroform/methanol/HCl to measure TG, diacylglycerol (DAG), ceramide, and fatty acyl-coenzyme A (CoA). The extracts were quantified to ensure equal protein loading and incubated with triglyceride enzyme mix and developer for 30 min using a PicoProbe TG fluorometric assay kit (Biovision). Cardiac TG levels were measured at Ex/Em (535/587 nm) in a microplate reader (BioTek-SYNERGY H1). The heart tissue samples were fixed in 10% phosphate-buffered formalin for 10 h. The tissue slides were rinsed in 60% isopropanol for 1 min and stained with filtered Oil Red O solution at 37 °C for 1 min. Positive-staining (red) cells were observed using a light microscope (Olympus). DAG levels were measured using a DAG assay kit from Cell biolabs (MET-5028, USA). Ceramide and fatty acyl-CoA analyses were performed using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) technique using equipment from mass spectrometry portal company (South Korea).
2.6 Echocardiographic assessment
Echocardiography was performed using a Vevo 2100 machine at the Cardiovascular Research Center in Seoul. Mice were anesthetized with 2% isoflurane and maintained with 1.5% isoflurane. Next, depilatory cream was applied to the mouse’s chest and wiped clean to remove all hair. The scanning probe (20 MHz) was used to obtain two-dimensional (2D) images of the parasternal long axis. These 2D images was converted to M-mode.
2.7 Isolated heart perfusion
Mice were anesthetized using 2,2,2-tribromoethanol (375 mg/kg; intraperitoneal injection) and the hearts were carefully excised. After cannulation of the aorta, the hearts were retrogradely perfused with Krebs-Henseleit buffer as previously described.20,35 Briefly, hearts from Lep+/+, Lepob/ob, Myh6-cre, or Myh6-cre/ulk1fl/fl mice were isolated and perfused in the non-recirculating retrograde mode with Krebs buffer or heparin (2 units/mL) containing fatty acid-free bovine serum albumin (BSA) (1%) after a 10-min stabilization period. This concentration of heparin can maximally release cardiac LPL from its heparan sulfate proteoglycan (HSPG) binding sites. Coronary effluents were collected at different time points over 20 min and assayed for LPL activity by measuring the hydrolysis of a fluorescence–LPL substrate emulsion (Biovision Inc).
2.8 Isolation of adult cardiomyocytes, fibroblasts, endothelial, and smooth muscle cells from heart tissue
Ventricular cardiomyocytes were prepared using methods adapted from those described previously.35,36 Briefly, 3-month-old mice were injected with 0.05 mL of 1000 USP/mL heparin for 15 min and then anesthetized using 2,2,2-tribromoethanol (375 mg/kg; intraperitoneal injection). After opening the chest cavity, the heart was quickly excised and perfused using a Langendorff system. After perfusion for 2 min, the heart was digested with 2 mg/mL collagenase II (Cellutron Inc) for 15 min. After sufficient digestion, the ventricles were isolated and gently agitated at 100 rpm for 10 min. The tissues were gently dissociated by repeated pipetting and resuspended in stopping buffer (20% fetal bovine serum [FBS] in Leibovitz Media; Life Technology). To induce calcium tolerance, cells were exposed to increasing calcium concentrations (100 μM, 400 μM, and 900 μM) for 7 min each. The calcium-tolerant myocytes were plated onto laminin-coated (Roche Applied Science) and Matrigel-coated (Corning Inc) culture dishes with 10% FBS in minimum essential medium (MEM) (Cellutron Inc). One hour after plating, the buffer was replaced by serum-free MEM and incubated for 4 h before treatment. In a separate experiment, mouse hearts were perfused with Hank’s buffered salt solution (HSBB) (5 mM KCl, 0.3 mM KH2PO4, 138 mM NaCl, 4 mM NaHCO3, 0.3 mM Na2HPO4 • 7H2O, 5.6 mM D-glucose, and 10 mM HEPES) for 5 min. HBSS was replaced with a warm enzyme solution (1 mg/mL collagenase II). Perfusion fluid was collected at 30-, 60-, and 90-min and centrifuged at 1000 rpm for 10 min. To obtain endothelial cells, samples were incubated with Medium 199-F-12 medium (1:1) with 10% FBS and 2% antibiotics and confirmed with RECA-1 antibody (endothelial cell positive marker). To obtain smooth muscle cells, cardiac tissue was incubated with Dulbecco’s Modified Eagle’s medium (DMEM) with 10% FBS, 10% mouse serum, and 2% antibiotics and confirmed with smooth muscle alpha-actin antibody (smooth muscle positive marker). To obtain fibroblasts, cardiac tissue was digested with collagenase II and trypsin. Cells were incubated in a gelatin-coated dish with 10% FBS-DMEM for 6 days. Cells were confirmed with Thy1 antibody (fibroblast-specific marker). Samples were then prepared for, and analyzed using, real-time PCR and western blot.
2.9 Glucose uptake in primary cultures of cardiomyocytes
Isolated cardiomyocytes were treated with 0.1 μM insulin for 30 min and washed twice with 2 mL of phosphate-buffered saline (PBS) to remove serum and glucose. Cells were then incubated for 30 min in 1 mL of fresh Krebs-Ringer (KR) buffer, and a glucose uptake assay was performed over a period of 30 min after the addition of 0.5 μCi/mL 2-deoxyglucose. The assay was terminated by rapid washes with 1 mL of ice-cold PBS buffer. Cells were disrupted with 1 mL of 0.5 M NaOH for 60 min at 37 °C, and cell-associated radioactivity was determined by scintillation counting. Glucose uptake was expressed as a percentage of basal uptake.
2.10 Autophagosome assay kit
Cardiomyocytes were isolated from hearts and incubated with serum-free MEM. Cells were washed with PBS and incubated with detection reagent from an autophagosome detection kit (MAK138; Sigma-Aldrich), sarcomeric alpha actinin antibody (ab9465) and 4',6-diamidino-2-phenylindole for 15 min. Following three washes, fluorescence intensity was measured at Ex/Em (360/520 nm) using a microplate reader (BioTek-SYNERGY H1) and Zeiss LSM 510 confocal microscope.
2.11 Real-time PCR
Total ribonucleic acid (RNA) was isolated using TRIzol reagent (Invitrogen) from ventricles (50 mg) or plated myocytes (0.4 × 106). The isolated RNA was treated with DNase I (Invitrogen) and then converted to complementary deoxyribonucleic acid (cDNA) using a reverse transcription kit (Clontech). Using LPL (Mm01345523_m1), cluster of differentiation 36 (CD)36 (Mm00432403_m1), glycerol-3-phophate acyltransferase (GPAT) (Mm00833328_m1), diacylglycerol acyltransferase (DGAT) (Mm00515643_m1), and adipose tissue triglyceride lipase (ATGL) (Mm00503040_m1) primer/probe set from Life Technology, real-time PCR was performed with TaqMan PCR Master Mix on an ABI Real time PCR System 7000 (Applied Biosystems). PCR conditions were 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. For each experimental sample, the relative abundance value was normalized to the value derived from the endogenous control (18s ribosomal ribonucleic acid [rRNA]) of the same sample. Relative mRNA levels were quantified using the comparative 2-ΔΔCT method.
2.12 Northern blot of LPL mRNA
A total of 25 ug of RNA was isolated from the ventricles using TRI reagent (Molecular Research Center). After quantification using a Quibit 2.0 fluorometer (Invitrogen), the RNA was separated on a formaldehyde-agarose gel and transferred to a Nylon membrane (Biodyne). A 32P-labeled oligonucleotide probe was produced using LPL cDNA clone (NM_008509; OriGene Technologies, Inc). After prehybridization for 1 h at 48 °C, membranes were hybridized with an internal 32P-labeled probe overnight at 48 °C in hybridization buffer (5% dextran sulfate, 1 mol/l NaCl and 1% sodium dodecyl sulfate [SDS]). Blots were washed and exposed to X-ray film.
