




Abstract
It is widely believed that nootropic (cognition-enhancing) agents produce their therapeutic effects by augmenting excitatory synaptic transmission in cortical circuits, primarily through positive modulation of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptors (AMPARs). However, GABA-mediated inhibition is also critical for cognition, and enhanced GABA function may be likewise therapeutic for cognitive disorders. Could nootropics act through such a mechanism as well? To address this question, we examined the effects of nootropic agents on excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs) recorded from layer V pyramidal cells in acute slices of somatosensory cortex. Aniracetam, a positive modulator of AMPA/kainate receptors, increased the peak amplitude of evoked EPSCs and the amplitude and duration of polysynaptic fast IPSCs, manifested as a greater total charge carried by IPSCs. As a result, the EPSC/IPSC ratio of total charge was decreased, representing a shift in the excitation–inhibition balance that favors inhibition. Aniracetam did not affect the magnitude of either monosynaptic IPSCs (mono-IPSCs) recorded in the presence of excitatory amino acid receptor antagonists, or miniature IPSCs (mIPSCs) recorded in the presence of tetrodotoxin. However, the duration of both mono-IPSCs and mIPSCs was prolonged, suggesting that aniracetam also directly modulates GABAergic transmission. Cyclothiazide, a preferential modulator of AMPAR function, enhanced the magnitude and duration of polysynaptic IPSCs, similar to aniracetam, but did not affect mono-IPSCs. Concanavalin A, a kainate receptor modulator, had little effect on EPSCs or IPSCs, suggesting there was no contribution from kainate receptor activity. These findings indicate that AMPAR modulators strengthen inhibition in neocortical pyramidal cells, most likely by altering the kinetics of AMPARs on synaptically connected interneurons and possibly by modulating GABAA receptor responses in pyramidal cells. This suggests that the therapeutic actions of nootropic agents may be partly mediated through enhanced cortical GABAergic inhibition, and not solely through the direct modification of excitation, as previously thought.
Introduction
Nootropic (cognitive-enhancing) agents are potent, positive modulators of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptor (AMPAR) function (Ito et al., 1990; Stäubli et al., 1994). Their enhancement of excitatory transmission is thought to occur through inhibition of AMPAR desensitization and deactivation, possibly via allosteric modulation of receptor–ionophore complexes and, in turn, altered channel kinetics (Trussell and Fischbach, 1989; Isaacson and Nicoll, 1991; Tang et al., 1991; Vyklicky et al., 1991; Yamada and Tang, 1993). In vivo, AMPAR modulators offset age-related memory deficits in animal models and improve mnemonic function in a variety of learning paradigms (Shors et al., 1995; Granger et al., 1996). Not surprisingly, nootropics are being investigated for the treatment of memory disorders such as Alzheimer's Disease (Danysz, 2002).
Inhibition is also important in the regulation of cognitive operations, possibly by governing the timing of synaptic inputs (McBain et al., 1999). Although inhibition's exact role in higher cortical functions remains unresolved, converging lines of evidence indicate that local inhibitory circuits are critical for information processing, particularly during memory and other cognitive tasks (Rao et al., 2000; Constantinidis et al., 2002). Selective alterations in GABAergic inhibition in specific cortical subregions are associated with cognitive disorders such as schizophrenia and Alzheimer's Disease (Solodkin et al., 1996; Benes and Beretta, 2001; Zhong et al., 2003). For example, schizophrenia subjects appear to have gene expression defects in a particular subclass of prefrontal cortex GABA neurons, which could directly contribute to the pathophysiology of cognitive decline characterizing this disease (Hashimoto et al., 2003). Moreover, it was recently demonstrated that administering GABA receptor agonists improves visual cortical dysfunction in aged monkeys, lending further support to the concept that loss of GABAergic inhibition underlies age-related decline in cortical function, and that restoration of GABA function might have therapeutic value for cognitive disorders (Leventhal et al., 2003). If agents that modulate GABAergic inhibition can improve cognition, could nootropics act through such a mechanism as well?
