





Abstract
Lipid homeostasis is reprogrammed in the presence of inflammation, which results in excessive lipid accumulation in macrophages, and leads to the formation of lipid–laden foam cells. We aimed to link an inflammation-responsive transcription factor CCAAT/enhancer-binding protein delta (CEBPD) with polarized macrophages and dissect its contribution to lipid accumulation.
We found that CEBPD protein colocalized with macrophages in human and mouse (C57BL/6, Apoe−/−) atherosclerotic plaques and that Cebpd deficiency in bone marrow cells suppressed atherosclerotic lesions in hyperlipidemic Apoe−/− mice. CEBPD was responsive to modified low-density lipoprotein (LDL) via the p38MAPK/CREB pathway, and it promoted lipid accumulation in M1 macrophages but not in M2 macrophages. CEBPD up-regulated pentraxin 3 (PTX3), which promoted the macropinocytosis of LDL, and down-regulated ATP-binding cassette subfamily A member 1 (ABCA1), which impaired the intracellular cholesterol efflux in M1 macrophages. We further found that simvastatin (a HMG–CoA reductase inhibitor) could target CEBPD to block lipid accumulation in a manner not directly related to its cholesterol-lowering effect in M1 macrophages.
This study underscores how CEBPD functions at the junction of inflammation and lipid accumulation in M1 macrophages. Therefore, CEBPD-mediated lipid accumulation in M1 macrophages could represent a new therapeutic target for the treatment of cardiovascular diseases.
1. Introduction
A key event in atherosclerosis is the maladaptive inflammatory response to subendothelial lipoproteins. Macrophages are a major cell type in atherosclerotic lesions that play a central role in atherosclerosis by producing oxidative stress and pro-inflammatory mediators. Lipid homeostasis is reprogrammed in the presence of inflammation. Moreover, excessive lipid accumulation in macrophages results from increased uptake of low-density lipoprotein (LDL) or impaired cholesterol efflux, which causes the formation of lipid–laden foam cells.1 Therefore, foam cells are an indication of a build-up of atherosclerotic plaques, which are commonly associated with an increased risk of heart attack and stroke.
Scavenger receptors, including scavenger receptor A1 (SR-A1, which is encoded by MSR1) and CD36 (also known as platelet glycoprotein 4), were originally characterized by their abilities to recognize and uptake modified LDL. In response to modified LDL, the toll-like receptors activate intracellular signaling pathways that lead to the induction of the transcription of genes that are critical for inflammatory responses and fluid-phase pinocytosis.2 This process results in the further accumulation of lipids. In contrast, the excessive accumulation of cellular lipids triggers compensatory mechanisms, such as the efflux of cholesterol from macrophages. ATP-binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1) mediate the efflux of free cholesterol to lipid-poor apolipoprotein A1 (APOA1), which leads to the formation of high-density lipoproteins.3 However, the mechanisms that link inflammation to increased uptake of LDL and impaired cholesterol efflux in macrophages are not fully understood.
Macrophages display tremendous heterogeneity in their activities and functions, and this heterogeneity primarily reflects their local metabolic and immune microenvironments. In response to specific external stimuli, macrophages can undergo M1 (classical) or M2 (alternative) activation, in a process that mirrors that of Th1–Th2 polarization in T cells.4 The activation of M1 macrophages is characterized by the high levels of production of reactive oxygen species and nitric oxide and promotes inflammatory immune responses and strong microbicidal and tumoricidal activities. In contrast, the activation of M2 macrophages is thought to be involved in parasite containment and the promotion of tissue remodeling and tumor progression and to have immunoregulatory functions. However, the association of M1 and M2 macrophages with lipid accumulation in response to LDL remains less well characterized.
Macrophage activation and inflammatory responses originate mainly at the transcriptional level.5 CCAAT/enhancer-binding protein delta (CEBPD), which is a member of a subfamily of basic leucine zipper domain transcription factors, is expressed at a relatively low level under normal physiological conditions but is upregulated by a variety of extracellular stimuli, such as interleukin-6 (IL-6), IL-1β, lipopolysaccharide (LPS), interferon α (IFNα), IFNγ, and tumor necrosis factor α (TNFα).6–9 Importantly, CEBPD is also reciprocally involved in the activations of IL-6, IL-1β, and TNFα.10 A growing number of studies have demonstrated that CEBPD is induced in inflammation-associated disorders, such as Alzheimer’s disease,11,12 rheumatoid arthritis,6,13 type 2 diabetes,14 and atherosclerosis,10 which suggests that CEBPD could be a critical regulator of inflammation. However, several issues, including the association between CEBPD and polarized macrophages (i.e. M1 and M2 macrophages) and whether CEBPD contributes to lipid accumulation in blood vessel lesions remain unexplored.
Statin compounds [3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors] can inhibit hepatic HMG–CoA reductase which is a rate-limiting enzyme in the cholesterol synthesis pathway, and ultimately lead to increased clearance of circulating LDL–cholesterol. Moreover, statins have been suggested to have anti-inflammatory effects beyond their cholesterol-lowering action.15 Despite extensive research on the molecular mechanisms by which statins act, little is known regarding the details of which transcription factors are affected by statins in macrophages.16 In this study, by up-regulating PTX3 and down-regulating ABCA1, we reveal the involvement of CEBPD in lipid accumulation in M1 macrophages and further demonstrate that simvastatin could suppress CEBPD expression by inactivating the p38MAPK/CREB pathway in M1 macrophages. These findings provide promising evidence demonstrating that simvastatin targets an inflammatory effector to block lipid accumulation.