2.13 Western blotting
Briefly, heart tissue or cells were isolated and homogenized in ice-cold lysis buffer. After centrifugation at 5000 g for 20 min, the protein content of the supernatant was quantified using a Bradford protein assay. Samples were diluted, boiled with sample loading dye, and 200 μg was used in SDS-polyacrylamide gel electrophoresis (PAGE). After blotting, membranes were blocked in 5% skimmed milk in Tris-buffered saline containing 0.1% Tween-20.20 Membranes were incubated with antisera directed against p62 (1:1000), CD36 (1:1000), ATGL (1:1000), GPAT (1:1000) MLCV2 (1:1000) DGAT (1:1000), Thy1 (1:1000), CD31 (1:1000), phospho-protein kinase D (p-PKD) (ser-744/748; 1:1000), p-Hsp25 (ser-86; 1:1000), total PKD (1:2000), total Hsp25 (1:3000), LPL (1:3000), LC3 I and II (1:1000), phospho-unc-51 like kinase-1 (p-ULK1) (ser-317; 1:1000) total ULK1 (1:3000), total ATG3 (1:3000), total ATG5 (1:3000), total ATG7 (1:3000), total ATG12 (1:3000), beta-actin (1:4000), platelet endothelial cell adhesion molecule-1 (PECAM-1) (1:1500), smooth muscle alpha-actin (1:1500) and then with secondary antibodies (mouse-specific horseradish peroxidase [HRP]-conjugated antibody or rabbit-specific HRP-conjugated antibody). Bands were visualized using an ECL detection kit and quantified by densitometry. The blots were stripped and re-exposed to reveal the housekeeping protein (beta-actin).
2.14 Materials
A Lipoprotein Lipase Activity fluorometric assay kit was obtained from Biovision. Heparin sodium and fatty acid-free BSA were purchased from Sigma Aldrich. Primers for genotyping were obtained from Integrated DNA Technologies. Total p62 (#7695), CD36 (#14347), ATGL (#2439), PKD (#2052), p-PKD (#2054), LC3 (#12741), p-ULK1 (#12753), total ULK1 (#8054), total ATG3 (#3415), total ATG5 (#12994), total ATG7 (#8558), total ATG12 (#4180), and beta-actin (#8457) antibodies were purchased from Cell Signaling Technology. Total Hsp25 (GTX12351) and p-Hsp25 (GTX17938) antibodies were obtained from GeneTex. GPAT (ABS764) was purchased from Millipore Corporation. MLCV2 (NBP1-30249) was purchased from Novus Biologicals. LPL (ab21356), DGAT (ab54037), Thy1 (ab92574), CD31 (ab24590), and smooth-muscle alpha-actin (ab124964) antibody were purchased from Abcam Inc. D-Tat-Beclin-1 was obtained from Calbiochem. The enhanced chemiluminescence detection kit was purchased from Advansta Inc.
2.15 Statistical analysis
Values are means ± standard errors (SEs). The significance of differences was determined by a two-way analysis of variance (ANOVA), or a one-way ANOVA followed by a Bonferroni post-hoc analysis where appropriate. Differences were considered significant when P < 0.05.
3. Results
3.1 Autophagy, TG and contractility in hearts of genetic and dietary obese models
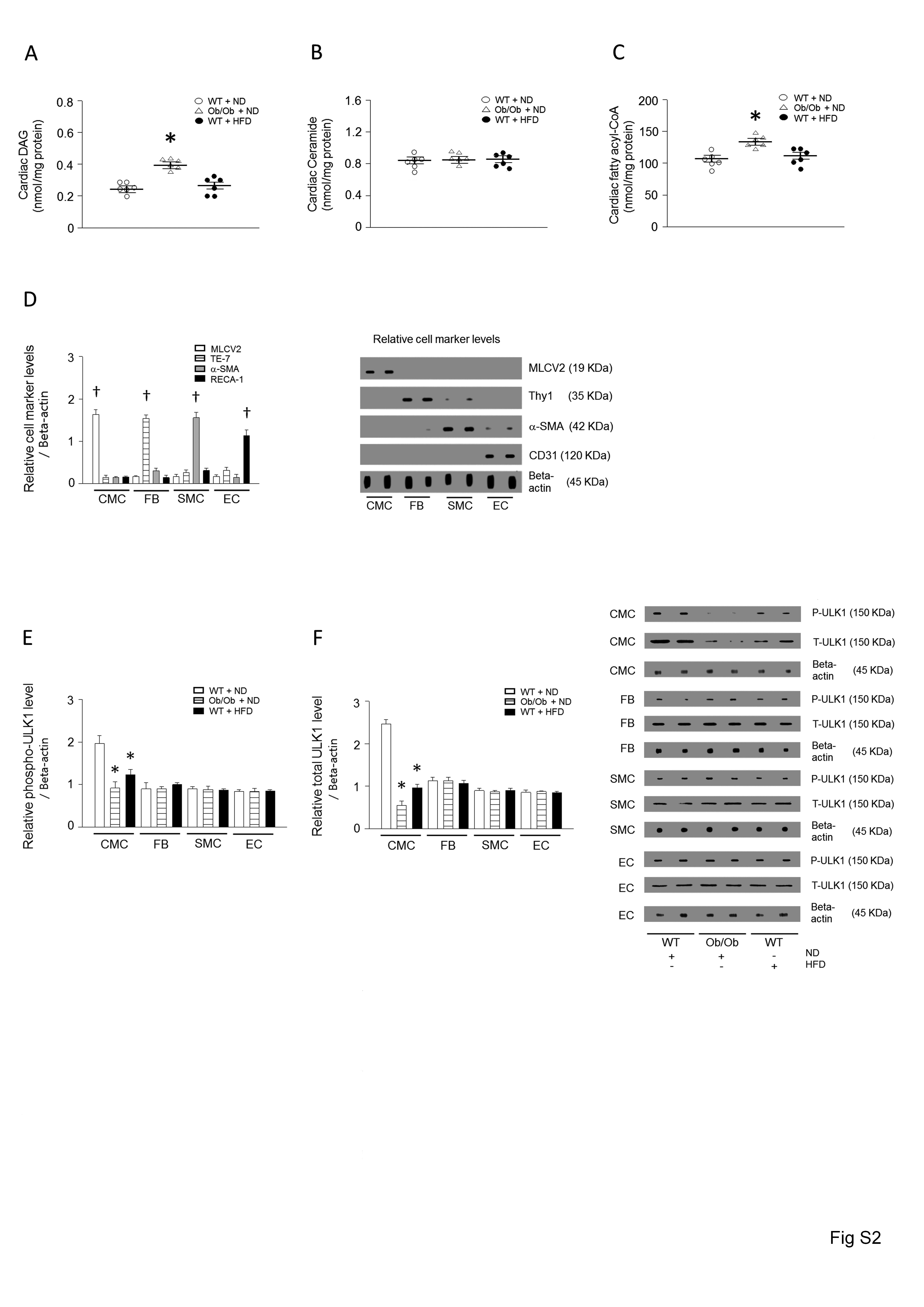
We assessed cardiac function in Ob/Ob (leptin-null) or WT mice after 28 weeks of HFD feeding by echocardiography in anesthetized mice. The hearts of Ob/Ob mice showed decreased fractional shortening (FS) and ejection fraction (EF), and an increase in left ventricular mass compared with the control group (Figure 1A–C, see Supplementary material online, Figure S1A). Furthermore, cardiac lipid accumulation was determined in these mice (Figure 1D, see Supplementary material online, Figure S2A–C). Among the lipid intermediates, DAG, fatty acyl-CoA, and TG were upregulated. Importantly, Ob/Ob mice had an accumulation of cardiac TG two-fold higher than that of the control group (Figure 1D, see Supplementary material online, Figure S3A–B). Although the hearts of HFD-fed WT mice showed only a slight reduction in FS and EF compared to ND-fed mice, they also exhibited a higher cardiac TG accumulation. We next evaluated autophagy in the hearts of Ob/Ob or HFD-fed WT mice by measuring the amount of microtubule-associated protein LC3-II, one of the selective substrates for autophagy.37 In addition, bafilomycin (autophagosome-lysosome fusion inhibitor) was infused to measure autophagic flux in each mouse. Compared with the hearts of control mice, the amount of LC3-II was decreased in the hearts of obese mice (Figure 1E). As autophagy is initiated by activation of Atg-related proteins, we also determined their expression levels38 and, remarkably, found a significant decrease only in ULK1 phosphorylation and protein levels without changes in other Atgs in the obese vs. control mice (Figure 1F–G). However, the ratio of phospho/total protein level was not altered compared to the control animals. This demonstrates that ULK1 expression level, rather than phosphorylation level, was dominantly regulated in both Ob/Ob mice and HFD-fed mice. To determine which cell types are involved in the downregulated ULK1 levels, we isolated multiple cells from the heart. Cell-specific markers were used to show the isolation purity for cardiomyocytes (CMC; MLCV2), fibroblasts (FB; Thy1), smooth muscle cells (SMC; alpha-smooth muscle actin), and endothelial cells (EC; RECA-1) (see Supplementary material online, Figure S2D). Among these cell types, only HFD-fed or Ob/Ob heart-derived cardiomyocytes had altered ULK1 phosphorylation and protein levels (see Supplementary material online, Figure S2E–F).
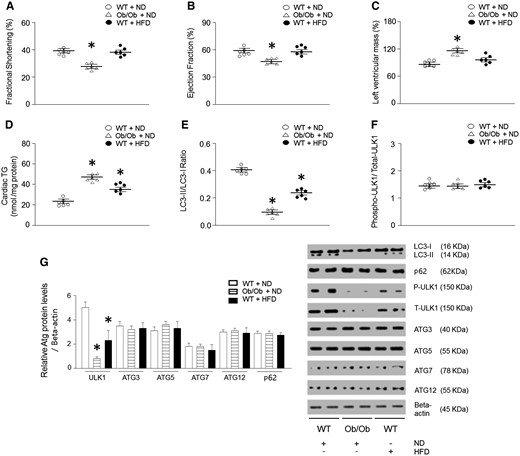
Genetically or long-term HFD-induced obesity resulted in altered cardiac autophagy, TG levels, and function. WT and Ob/Ob mice were generated from Ob/+ heterozygous intercrosses. Six-week-old male mice were fed either an HFD (60% of calories from fat) or an ND (10% of calories from fat) for 28 weeks. (A–C) FS, EF, and left ventricular mass were determined from the m-mode images (n = 6). (D) Cardiac lipids were extracted with organic solvent and TG was determined using a PicoProbe triglyceride fluorometric assay kit (n = 6). (E–G) Mouse hearts were isolated to evaluated autophagy-related proteins (p62, p-ULK1, ULK1, LC3, ATG3, ATG5, ATG7, and ATG12) using western blotting and densitometry (n = 6). Results were analyzed using one-way-ANOVA (graphs show means ± SE of 6 experiments in each group). ∗Significantly different from ND-treated WT mice, P < 0.05. +Significantly different from other cell lines, P < 0.05. ANOVA, analysis of variance; EF, ejection fraction; FS, fractional shortening; HFD, high-fat diet; ND, normal-chow diet; Ob/Ob, Lep°b/°b; SE, standard error; TG, triglyceride; WT, wild type.
3.2 Obesity increases cardiac LPL protein levels without corresponding changes in mRNA
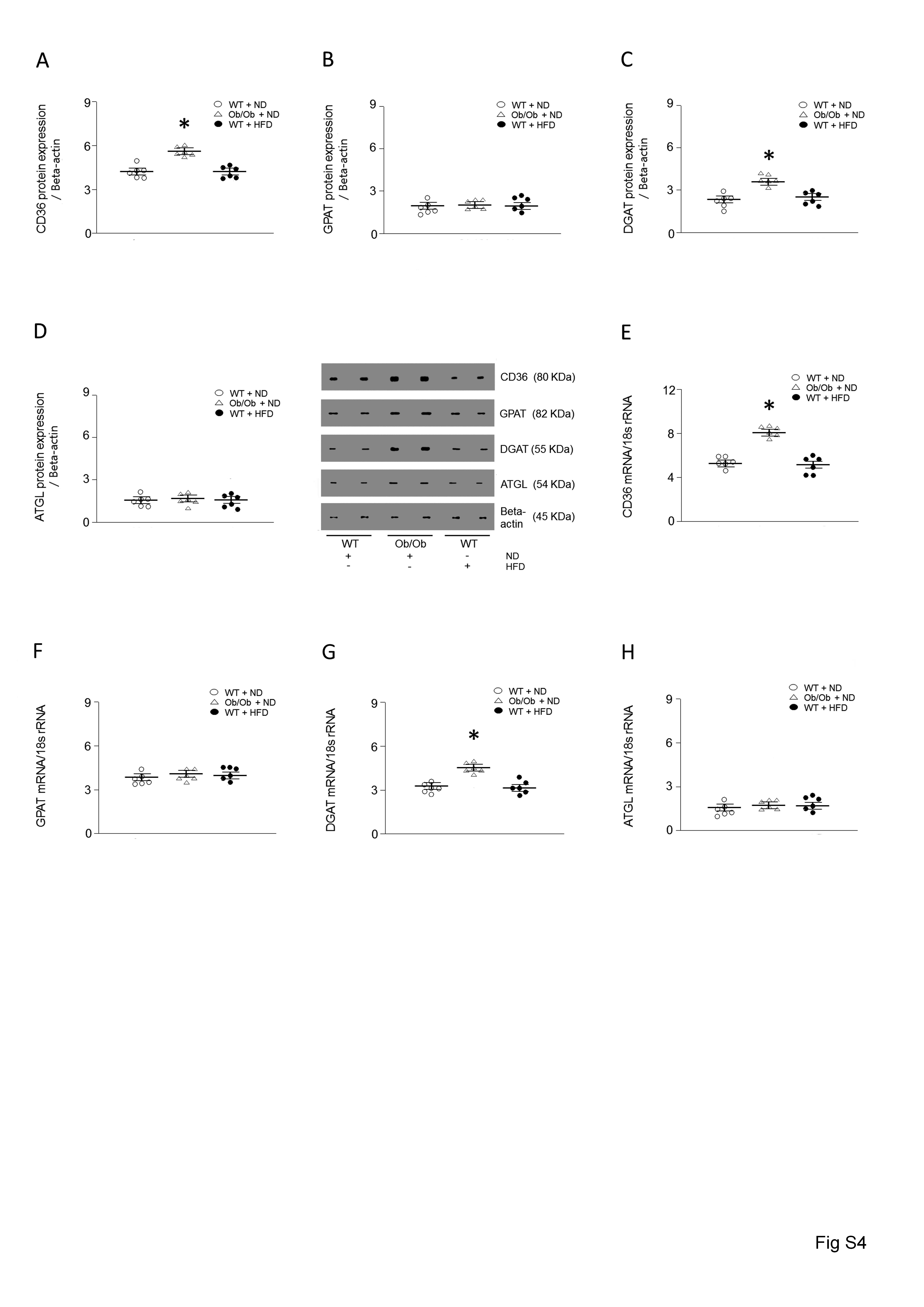
We next determined levels of protein that are involved in lipid metabolism. Among the proteins, Ob/Ob mouse hearts demonstrated elevated CD36, DGAT (see Supplementary material online, Figure S4A–H), and LPL levels (Figure 2A). Interestingly, there was no increase in LPL mRNA levels associated with obesity, as determined by real-time PCR (Figure 2B). To further confirm, RNA was transferred onto a nitrocellulose membrane and hybridized with a full-length probe for LPL (Figure 2C), which consistently showed no alterations in cardiac LPL mRNA levels in genetic or diet-induced obesity models. Moreover, there was a substantial increase in cardiac LPL activity (approximately 190–250%) observed in the vascular lumen of the hearts of both Ob/Ob and HFD-fed mice compared with the respective control groups (Figure 2D).
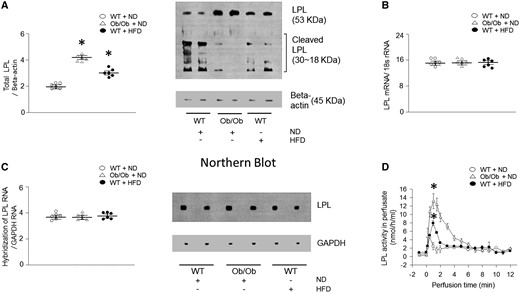
Cardiac LPL protein is increased but mRNA is unchanged in obese mice. WT and Ob/Ob mice were generated from Ob/+ heterozygous intercrosses. (A) Total LPL was determined using Western blotting (n = 6). (B–C) Total RNA was isolated from hearts, LPL mRNA was measured using real-time PCR or northern blot with an internal 32P-labeled probe (n = 6). (D) Ob/Ob, HFD- or ND-fed WT mouse hearts were isolated and perfused with heparin (5 units/mL) to release LPL from the coronary lumen. The collected LPL activity was assayed using a fluorescence-LPL substrate emulsion (n = 6). Results were analyzed using two-way-ANOVA (graphs show means ± SE of 6 mice in each experiment). ∗Significantly different from ND-treated WT hearts, P < 0.05. ANOVA, analysis of variance; HFD, high-fat diet; LPL, lipoprotein lipase; mRNA, messenger ribonucleic acid; ND, normal-chow diet; Ob/Ob, Lep°b/°b; PCR, polymerase chain reaction; RNA, ribonucleic acid; SE, standard error; WT, wild type.