2. Methods
2.1 Cell preparations
THP-1 (human monocytic leukemia cell line) and AMJ2 (mouse alveolar macrophage cell line) were cultured in RPMI-1640 and DMEM, respectively, containing 10% fetal bovine serum (FBS), 100 µg/mL streptomycin, and 100 units/mL penicillin. For differentiation of macrophages, THP-1 cells were treated with 5 ng/mL phorbol 12-myristate 13-acetate (PMA) for 48 h and then subsequently allowed to differentiate to macrophages for 5 days without PMA treatment. For further activation, PMA-differentiated THP1 macrophages were treated with 100 ng/mL LPS (for M1 activation) or 20 ng/mL IL-4 plus 20 ng/mL IL-13 (for M2 activation) for additional 2 days. Human peripheral blood mononuclear cells (PBMCs) were isolated using density gradient centrifugation. After sacrificed with carbon dioxide, mouse bone marrow (BM) mononuclear cells were obtained from femurs and tibias. For differentiation of macrophages, these monocytes cultured in RPMI-1640 were differentiated with 50 ng/mL recombinant granulocyte–macrophage colony-stimulating factor (GM–CSF) (Cell Guidance Systems, Babraham, Cambridge, UK) or 25 ng/mL recombinant macrophage colony-stimulating factor (M-CSF) (Cell Guidance Systems, Babraham, Cambridge, UK) for 5 days to macrophages. Any floating cells were discarded and the adherent macrophages were then cultured for 2 days without GM-CSF or M-CSF treatment. For further activation, GMCSF-differentiated macrophages were treated with 100 ng/mL LPS (for M1 activation) and MCSF-differentiated macrophages were treated with 20 ng/mL IL-4 plus 20 ng/mL IL-13 (for M2 activation) for additional 2 days.
2.2 Immunofluorescence (IF) staining
All human studies were carried out by procedures approved by the Institutional Review Board (IRB) (B-BR-101-147) of National Cheng Kung University Hospital (NCKUH). Written informed consent was obtained from all patients. The study was also performed conform the declaration of Helsinki. The coronary rupture plaques had been retrieved successfully from the enrolled ST elevation myocardial infarction (STEMI) patients receiving primary percutaneous coronary intervention and being aspirated by Pronto V3 aspiration catheter. The available human specimens were well-prepared with 5-μm-thick paraffin sections mounted on slides, dried in an oven at 65°C for 15 min, and deparaffinized in xylene and ethanol. The sections were then immersed in citrate buffer to perform heat-induced epitope retrieval (HIER). The slides were stained with CEBPD (LifeSpan BioSciences), MAC387 (AbD Serotec), F4/80 (GeneTex), or Nile Red (Sigma, St. Louis, MO, USA) primary antibody at 4°C overnight. The slides were then incubated with TRITC- or Alexa 488-conjugated secondary antibody (Jackson ImmunoResearch) at room temperature for 1 h. The slides were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI) (Enzo Life Sciences) and then photographed under a C1Si confocal laser scanning microscope (Nikon).
2.3 Animal studies
All animal studies were carried out by procedures approved by the Institutional Animal Care and Use Committee (IACUC) (103209) of National Cheng Kung University (NCKU). The animal experiments were also performed conform the NIH guidelines (guide for the care and use of laboratory animals). To assess the involvement of Cebpd in bone marrow cells in the pathogenesis of atherogenesis, we performed bone marrow transplantation in male Apoe−/− mice on C57BL/6 genetic background. First, male Apoe−/− recipient mice underwent irradiation (9.6 Gy) (RS 2000 X-ray Irradiator, Rad Source, Suwanee, GA, USA) at 8 weeks of age and then received 4 × 106 freshly prepared sterile BM cells obtained from 8-week-old male Cebpd+/+ or Cebpd−/− donor mice on C57BL/6 genetic background via tail vein injection. Two weeks later, anesthesia was induced by intraperitoneal (IP) injection of ketamine (80 mg/kg) and xylazine (10 mg/kg) mixture. Then, atherosclerosis was induced using angiotensin II (Sigma, St. Louis, MO, USA) subcutaneously released by osmotic pumps (ALZET) in a rate of 500 ng/min/kg/day for 30 days, and fed the Western diet (D12079B: 0.21% cholesterol, Research Diets, New Brunswick, NJ, USA) for 2 months following osmotic pump implantation. The mice were then killed with carbon dioxide for analyses. BM cells obtained from recipient mice were used to confirm BM reconstitution by PCR analyses. In male Cebpd−/− mice, the coding region of Cebpd was replaced by a neomycin-resistance gene.17 To detect the neomycin-resistance gene and 3′ UTR of Cebpd, forward primer (5′-GCTCCAGACTGCCTGGGAAAAGC-3′) and reverse primer (5′-CAGTCCAGTGCCCAAGCTGC-3′) were used for PCR amplification, and the expected PCR product was 305 bp.
2.4 Pinocytosis assay
Pinocytic activity of macrophages was assessed as described previously.18 In brief, Lucifer Yellow CH (Sigma, St. Louis, MO, USA) was dissolved in 10% FBS/RPMI medium at 0.5 mg/mL. The Lucifer Yellow medium was then added to macrophages (from wild-type and Cebpd−/− mice) cultured in 12-well plates (1 mL/well). The culture plates were then either maintained at 0°C or at 37°C for 2 h. The wells were drained, washed with 0.2% BSA/RPMI medium three times, and washed with PBS five times all at 4°C. Triton X-100 (0.05%, 600 μl/well) was added to each well to lyse cells. Fluorescence of the lysates was detected using a Fluoroskan Ascent FL microplate fluorometer (Thermo, Waltham, MA, USA) (excitation at 430 nm and emission at 540 nm). Standard curve of Lucifer Yellow was prepared in Triton X-100 and was found to be linear from 0.1 to 100 ng/mL.
2.5 BODIPY-cholesterol efflux assay
BODIPY-cholesterol efflux from macrophages was assessed as described previously.19 In brief, macrophages (from wild-type and Cebpd−/− mice) were labeled with RPMI medium containing 0.2% BSA, 50 µg/mL oxLDL, and 5 mg/L BODIPY-cholesterol (Avanti Polar Lipids, Alabaster, AL, USA) for 1 h. The cells were then washed and equilibrated with 0.2% BSA in RPMI medium for 1 h. Following equilibration, the cells were then incubated with RPMI medium containing 10% FBS or 10 µg/mL APOA1 (Sigma, St. Louis, MO, USA) as cholesterol acceptor for 4 h. Fluorescence of the cells and the media was detected using a Fluoroskan Ascent FL microplate fluorometer (Thermo, Waltham, MA, USA) (excitation at 495 nm and emission at 507 nm). BODIPY–cholesterol efflux was determined by dividing the fluorescence intensity in the media by the sum of the fluorescence intensity in the cells and the media.