3.3 ULK1 is a key autophagy protein to mediate proteolytic degradation of LPL
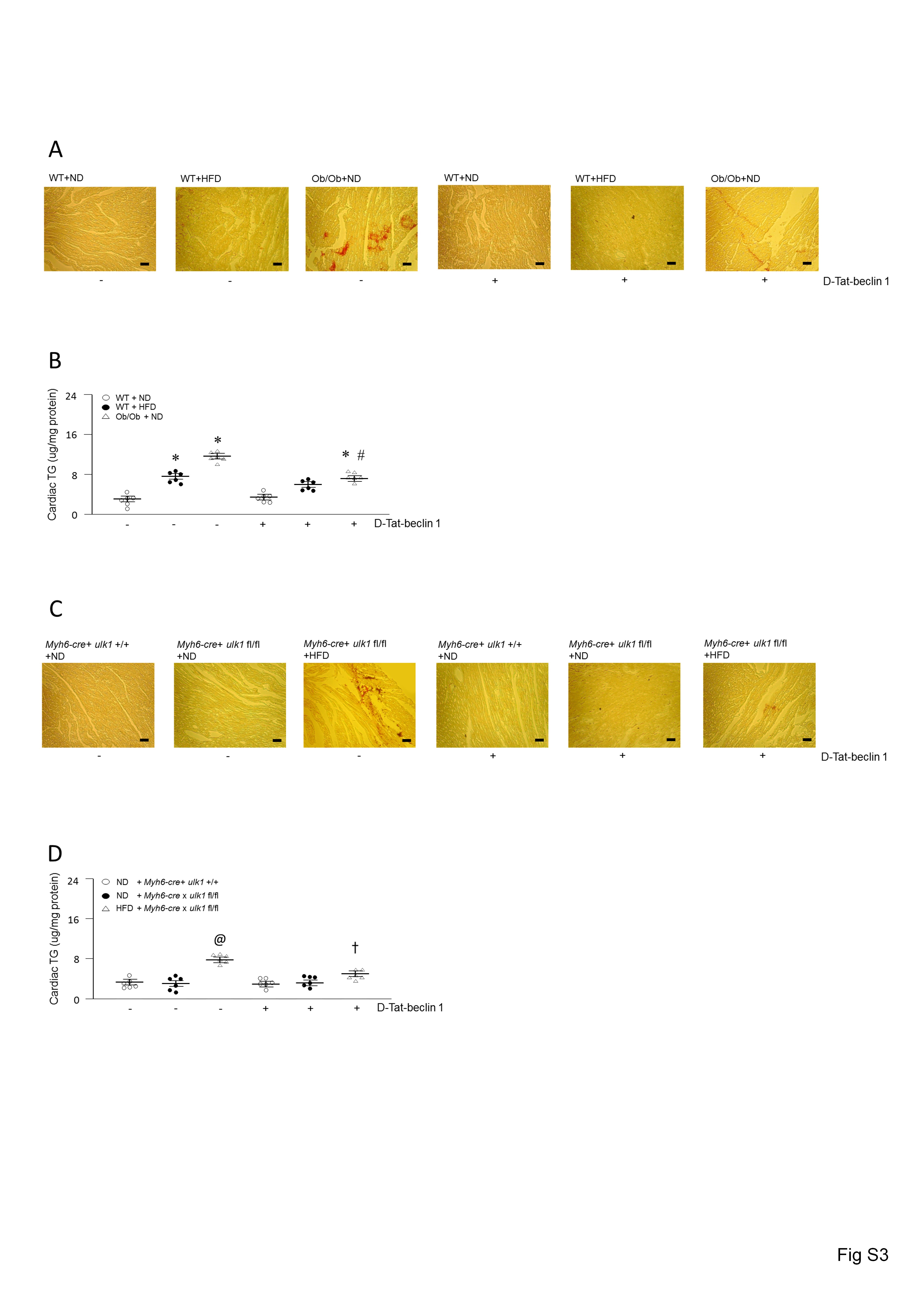
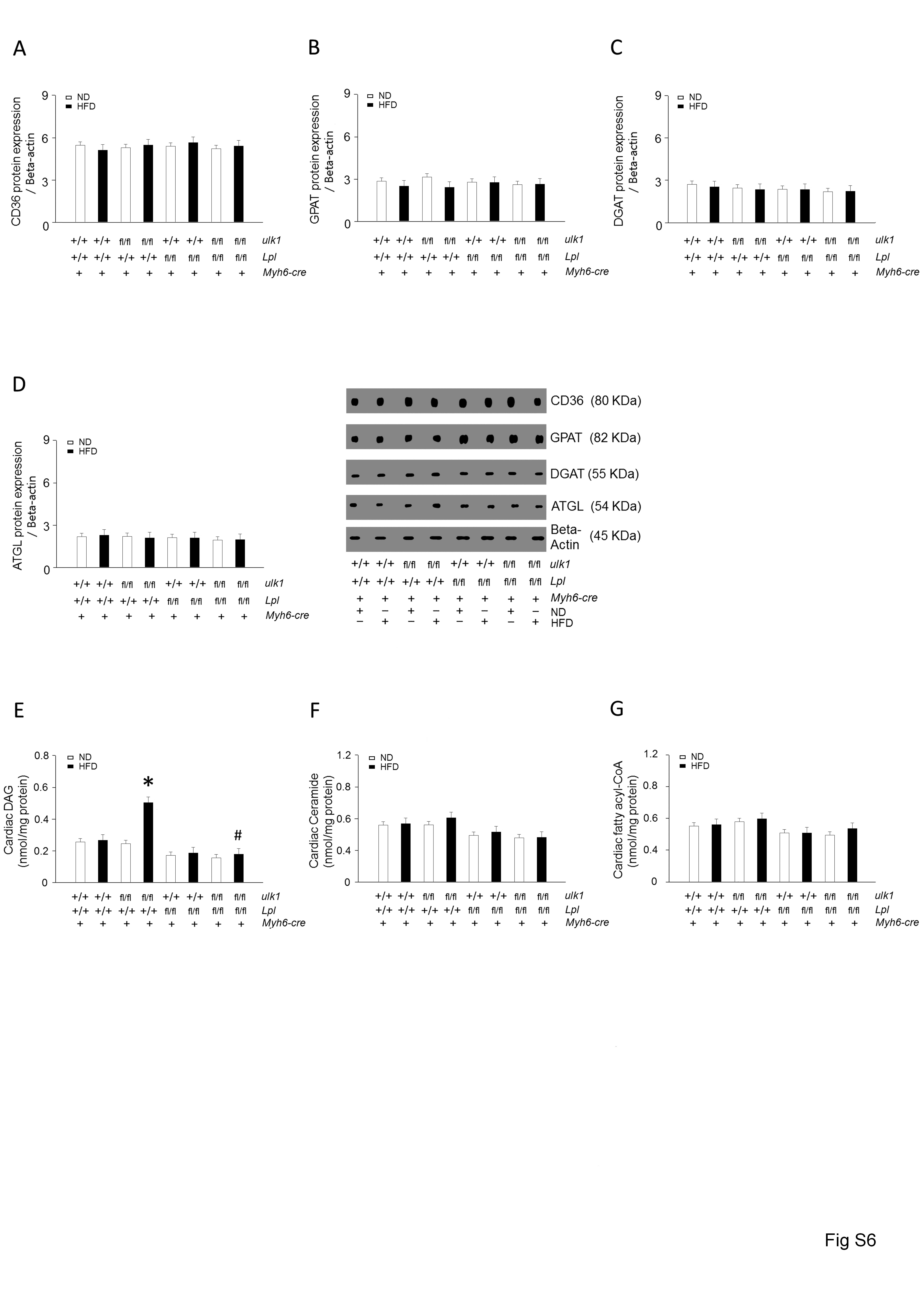
In protein lysates of WT hearts, cleavage products of LPL were evident (Figure 2A). However, these cleaved bands were not found in the lysates of hearts from Ob/Ob and HFD-fed mice, suggesting that some protein may be involved in preventing LPL cleavage in obese hearts. To determine whether ULK1 might play an essential role in regulating cardiac LPL cleavage and cardiac function, we generated cardiomyocyte-specific ulk1, Lpl, and double knockout mouse models, referred to as Myh6-cre/ulk1fl/fl, Myh6-cre/Lplfl/fl, and Myh6-cre/ulk1fl/flLplfl/fl, respectively. Efficient deletion of ulk1 and Lpl were confirmed using western blot of protein lysates of the hearts of Myh6-cre/ulk1fl/fl, Myh6-cre/Lplfl/fl, and Myh6-cre/ulk1fl/flLplfl/fl mice (Figure 3A). Remarkably, cardiac deficiency of ulk1 abrogated autophagy was assessed by low levels of LC3-II and this was associated with increased LPL protein levels and activity in the heart, without any changes in Lpl mRNA (Figure 3B–E, see Supplementary material online, Figure S5A). Cardiac deficiency of Lpl did not, however, affect ULK1 protein levels in Myh6-cre/Lplfl/fl (Figure 3A). Moreover, there were no alterations in the levels of CD36, GPAT, DGAT, or ATGL in these mice compared with control littermates (see Supplementary material online, Figure S6A–D). Similar to Ob/Ob and HFD-fed WT mice, Myh6-cre/ulk1fl/fl mice fed a HFD for 16 weeks showed a striking accumulation of TG and DAG (Figure 3F, see Supplementary material online, Figure S6E–G, see Supplementary material online, Figure S3C–D) and decreased cardiac function (Figure 3G–I, see Supplementary material online, Figure S1B). However, HFD-fed Myh6-cre/ulk1fl/flLPLfl/fl mice did not show alterations in TG and DAG levels (Figure 3F, see Supplementary material online, Figure S6E–G) or cardiac function (Figure 3G–I, see Supplementary material online, Figure S1C–D). In addition, cardiac ulk1 deficiency under basal conditions on ND did not affect cardiac TG or DAG levels (Figure 3F, see Supplementary material online, Figure S6E, see Supplementary material online, Figure S3C–D) or cardiac function compared to WT littermates (Figure 3G–I, see Supplementary material online, Figure S1B).
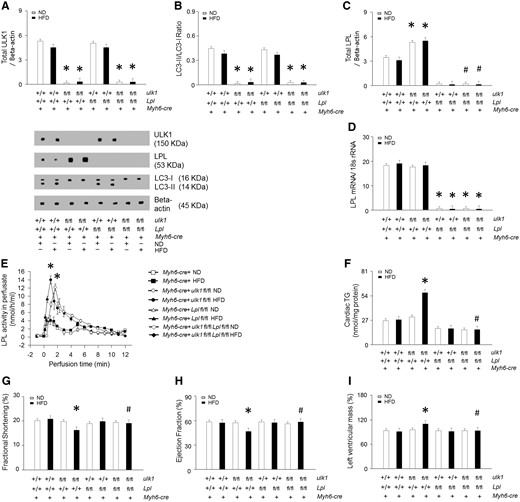
Heart-specific ulk1 knockout promotes cardiac LPL activity. WT mice were generated from ulk1fl/+ heterozygous intercrosses. (A–C) Hearts were isolated and homogenized from WT, Myh6-cre, Myh6-cre/Lplfl/fl, Myh6-cre/ulk1fl/fl, or Myh6-cre/ulk1fl/flLplfl/fl mice. All samples were then subjected to Western blotting to evaluate ULK1, LC3, or LPL (n = 6). (D) From the same groups, LPL mRNA levels were determined using real-time PCR (n = 6). (E) Hearts were also isolated and perfused with heparin (5 units/mL) to release LPL from the coronary lumen. LPL activity was assayed using a fluorescence-LPL substrate emulsion (n = 6). (F) Cardiac TG was extracted and determined using a PicoProbe triglyceride fluorometric assay kit (n = 6). (G–I) Following M-mode image analysis, FS, EF, and left ventricular mass were determined (n = 6). Results were analyzed using two-way-ANOVA (graphs show means ± SE of 6 mice in each experiment). ∗Significantly different from Myh6-cre mice, P < 0.05. #Significantly different from HFD-treated Myh6-cre/ulk1fl/fl mice, P < 0.05. ANOVA, analysis of variance; EF, ejection fraction; FS, fractional shortening; HFD, high-fat diet; LC3, light chain 3; LPL, lipoprotein lipase; mRNA, messenger ribonucleic acid; ND, normal-chow diet; Ob/Ob, Lep°b/°b; PCR, polymerase chain reaction; SE, standard error; TG, triglyceride; ULK1, unc-51 like kinase-1; WT, wild type.
3.4 Tat-beclin1 reduces cardiac TG accumulation by lowering LPL level
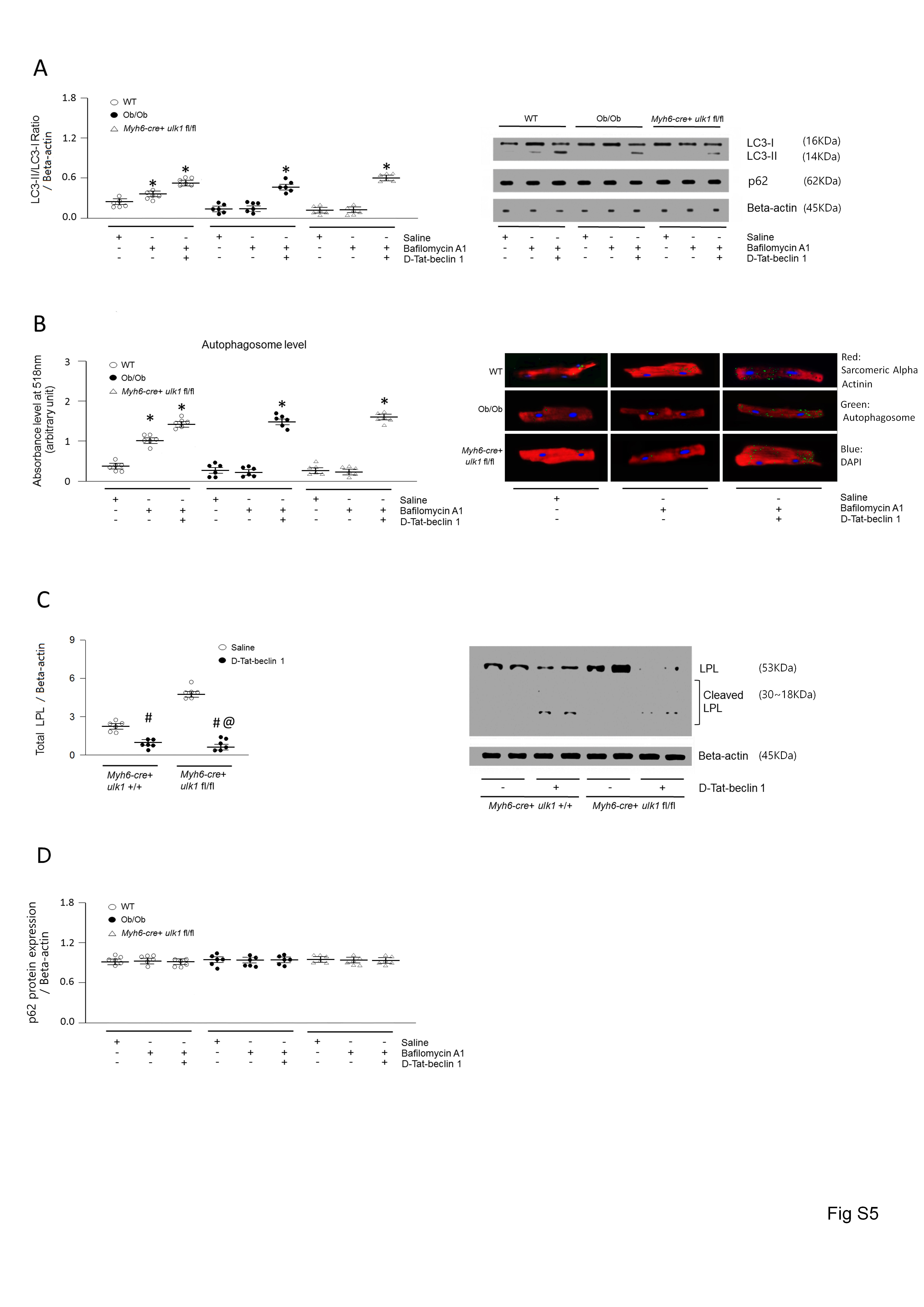
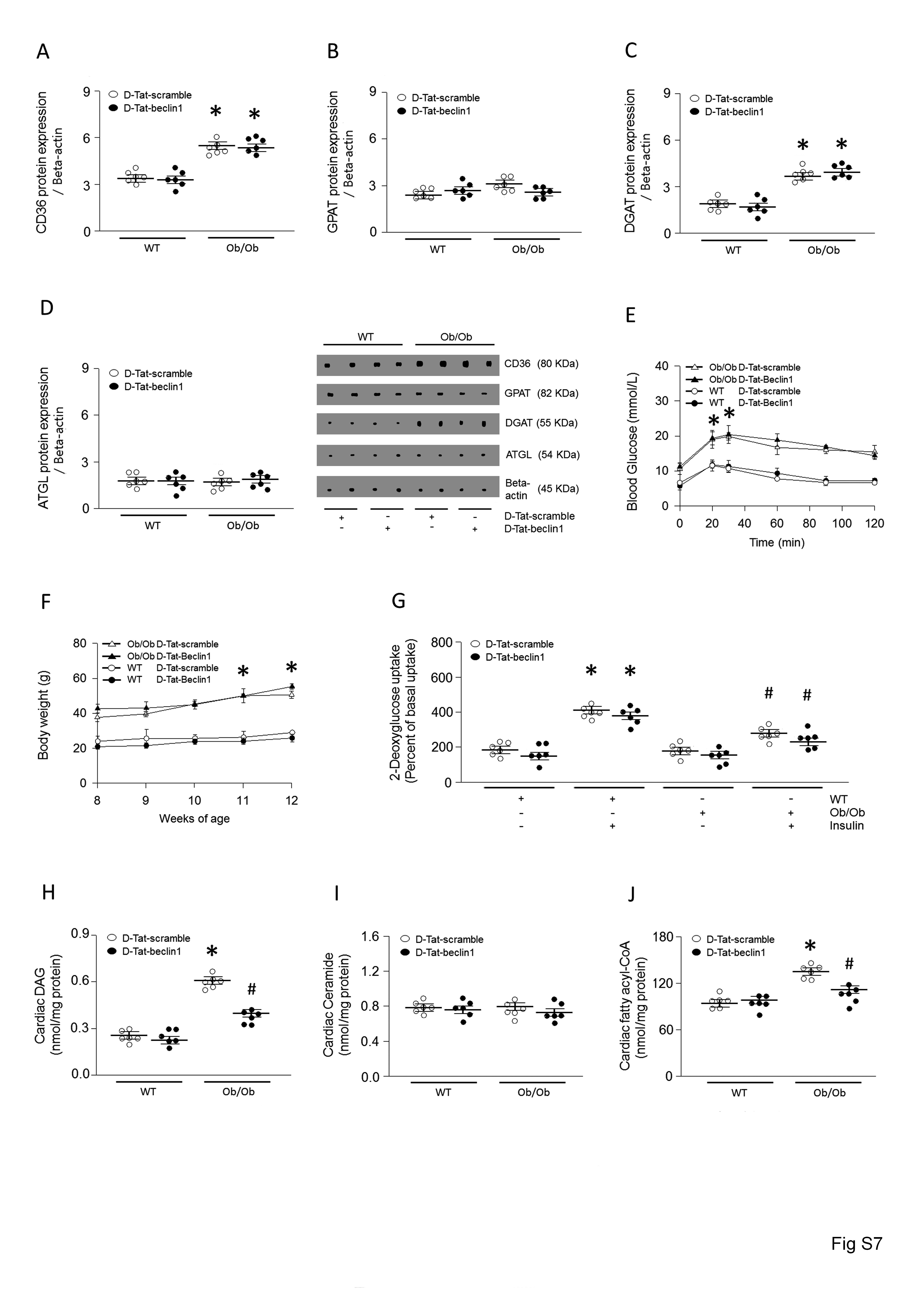
To further evaluate the role of autophagy in LPL regulation, we next activated autophagy by overexpressing Beclin1, another essential autophagy gene that functions downstream of ULK1.39 We designed a Tat-beclin1 fusion protein composed of a cell-permeable Tat protein domain that was attached to 18 amino acids derived from the active domain (amino acids 267–284) of beclin1.40 To render the protein resistant to proteolytic degradation in vivo, Tat-beclin1 was incorporated with retro-inverso-sequenced D-amino acids (D-Tat-beclin1 and D-Tat-scramble) (Figure 4A).40 To examine the effects of D-Tat-beclin1 on autophagy in vivo, we examined cardiac LC3-II levels in obese mice infused with D-Tat-beclin1 or D-Tat-scramble for 3 weeks. Compared with D-Tat-scramble-infused mice, D-Tat-beclin1-infused mice showed elevated conversion of LC3-I to LC3-II (Figure 4B). Moreover, bafilomycin A1 further increased amounts of LC3-II and autophagosome levels in D-Tat-beclin1-treated WT and Ob/Ob mouse hearts or cardiomyocytes (see Supplementary material online, Figure S5A and B). As predicted, following D-Tat-beclin1-infusion, the robust increases in LPL protein levels and the heparin-releasable LPL activity of the obese hearts were attenuated without any alterations in LPL mRNA levels (Figure 4C–F). Although Ob/Ob mice showed elevated CD36 and DGAT protein levels, D-Tat-beclin1 did not alter these