2.6 Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was carried out essentially as described previously.20 In brief, THP-1 cells, with or without prior stimulation with oxLDL, were treated with 1% formaldehyde for 15 min. The cross-linked chromatin was then prepared and sonicated to an average size of 300–500 bp. The fragmented DNA–protein complex was immunoprecipitated with antibodies specific for CEBPD or control rabbit IgG at 4°C overnight. After reversal of the cross-linking, the immunoprecipitated chromatin was amplified by PCR of PTX3 gene genomic locus with specific primers: PTX3/-503, 5′-GCTCGGATTGGACTTGACTT-3′ and PTX3/-143, 5′-GAGGGAAATGTGGAAGTTGC-3′. The amplified DNA products were resolved by agarose gel electrophoresis and confirmed by sequencing.
2.7 Statistical analysis
All experiments were repeated at least three times. The use of parametric or non-parametric tests was based on results from analyses of distributions (D’Agostino & Pearson omnibus normality test). For two groups of experiment, the data were analyzed by Mann–Whitney test (non-parametric data) using GraphPad Prism. For three or more groups of experiment, the data were analyzed by Kruskal–Wallis test followed by Dunn’s post-hoc test (non-parametric data) for multiple comparisons using GraphPad Prism. The data were expressed as mean ± SEM. Differences were considered statistically significant when indicated by asterisks. (*P ≤ 0.05, **P ≤ 0.01, *** P ≤ 0.001).
3. Results
3.1 CEBPD is expressed in macrophages in atherosclerotic plaques, and Cebpd deficiency in BM cells suppresses atherosclerotic lesions in hyperlipidemic Apoe−/− mice
To examine the expression of CEBPD in blood vessel lesions in patients with atherosclerosis, immunofluorescence staining was performed using specimens of coronary rupture plaques retrieved from patients who were admitted with diagnosis of ST elevation myocardial infarction (STEMI). The data revealed that the CEBPD protein colocalized with the macrophage marker (MAC387) in these plaques (Figure 1A). We also found that the mouse CEBPD (Cebpd) protein colocalized with the macrophage marker (F4/80) in Apoe−/− atherosclerotic plaques (Figure 1B). We thus performed BM transplantation in Apoe−/− mice to assess the involvement of Cebpd in BM cells in the pathogenesis of atherogenesis. The areas of the atherosclerotic lesions and the proportions of macrophages containing lipids in the aortae were significantly reduced in the Apoe−/− mice that received Cebpd−/− BM cells (Figure 1C and D). These results suggest that macrophage CEBPD/Cebpd plays a functional role in the pathogenesis of atherosclerosis.
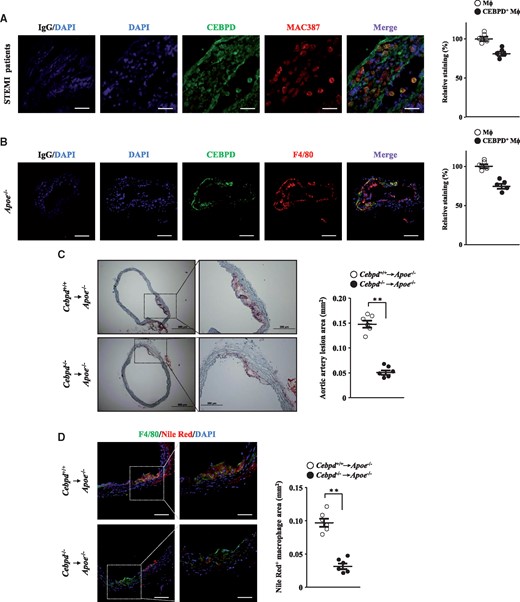
CEBPD is expressed in macrophages in atherosclerotic plaques, and atherosclerotic lesions are attenuated in hyperlipidemic Apoe−/− mice receiving Cebpd-deficient bone marrow cells. (A) Representative images showing CEBPD (green) and MAC387 (red) expression in atherosclerotic plaques from STEMI patients (scale bar: IgG/DAPI=120 µm, the others=20 µm) (n=5). MAC387-positive cells indicate macrophages and their signals are set as 100%. (B) Representative images showing CEBPD (green) and F4/80 (red) expression in carotid specimens from Apoe−/− mice (scale bar: 120 µm) (n=5). F4/80-positive cells indicate macrophages and their signals are set as 100%. (C) Apoe−/− recipient mice (n = 6 per group) underwent irradiation and then received sterile bone marrow cells obtained from Cebpd+/+ or Cebpd-/- donor mice. Two weeks later, recipient mice were treated with angiotensin II and fed the western diet for 2 months. For each mouse, ten slides from different layers of the ascending aorta were taken for analyses. Oil Red O stainings in cross-sections of the proximal ascending aortae are representative of six experimental animals. Oil Red O-positive areas indicate lesion areas. Statistical significance was determined using Mann–Whitney test. (D) Images showing F4/80 (green) and nile red (red) expression in cross-sections of the proximal ascending aortae are representative of six experimental animals (scale bar: left=120 µm, right=60 µm) (n = 6 per group). F4/80-positive cells indicate macrophages and nile red-positive cells indicate lipids. Statistical significance was determined using Mann–Whitney test. Quantification of the immunofluorescence images was analyzed by the TissueGnostics software.
3.2 CEBPD is responsive to modified LDL via the p38MAPK/CREB pathway
The level of Cebpd is associated with atherosclerotic lesion development and macrophage lipid accumulation, and RT-qPCR assays demonstrated that modified LDL could induce the transcription of the Cebpd gene in mouse primary macrophages (Figure 2A). Our previous studies indicated that the p38MAPK/cAMP-responsive element binding protein (CREB) pathway is important for the transcriptional activation of CEBPD and Cebpd genes.21 To test whether p38MAPK mediated the modified LDL-induced transcription of CEBPD gene, the p38MAPK inhibitor SB203580 was used. RT-qPCR assays revealed that SB203580 suppressed the level of oxLDL- or acLDL-induced Cebpd mRNA in mouse primary macrophages (Figure 2A). To determine whether common mechanisms involving p38MAPK/CREB-activated CEBPD gene transcription existed between mice and humans, we tested whether modified LDL could induce CEBPD expression in human THP-1 cells. RT-PCR assays and western blot analyses revealed that CEBPD was responsive to modified LDL in PMA-differentiated THP1 macrophages (Figure 2B). The results also revealed that SB203580 suppressed oxLDL- or acLDL-induced phospho-p38 (p-p38), phospho-CREB (p-CREB), and CEBPD expression in PMA-differentiated THP1 macrophages (Figure 2C). Moreover, CEBPD reporters (pCEBPD, −1000/+18) that were co-transfected with a dominant negative form of a p38MAPK (DN-p38α) expression vector or a DN–CREB expression vector repressed oxLDL−, acLDL−, or p38MAPK upstream activators [constitutively active form of an MKK3 (MKK3Ac) or MKK6 (MKK6Ac) expression vector]-induced CEBPD reporter activity in THP-1 cells (Figure 2D). Taken together, these results suggest that p38MAPK plays a functional role in CEBPD activation in macrophages and further support the importance of p38MAPK in the transcriptional activation of CEBPD gene.
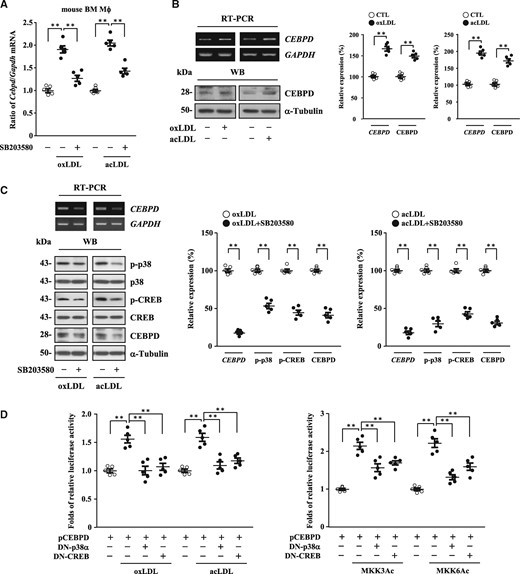
Modified LDL induces CEBPD expression via the p38MAPK/CREB signaling pathway in macrophages. (A) Following pre-treatment with or without p38MAPK inhibitor SB203580, mouse primary macrophages (2×105/mL) were then treated with or without oxLDL or acLDL (n=5). Total RNA was harvested for RT-qPCR assays. Statistical significance was determined using Kruskal-Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (B) PMA-differentiated THP1 macrophages (2×105/mL) were treated with or without oxLDL or acLDL. Total RNA and whole cell lysates were harvested for RT-PCR assays and western blot analyses, respectively. One of five independent experiments is shown. Statistical significance was determined using Mann–Whitney test. (C) Following pre-treatment with or without SB203580, PMA-differentiated THP1 macrophages (2×105/mL) were then treated with oxLDL or acLDL. Total RNA and whole cell lysates were harvested for RT-PCR assays and western blot analyses, respectively. One of five independent experiments is shown. Statistical significance was determined using Mann–Whitney test. (D) THP-1 cells (1×106/mL) were co-transfected with CEBPD reporters (pCEBPD, −1000/+18) with or without dominant negative form of p38MAPK (DN-p38α) expression vectors or DN-CREB expression vectors. The transfectants were then treated with or without oxLDL or acLDL or co-transfected with or without constitutively active form of MKK3 (MKK3Ac) or MKK6 (MKK6Ac) expression vectors (n=5). The lysates of the transfected cells were harvested for luciferase assays. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons.
3.3 Activation of CEBPD contributes to lipid accumulation in M1 macrophages
Both M1 and M2 macrophages have been suggested to be involved in lipid accumulation.22,23 We first examined whether CEBPD was involved in macrophage polarization. Macrophages can undergo classical M1 activation when stimulated with LPS or IFNγ, or alternative M2 activation following stimulated with IL-4/IL-13 or PGE2.24 The transcription of the mouse Cebpd and human CEBPD genes was activated in both M1 and M2 macrophages (Figure 3A and B, and Supplementary material online, FigureS1A). We next assessed whether CEBPD contributed to the accumulation of lipids in polarized macrophages. Interestingly, following the confirmation of the M1 and M2 macrophage markers (Supplementary material, FigureS2A), lipid accumulation was significantly reduced in Cebpd−/− M1 macrophages (activated by LPS) but not in Cebpd−/− M2 macrophages (activated by IL-4/IL-13; Figure 3C). These results suggest that lipids are accumulated in both M1 and M2 macrophages. However, CEBPD specifically contributes to lipid accumulation in M1 macrophages. Excessive lipid accumulation in macrophages is mainly the result of the uncontrolled uptake of LDL or impaired cholesterol efflux. We next explored the processes that were affected by CEBPD in M1 macrophages. Pinocytosis assays revealed that LDL uptake was reduced in Cebpd−/− M1 macrophages (Figure 3D). Moreover, BODIPY–cholesterol efflux assays revealed that cholesterol efflux was increased in the Cebpd−/− M1 macrophages (Figure 3E). These results suggest that CEBPD activation contributes not only to the increased uptake of LDL but also to the impaired cholesterol efflux in M1 macrophages.