protein levels (see Supplementary material online, Figure S7A–D). In addition, D-Tat-beclin1 did not have any effects on glucose tolerance, body weight gain, or glucose uptake (see Supplementary material online, Figure S7E–G). Furthermore, the reduction in the LPL protein levels in D-Tat-beclin1-infused obese mice was accompanied with reductions in cardiac TG, DAG, and fatty acyl-CoA levels approximately 40–65% lower than that of D-Tat-scramble-treated obese mice (Figure 4G, see Supplementary material online, Figure S7H–J, see Supplementary material online, Figure S3A–B). Moreover, D-Tat-beclin1 restored cardiac function in Ob/Ob mice (Figure 4H–J, see Supplementary material online, Figure S1E). Hence, we clearly show that defects in autophagy attenuate LPL degradation, which in turn leads to accumulation of LPL protein levels, which then promotes lipid accumulation. These excessive cardiac lipids ultimately contribute to cardiac dysfunction in obesity. Thus, augmenting autophagy is a novel therapeutic strategy for the prevention or treatment of lipotoxic cardiomyopathy.
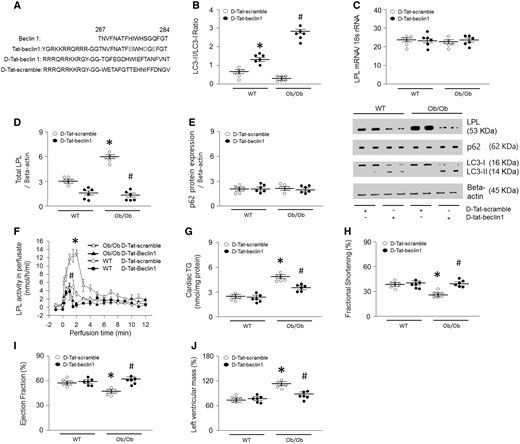
D-Tat-beclin1 decreases cardiac TG by lowering LPL in Ob/Ob mouse hearts. WT and Ob/Ob mice were generated from Ob/+ heterozygous intercrosses. Eight-week-old male Ob/Ob mice were infused with D-Tat-scramble or D-Tat-beclin1 (4 μg/min per kg body weight for 3 weeks using osmotic pumps). After the infusion had finished, the hearts were isolated and homogenized. (A) Sequences of cell-permeable Tat protein and beclin1 amino acids 267–284. Red letters indicate amino acid substitutions to enhance hydrophilicity. To obtain stability against in vivo proteolysis, it was composed of retro-inverso sequenced D-amino acids (D-Tat-beclin-1). (B–E) LPl, p62, and LC3 levels were determined using real-time PCR or Western blotting (n = 6). (F) Hearts were isolated and perfused with heparin (5 units/mL) to collect LPL and its activity was assayed using a fluorescence-substrate (n = 6). (G) Cardiac TG was extracted with organic solvent and determined using a PicoProbe TG fluorometric assay kit (n = 6). (H–J) Following M-mode image analysis, FS, EF, and left ventricular mass were determined (n = 6). Results were analyzed using two-way-ANOVA (graphs show means ± SE of 6 experiments in each group). ∗Significantly different from D-Tat-scramble infused WT groups, P < 0.05. #Significantly different from D-Tat-scramble infused Ob/Ob groups, P < 0.05. ANOVA, analysis of variance; EF, ejection fraction; FS, fractional shortening; LPL, lipoprotein lipase; PCR, polymerase chain reaction; SE, standard error; TG, triglyceride; Ob/Ob, Lep°b/°b; WT, wild type.
4. Discussion
Cardiomyocytes, located within the cardiac chambers, are essential functional cells for maintaining whole-body circulation. More than half of all cardiomyocytes continuously beat and contract without replacement throughout the organism’s life.41 Maintenance of this capacity in such long-lived cells requires a mechanism for the removal and recycling of maladaptive organelles, as well as oxidized, aggregated, and excess proteins.10,41 This self-destructive and self-preserving mechanism is known as autophagy.10–12
We observed a general lack of autophagy activation in the hearts of obese mice, motivating us to further explore the specific autophagy-related proteins potentially contributing to cardiac dysfunction in obesity. We found that ULK1 is down-regulated in the hearts of both the genetic and dietary mouse models of obesity. To identify which cell types have insufficient levels of ULK1, the heart tissue was separated into cardiomyocytes, fibroblasts, smooth muscle cells, and endothelial cells. Among these cell types, ULK1 expression was decreased predominantly in cardiomyocytes. To examine the role of ULK1 in cardiac function, we generated cardiomyocyte-specific ulk1 knockout mice. When these mice were fed an HFD to induce obesity, both the FS and EF values decreased in ulk1 deficient hearts. This was accompanied by a significant increase in cardiac TG and DAG levels, almost two-fold higher than that of WT control littermates. On the other hand, mice under basal conditions fed ND did not show any defects in cardiac function. Given the well-known association of lipid toxicity with cardiac dysfunction, we further focused on the mechanisms by which ULK1 might contribute to cardiac lipid metabolism.18
Inhibition of ULK1 increases lipid accumulation through fatty acid uptake in adipocytes.17 In the heart, LPL plays an important role in supplying fatty acids through intravascular TG hydrolysis.21 Our results showed that ulk1 deletion in the myocardium elevated LPL protein levels and activity in the heart. The elevated LPL protein levels were abrogated following D-Tat-Beclin1 (which activates downstream of ULK1) infusion (see Supplementary material online, Figure S5C). In addition, increased LPL levels were observed in the hearts of both Ob/Ob and HFD-fed WT mice. As this increase in protein levels was not a result of enhanced transcription at the mRNA level, we concluded that the suppression of LPL proteolysis in obesity resulted in the abnormal augmentation of LPL levels (Figure 5). In addition, as ULK1 signals have been known to activate non-selective macro-autophagy, LPL degradation is not likely to be selectively mediated by ULK1-initiated autophagy. However, among lipid metabolism proteins, only LPL protein levels were up-regulated following ULK1 deficiency. Therefore, we conclude that ULK1-provoked proteolysis of LPL occurs by non-selective macro-autophagy.
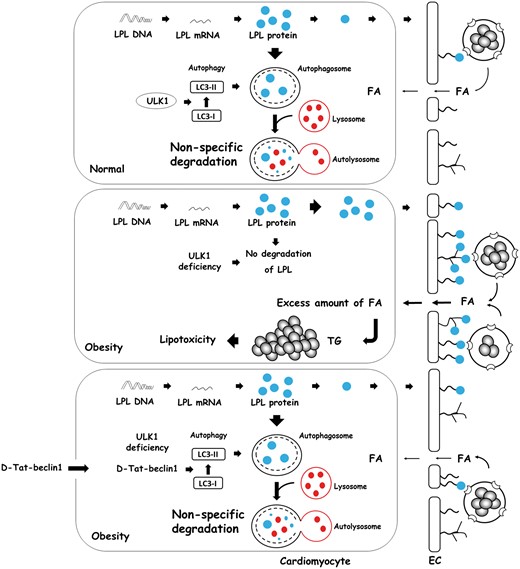
Autophagy is essential for impeding cardiac TG accumulation through LPL degradation. An excess amount of LPL is degraded by ULK1-induced non-specific autophagy in normal hearts. Following obesity or deficiency of ULK1, most cardiac LPL is not degradable and is transported to the coronary lumen. This increase in luminal LPL is associated with a striking cardiac TG accumulation. To induce autophagy in obesity, D-Tat-beclin1 is infused to prevent cardiac TG accumulation by lowering LPL activity. FA, fatty acids; LPL, lipoprotein lipase; TG, triglyceride; ULK1, unc-51 like kinase-1.
To determine the relationship between ULK1 and LPL up-regulation, and their effects on cardiac function, we generated Myh6-cre/Lplfl/fl and Myh6-cre/ulk1fl/flLplfl/fl double knockout mice. Although decreased CD36 mRNA has been reported in Myh6-cre/Lplfl/fl, we did not observe any alteration in CD36 protein levels in cardiac-specific LPL knockout mice.42 Our results show that Lpl deletion abrogated the cardiac dysfunction that occurred in cardiomyocyte-specific ulk1 knockout mice fed an HFD. Therefore, increased LPL is an important mediator of cardiac dysfunction in ULK1-deficient models.
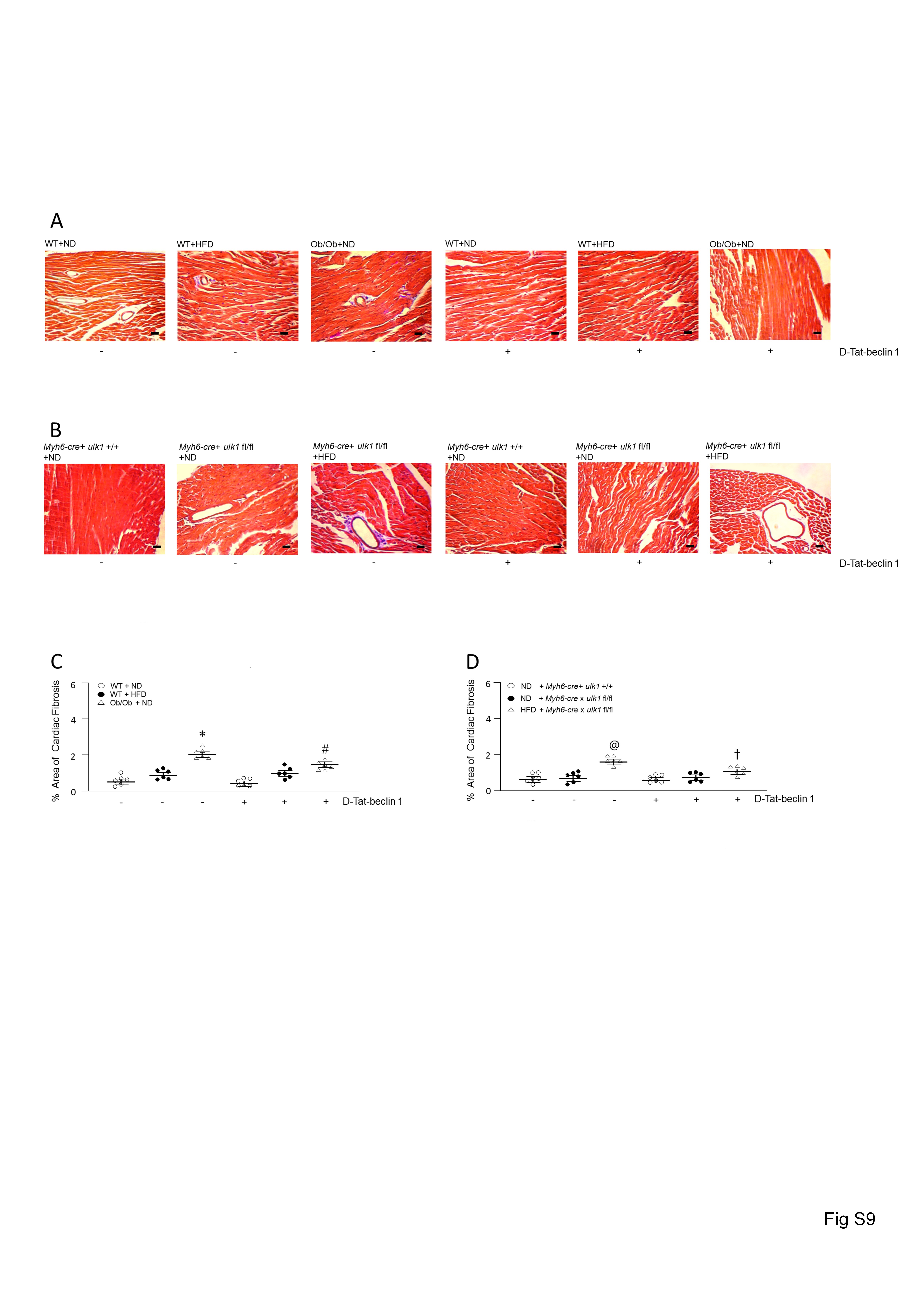
It has been reported that apoptosis and fibrosis are markedly elevated in genetic and diet-induced obese animals. However, we observed minimal apoptosis in the obese animals (see Supplementary material online, Figure S8). Only cardiac fibrosis was slightly increased in the hearts of HFD-fed WT, HFD-fed ulk1-deficient, and Ob/Ob mice (see Supplementary material online, Figure S9). Importantly, fibrosis was attenuated following D-Tat-beclin-1 infusion, suggesting an anti-fibrotic effect of autophagy in the hearts of obese models.