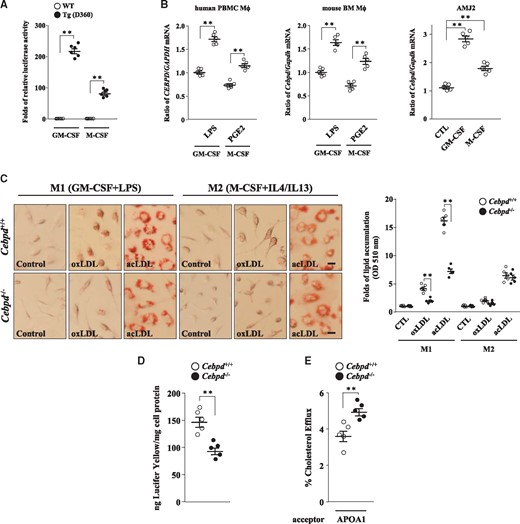
Activation of CEBPD contributes to lipid accumulation in M1 macrophages. (A) Monocytes (1×106/mL) were isolated from the bone marrow of WT and humanized CEBPD reporter mice and then differentiated using GM-CSF or M-CSF (n=6). Whole cell lysates were harvested for luciferase assays. Statistical significance was determined using Mann–Whitney test. (B) GMCSF- or MCSF-differentiated human PBMCs and mouse bone marrow macrophages (2×105/mL) were treated with or without LPS (100 ng/mL) or PGE2 (15 ng/mL), respectively. Mouse immortalized macrophage AMJ2 cells (2×105/mL) were treated with or without GM-CSF (50 ng/mL) or M-CSF (25 ng/mL). Total RNA was harvested for RT-qPCR assays. All experiments were repeated five times. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (C) Monocytes were isolated from the bone marrow of WT and Cebpd-/- mice and polarized into M1 or M2 macrophages. These macrophages (2×105/mL) were treated with or without oxLDL or acLDL for 3 days and then subjected to Oil Red O stainings. Images are representative of five independent experiments (scale bar: 5 µm). Statistical significance was determined using Mann–Whitney test. (D) Lucifer Yellow-dissolved medium was added to WT and Cebpd-/- M1 macrophages (2×105/mL) (n = 5). Statistical significance was determined using Mann–Whitney test. (E) Following labeling with medium containing oxLDL and BODIPY-cholesterol, WT and Cebpd-/- M1 macrophages (2×105/mL) were then treated with APOA1 which was used as a cholesterol acceptor (n = 5). Statistical significance was determined using Mann–Whitney test.
3.4 Ptx3 and Abca1 are involved in Cebpd deficiency-attenuated lipid accumulation in M1 macrophages
To identify the genes that were regulated by Cebpd in M1 macrophages, a next generation sequencing (NGS) analysis was performed. In the NGS analysis, in response to Cebpd, over 500 genes (>1.5-fold, P < 0.01) were identified, and the top 60 significantly up-regulated and down-regulated genes were listed in Supplementary material online, Table S1. In the NGS profiling assay, the genes related to potent LDL uptake [mouse PTX3 (Ptx3)] and cholesterol efflux [mouse ABCA1 (Abca1)] were selected, and their mRNA levels were verified in mouse M1 macrophages. Ptx3 transcripts were reduced, and Abca1 transcripts were increased in Cebpd−/− M1 macrophages (Supplementary material online, Figure S3A). Meanwhile, the expression patterns of PTX3 and ABCA1 in human THP-1 M1 macrophages were also the same as those observed in the mouse M1 macrophages (Supplementary material online, Figure S3B). These results indicate that common mechanisms involving the CEBPD-mediated PTX3 and ABCA1 gene transcription exist in mice and humans. We also examined the responses of SR-A and CD36 (which indicate LDL uptake) and ABCG1 and SR-BI (which indicate cholesterol efflux) in THP-1 M1 macrophages. Consistent with the results of the NGS profiling assay, the knockdown of CEBPD in THP-1 M1 macrophages also increased CD36 gene transcription, but the transcriptions of the other genes that were examined were not significantly altered (Supplementary Material, Figure S3C).
In addition, probucol, which is an ABCA1 inhibitor, and recombinant PTX3 protein (baculovirus-expressed PTX3, bvPTX3) were used to assess the involvements of Abca1 and Ptx3 in Cebpd-induced lipid accumulation. The results revealed that treatment with probucol or bvPTX3 restored Cebpd deficiency-attenuated lipid accumulation in mouse M1 macrophages (Figure 4A). Moreover, pinocytosis assays revealed that bvPTX3 promoted Cebpd deficiency-reduced LDL uptake in a dose-dependent manner, and BODIPY–cholesterol efflux assays revealed that probucol attenuated Cebpd deficiency-increased cholesterol efflux in mouse M1 macrophages (Figure 4B and C). The above results suggest that Ptx3 and Abca1 participate in Cebpd-induced lipid accumulation in M1 macrophages.
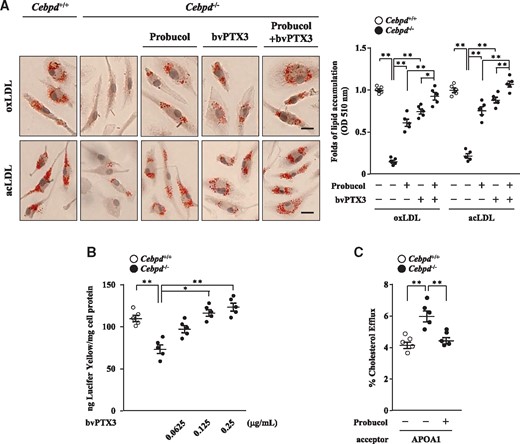
Ptx3 and Abca1 are involved in Cebpd deficiency-attenuated lipid accumulation in mouse M1 macrophages. (A) Monocytes were isolated from the bone marrow of WT and Cebpd-/- mice and then polarized into M1 macrophages. Cebpd-/- M1 macrophages were treated with or without ABCA1 inhibitor probucol, recombinant PTX3 protein bvPTX3 (0.125 µg/mL), or probucol combined with bvPTX3 (0.125 µg/mL). The macrophages (2×105/mL) were then treated with oxLDL or acLDL for 3 days and subjected to Oil Red O stainings. Images are representative of five independent experiments (scale bar: 5 µm). (B) WT and Cebpd-/- M1 macrophages (2×105/mL) were added with Lucifer Yellow-dissolved medium and treated with or without bvPTX3 at the indicated concentrations (n = 5). (C) Following labeling with medium containing oxLDL and BODIPY-cholesterol, WT and Cebpd-/- M1 macrophages (2×105/mL) were pre-treated with or without probucol. The macrophages were then treated with APOA1 which was used as a cholesterol acceptor (n = 5). Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons.