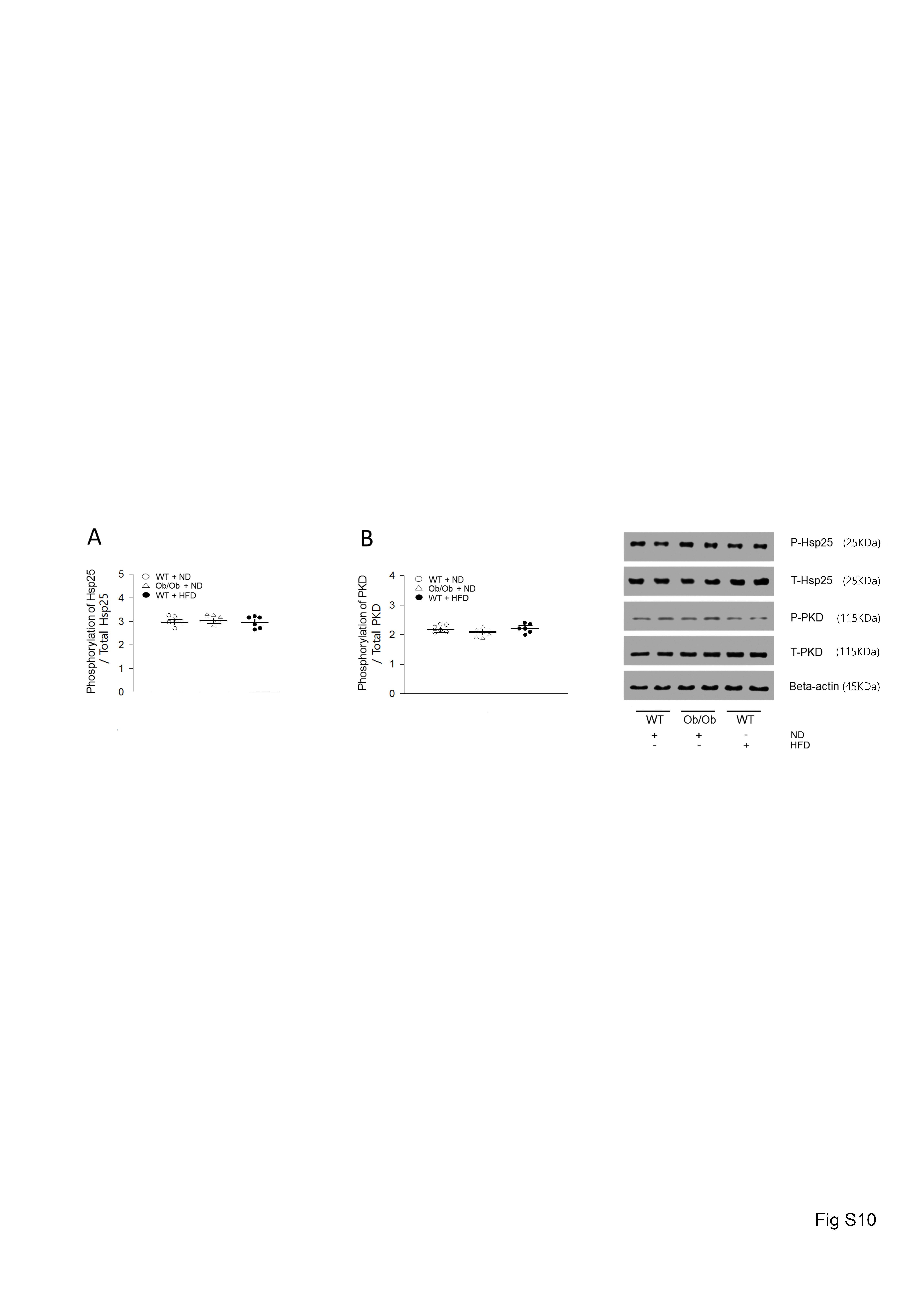
Coronary LPL activity is increased by protein transportation from cardiomyocytes into the vascular lumen.20 In this process, phosphorylation of Hsp25 or PKD are key components that increase LPL vesicular trafficking from Golgi membranes. However, there was no alteration in the phosphorylation of Hsp25 or PKD in the obese or ulk1-deficient hearts compared to control groups (see Supplementary material online, Figure S10). Taken together, these results demonstrate that LPL activity is elevated in conjunction with enhanced LPL protein levels rather than due to an accelerated transportation rate in obese or ulk1-deficient hearts.
Interestingly, the HFD-fed WT mice did not show cardiac dysfunction despite augmented LPL and TG levels. One possible reason for this is that the mice still had sufficient ULK1 to maintain cardiac function. Furthermore, the model may not mimic all aspects of the condition, although many of the general characteristics of HFD-fed animals do mirror human obesity.43 To examine the role of autophagy in cardiac dysfunction in Ob/Ob mice, they were infused with D-Tat-beclin1 that, remarkably, restored cardiac function and normalized LPL and TG levels in their hearts.
Moreover, p62 protein levels were not altered in our flux experiments (see Supplementary material online, Figure S5D) in obese models such as Ob/Ob, HFD-fed mice (Figure 1G), or D-Tat-beclin1-treated mice (Figure 4E). Whereas it is not clear why we did not see a change in p62 levels, this phenomenon has been described by others, possibly through restoration by serum starvation or energy deficiency.44 As adult cardiomyocytes were incubated without serum, this may explain the lack of alterations in p62 protein levels in our flux experiments. Similar phenomena may be at play after treatment with D-Tat-beclin1 (Figure 4E).
Altogether, our results clearly demonstrate that activation of autophagy can attenuate accumulation of LPL, thereby limiting fatty acid excess, and prevent cardiac dysfunction in obese hearts. Importantly, D-Tat-beclin1 did not alter insulin resistance or body weight gain, further supporting a direct role of autophagy in cardiac lipid metabolism. Our results identified a novel ULK1-LPL axis and thereby clarify our understanding of the essential role of autophagy in the heart. Further refinement of the metabolic and cardiovascular importance of ULK1 may lead to the development of improved therapeutic strategies for obesity-related cardiomyopathy.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education (NRF-2014R1A1A2054810). The echocardiography was supported by the Cardiovascular Research Center at Republic of Korea. This work was also supported by the Ewha Womans University Research Grant of 2014.
Conflict of interest: none declared.
References
Author notes
Time for primary review: 39 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}