3.5 CEBPD activates PTX3 to promote macropinocytosis of LDL and represses ABCA1 gene transcription in M1 macrophages
We previously found that CEBPD activates PTX3 gene transcription in response to IL-1β in astrocytes.25 Here, we tested whether CEBPD activated PTX3 gene transcription in response to oxLDL or acLDL in M1 macrophages. Western blot analyses revealed that the loss of Cebpd and CEBPD in mouse and THP-1 M1 macrophages, respectively, suppressed oxLDL-induced Ptx3 and PTX3 expression, respectively (Figure 5A). Concerning the low transfection efficiency in mouse primary macrophages, we tested whether CEBPD could regulate PTX3 gene transcription in human THP-1 cells. The overexpression of CEBPD promoted PTX3 reporter (pPTX3, −473/+60) activity in THP-1 cells (Figure 5B, left panel), whereas the knockdown of CEBPD repressed oxLDL- or acLDL-induced PTX3 reporter activity in THP-1 cells (Figure 5B, right panel). Furthermore, a ChIP assay revealed that CEBPD bound to the PTX3 gene promoter (-503/-143) in vivo (Figure 5C). The above results suggest that CEBPD activates PTX3 expression by directly activating PTX3 gene transcription in M1 macrophages.
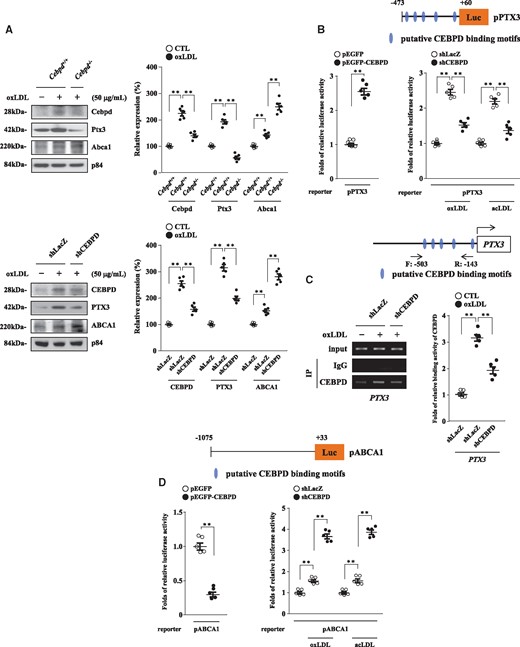
CEBPD activates PTX3 gene transcription and represses ABCA1 gene transcription in M1 macrophages. (A) WT and Cebpd-/- M1 macrophages (2×105/mL) were treated with or without oxLDL. THP-1 cells were pre-infected with lentiviruses encoding shLacZ or shCEBPD and then differentiated into M1 macrophages. The macrophages (2×105/mL) were then also treated with or without oxLDL. Whole cell lysates were harvested for western blot analyses. All images are representative of five independent experiments. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (B) PTX3 reporters (pPTX3, −473/+60) were co-transfected with or without CEBPD expression vectors. THP-1 cells (1×106/mL) were pre-infected with lentiviruses encoding shLacZ or shCEBPD and then transfected with PTX3 reporters with or without oxLDL or acLDL (n=5). The lysates of the transfected cells were harvested for luciferase assays. Statistical significance was determined using Mann–Whitney test and Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (C) THP-1 cells (1×106/mL) were pre-infected with lentiviruses encoding shLacZ or shCEBPD and then treated with or without oxLDL. As a positive control, PCR amplification was also performed using 1/20 of the input DNA from chromatin before the immunoprecipitation (IP) step. Chromatin was isolated from cells, and the IP step was performed using a CEBPD antibody. “−503/−143” indicates the PCR product following specific primer amplification using the purified template from the IP step. Images are representative of five independent experiments. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (D) The ABCA1 reporters (pABCA1, −1075/+33) were co-transfected with or without CEBPD expression vectors. THP-1 cells (1×106/mL) were pre-infected with lentiviruses encoding shLacZ or shCEBPD and then transfected with ABCA1 reporters with or without oxLDL or acLDL (n=5). The lysates of the transfected cells were harvested for luciferase assays. Statistical significance was determined using Mann–Whitney test and Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons.
Pinocytosis has been suggested to participate in the uptake of LDL. Although PTX3 has been suggested to facilitate the phagocytosis of recognized pathogens by macrophages, the effect of PTX3 on the regulation of LDL uptake remains unknown. We thus assessed whether PTX3 functioned in LDL uptake by pinocytosis. Oil Red O stainings revealed that the knockdown of Ptx3 reduced lipid accumulation and that treatment with bvPTX3 restored macropinocytosis inhibitor AS605240-attenuated lipid accumulation in mouse M1 macrophages (Supplementary Figure S4A and B). These data are the first evidence to indicate the involvement of PTX3 in macropinocytosis.
In addition, the formation of macrophage foam cells is tightly controlled by ineffective ABCA1-promoted cholesterol efflux. Western blot analyses revealed that the loss of Cebpd and CEBPD in mouse and THP-1 M1 macrophages, respectively, strengthened oxLDL-induced Abca1 and ABCA1 expression, respectively (Figure 5A). Considering the low transfection efficiency in mouse primary macrophages, we tested whether CEBPD could regulate ABCA1 gene transcription in human THP-1 cells. The overexpression of CEBPD repressed ABCA1 reporter (pABCA1, −1075/+33) activity in THP-1 cells (Figure 5D, left panel), whereas the knockdown of CEBPD strengthened oxLDL- or acLDL-induced ABCA1 reporter activity in THP-1 cells (Figure 5D, right panel). The above results suggest that CEBPD inhibits ABCA1 expression by negatively regulating ABCA1 gene transcription in M1 macrophages.
3.6 Simvastatin suppresses CEBPD expression by inhibiting the p38MAPK/CREB pathway, and it attenuates lipid accumulation in M1 macrophages
As mentioned above, statins have anti-inflammatory effects in addition to their cholesterol-lowering activities. As illustrated in Figure 2, p38MAPK is an upstream regulator of CEBPD gene transcriptional activation in response to modified LDL in macrophages. It has been reported that statins suppress oxLDL-induced macrophage proliferation via the inactivation of the p38MAPK pathway.26 We thus examined whether statins regulated p38MAPK activity to further affect the modified LDL-induced expression of CEBPD in M1 macrophages. RT-PCR assays and western blot analyses revealed that simvastatin (moderate intensity, lipophilic) suppressed oxLDL- or acLDL-induced CEBPD expression in THP-1 M1 macrophages in a dose-dependent manner (Figure 6A). Western blot analyses also revealed that simvastatin suppressed oxLDL- or acLDL-induced phospho-p38 (p-p38), phospho-Creb (p-Creb), and Cebpd expression in mouse M1 macrophages (Figure 6B). Given the low transfection efficiency in mouse primary macrophages, we tested whether simvastatin could regulate CEBPD gene transcription in human THP-1 cells. Treatment with simvastatin repressed oxLDL-, acLDL-, or p38MAPK upstream activators (MKK3Ac or MKK6Ac expression vector)-induced CEBPD reporter activity in THP-1 cells (Figure 6C). In addition, the results revealed that simvastatin repressed oxLDL-induced CEBPD reporter activity in mouse M1 macrophages that were isolated from humanized CEBPD reporter mice (Figure 6D). RT-qPCR assays and western blot analyses revealed that simvastatin suppressed oxLDL- or acLDL-induced Cebpd, Ptx3, and Abca1 expression in mouse M1 macrophages (Supplementary material online, Figure S5A), whereas the overexpression of CEBPD restored Ptx3 transcripts and further attenuated Abca1 transcripts under simvastatin treatment (Figure 6E). Moreover, treatment with simvastatin attenuated lipid accumulation upon the addition of modified LDL to mouse M1 macrophages (Supplementary material online, Figure S5B). These results suggest that simvastatin suppresses CEBPD expression by inhibiting the p38MAPK/CREB pathway and attenuates lipid accumulation in M1 macrophages.
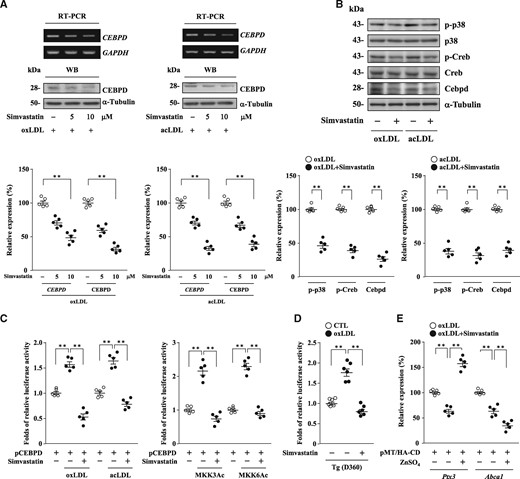
Simvastatin suppresses CEBPD expression by inhibiting the p38MAPK/CREB signaling pathway in M1 macrophages. (A) Following pre-treatment with simvastatin at the indicated concentrations, THP-1 M1 macrophages (2×105/mL) were then treated with oxLDL or acLDL. Total RNA and whole cell lysates were harvested for RT-PCR assays and western blot analyses, respectively. One of five independent experiments is shown. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (B) Following pre-treatment with or without simvastatin (10 µM), mouse M1 macrophages (2×105/mL) were then treated with oxLDL or acLDL. Whole cell lysates were harvested for western blot analyses. Images are representative of five independent experiments. Statistical significance was determined using Mann–Whitney test. (C) THP-1 cells (1×106/mL) were transfected with CEBPD reporters with or without simvastatin (10 µM). The transfectants were then treated with or without oxLDL or acLDL, or co-transfected with or without MKK3Ac or MKK6Ac expression vectors (n = 5). The lysates of the transfected cells were harvested for luciferase assays. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (D) Monocytes were isolated from the bone marrow of humanized CEBPD reporter mice and then differentiated into macrophages. Following pre-treatment with or without simvastatin (10 µM), mouse primary macrophages (2 × 105/mL) were then treated with or without oxLDL (n = 6). Whole cell lysates were harvested for luciferase assays. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. (E) Mouse M1 macrophages (2 × 105/mL) were transfected with zinc-inducible CEBPD expression vectors with or without simvastatin (10 µM). The transfectants were then treated with or without ZnSO4 under oxLDL treatment (n = 5). Total RNA was harvested for RT-qPCR assays. Statistical significance was determined using Kruskal–Wallis test followed by Dunn’s post-hoc test for multiple comparisons.
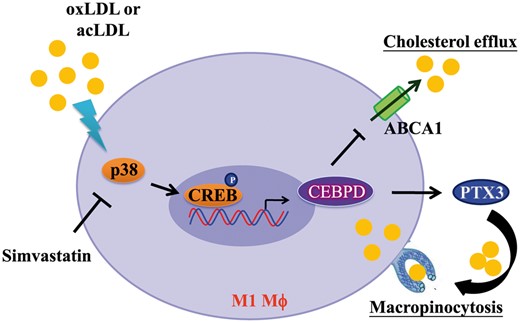
Mechanisms underlying the lipid accumulation in M1 macrophages. In response to modified LDL, the p38MAPK/CREB pathway contributes to CEBPD activation which promotes lipid accumulation in M1 macrophages. The underlying mechanisms involved in this process includes an increase in pentraxin 3 (PTX3)-mediated macropinocytosis of LDL and a reduction in ATP-binding cassette subfamily A member 1 (ABCA1)-mediated cholesterol efflux. Also, simvastatin (a HMG–CoA reductase inhibitor) suppresses CEBPD expression by inhibiting the p38MAPK/CREB pathway and attenuates lipid accumulation in M1 macrophages.
4. Discussion
We find that CEBPD protein colocalizes with macrophages in human and mouse atherosclerotic plaques and that Cebpd deficiency in bone marrow cells suppresses atherosclerotic lesions in hyperlipidemic Apoe−/− mice. In response to modified LDL, the p38MAPK/CREB pathway contributes to CEBPD activation, which promotes lipid accumulation in M1 macrophages but not in M2 macrophages. Moreover, CEBPD activates PTX3 to promote the macropinocytosis of LDL and represses ABCA1 to impair the intracellular cholesterol efflux in M1 macrophages. Taken together, our results link CEBPD with the increased uptake of LDL and impaired cholesterol efflux, which lead to lipid accumulation in polarized macrophages, especially M1 macrophages. We also demonstrate that simvastatin could target CEBPD to block lipid accumulation in a manner not directly related to cholesterol lowering in M1 macrophages (Figure 7).
4.1 CEBPD plays a functional role in lipid accumulation in M1 macrophages
The expression of CEBPD is detectable in cells, including macrophages,13 smooth muscle cells,10 and endothelial cells,27 and is involved in the regulation of multiple biological functions. For these reasons, CEBPD signals seem ubiquitous in human atherosclerotic plaques (Figure 1A). In this study, we do not rule out the possibility that CEBPD plays functional roles in other cell types and contributes to atherosclerotic pathogenesis. However, at least in part, we discover a new insight into CEBPD biology in the macrophage lipid accumulation of atherosclerotic plaques. In addition, in response to different microenvironmental signals, macrophages can be polarized and acquire specific phenotypes such as M1 or M2. Some studies have indicated that CREB might be a pivotal transcription factor in macrophage polarization promoting M2-associated genes while repressing M1 activation.28–31 Interestingly, in this study, we find that the p38MAPK/CREB pathway-mediated CEBPD is activated in both M1 and M2 macrophages (Figure 3A and B, Supplementary material online, Figure S1A). M1 activation is stimulated by LPS or IFNγ, and M2 activation is stimulated by IL-4/IL-13 or PGE2.24 Although there are a variety of stimuli for each type, this study at least partially demonstrate the influence of CEBPD on M1 macrophages (which are activated by LPS) but not on M2 macrophages (which are activated by IL-4/IL-13) in lipid accumulation. These findings again illustrate the complexity and heterogeneity of macrophage phenotypes and functions.
4.2 PTX3 promotes lipid accumulation via macropinocytosis of LDL in M1 macrophages
Several studies have demonstrated that PTX3 might represent an early marker of myocardial lesions32,33 and that it is expressed in macrophages in advanced human atherosclerotic plaques.34–37 In addition to activating the classical pathway for complement activation and facilitating the phagocytosis of recognized pathogens by macrophages and dendritic cells,38–40 PTX3 also restricts the cross-presentation of antigens that are derived from apoptotic cells.41 However, the potential involvement of PTX3 in macropinocytosis remains unknown. Macropinocytosis is a signal-dependent process that could respond to phorbol myristate acetate,42 major histocompatibility complex presentation,43,44 and the activations of PI3K/AKT,45 PAK1,46 and Rac.47 PTX3, as a soluble pattern recognition receptor, recognizes bacteria and promotes both phagocytosis and immune activation by interacting with complement proteins and IgG receptors (FcγRs) that are expressed on innate effector cells.48 FcγRs are detectable in macrophages and could directly activate Rac,49,50 PI3K/AKT,51 and PAK1.52 However, the functions and links among PI3K/AKT, Rac, and PAK1 in M1 macrophages remain unclear. Although AS605240 has been suggested to inhibit PI3Kγ-induced macropinocytosis,53 we cannot rule out the possibility that PTX3 could restore AS605240-inhibited macropinocytosis via a PI3Kγ-independent pathway. Here, we provide the first evidence demonstrating that macropinocytosis is the mechanism underlying PTX3-promoted lipid accumulation in M1 macrophages, and the signaling pathways that are responsive to PTX3 in terms of contributing to the macropinocytosis of LDL need to be investigated in the future.
4.3 The repression of ABCA1 gene transcription might be mediated through a miR33-independent pathway
We found that CEBPD repressed ABCA1 gene transcription in M1 macrophages. However, using a promoter prediction program, we found that there was no CEBPD binding site on the ABCA1 gene promoter (−1075/+33) (Figure 5D). Although miR-33 has been suggested to repress ABCA1 gene transcription,54 miR-33 is not responsive to CEBPD in our NGS profiling assay (data not shown). Furthermore, we demonstrate that PTX3 could suppress ABCA1 expression through PPARγ down-regulation and reduce cholesterol efflux in M1 macrophages (Supplementary material online, Figure S6A and B). These findings agree with a study that demonstrated PTX3 could inhibit cholesterol efflux by down-regulating the PPARγ/LXRα/ABCA1 pathway in macrophages.55 However, the factors with which CEBPD cooperates to repress ABCA1 gene transcription in M1 macrophages remain to be resolved.
4.4 CEBPD-mediated lipid accumulation could represent a new therapeutic target for cardiovascular diseases
In this study, we find that modified LDL-induced CEBPD gene transcription is repressed by simvastatin via the inactivation of the p38MAPK pathway in macrophages. Interestingly, Ptx3 expression was positively regulated when simvastatin suppressed Cebpd expression in mouse M1 macrophages, which indicated that Cebpd is a critical up-regulator of Ptx3 expression. However, the change in Abca1 expression was not reversed when simvastatin was used to suppress Cebpd expression (Supplementary material online, Figure S5A), which implied that a Cebpd-independent pathway is responsive to simvastatin and that it suppresses Abca1 expression. The current findings suggested that statins lead to an increase in miR-33, which results in the partial repression of ABCA1 gene transcription.54 It has been speculated that the miR-33-dependent silencing of the sterol transporter ABCA1 evolved to ensure the minimal loss of sterols during conditions of very low intracellular cholesterol that could compromise cell viability. This speculation provides an explanation for why simvastatin suppresses Abca1 expression in this study (Supplementary material online, Figure S5A). However, simvastatin could still attenuate lipid accumulation upon modified LDL treatment in mouse M1 macrophages (Supplementary material online, Figure S5B). Therefore, whether Ptx3-promoted LDL macropinocytosis is involved in simvastatin-attenuated lipid accumulation in M1 macrophages deserves further investigation, and this process could serve as a new therapeutic target for the treatment of cardiovascular diseases.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Author contributions
H.-Y.L. designed and performed most of the experiments, analyzed the data, interpreted the results, and wrote the manuscript. L.-W.H. performed the immunofluorescence and the animal studies. H.-H.T. constructed the plasmids, produced the silencing RNAs, and performed the animal studies. Y.-C.L. produced the recombinant proteins. S.-H.Y. assisted with the animal studies. P.-Y.L. assisted with the human studies and provided helpful suggestions for designing the experiments. J.-M.W. designed the experiments, edited the manuscript, and directed the project.
Acknowledgements
We thank. Tzong-Shyuan Lee for kind consultation.
Conflict of interest: none declared.
Funding
This study was supported by grant MOST 106-2320-B-006-063-MY3 from the Ministry of Science and Technology and grant NHRI-EX106-10422BI from the National Health Research Institutes.
References
Author notes
The last two authors contributed equally to the study.
Time for primary review: 38 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}