The actions of AMPAR modulators on GABA inhibition have not been well-studied. Field recordings in hippocampal slices suggest that two nootropic agents, BDP-12 and IDRA 21, enhance disynaptic inhibitory events in CA1 pyramidal cells, presumably by acting on AMPARs on presynaptic interneurons (Arai et al., 1996a,b). However, the action of nootropics on local circuits remains largely unknown. We previously reported that inhibitory interneurons in neocortex, unlike those in hippocampus, can be driven solely and maximally by non-NMDA (AMPA/kainate) ionotropic glutamatergic transmission, independent of the level of NMDA receptor-mediated transmission (Ling and Benardo, 1995). Based upon these data, we posit that a major effect of nootropic agents in neocortex is enhancement of downstream GABAergic inhibition. To explore this possibility, we compared the effects of three non-NMDA receptor modulators on fast inhibitory and excitatory postsynaptic currents (IPSCs and EPSCs): (i) aniracetam, a pyrrolidinone which slows AMPA and kainate receptor desensitization and deactivation, possibly by slowing channel closing (Tang et al., 1991; Hestrin, 1992); (ii) cyclothiazide, a benzothiazide which is a potent, selective inhibitor of AMPAR desensitization that may act by stabilizing a closed channel state (Partin et al., 1993); and (iii) concanavalin A (Con A), a lectin which strongly and irreversibly blocks kainate receptor desensitization, but has only weak effects on AMPARs (Partin et al., 1993). Our findings indicate that positive modulation of AMPAR-mediated excitation enhances fast inhibitory strength, augmenting both the maximal magnitude and duration of fast GABAergic IPSCs in target pyramidal cells. The results of this study have important implications for the role of GABA inhibition in normal cognition and in the pathophysiology of degenerative states that target memory and higher cortical function, and perhaps can serve to focus future therapeutic interventions.
Materials and Methods
Slice Preparation
Cortical slices were prepared from 18- to 30-day old Sprague–Dawley rats (80–100 g) as previously described (Ling and Benardo, 1999). Rats were anesthetized by intramuscular injection of ketamine (87 mg/kg) and xylazine (13 mg/kg), then killed by decapitation. The brains were quickly removed and placed in cold physiological saline. Coronal slices (300–400 μm) were cut with a Vibratome tissue slicer (Vibratome Co., St Louis, MO) and then transferred to an antechamber where they rested submerged in warm (35.5 ± 1°C), oxygenated (95% O2–5% CO2) physiological saline. Slices were allowed to incubate for at least 1h prior to recording. Following incubation, a single slice was transferred to a recording chamber placed on the stage of an upright microscope (Zeiss Axioskop, Carl Zeiss, Inc., Thornwood, NY), and perfused with warm (34–35°C) oyxgenated saline at ∼5 ml/min. Normal, external physiological saline contained 124 mM NaCl, 5 mM KCl, 26 mM NaHCO3, 1.6 mM MgCl2, 2 mM CaCl2 and 10 mM glucose, and was continuously bubbled with a mixture of 95% O2–5% CO2 (pH between 7.35 and 7.4). All drugs were delivered directly through the perfusate. Stocks of aniracetam and cyclothiazide were prepared in dimethylsulfoxide (DMSO). Dilutions were made before each experiment and the highest concentration of DMSO used was 0.1%. Aniracetam, cyclothiazide, 6-cyano-7-nitroquinoxalone-2,3-dione (CNQX) and 3-(RS)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP) were obtained from Tocris Cookson (Ballwin, MO). All other chemicals were obtained from Sigma Chemical (St Louis, MO).
Electrophysiological Recording and Analysis
Whole-cell recordings were obtained from layer V pyramidal cells in cortical slices, visualized and accessed using infrared differential interference contrast (IR-DIC) optics with a ×60 water immersion objective. Patch electrodes were pulled to tip resistances of 2–5 MΩ and filled with a solution composed of (in mM): 130 Cs-methanesulfonate, 2 MgCl2, 2 CaCl2, 10 EGTA, 10 HEPES, 2 Na-ATP and 10 QX-314, pH 7.25, adjusted with CsOH. Cesium was used to block potassium currents, including slow GABAB IPSCs. QX-314 was included to block voltage-dependent sodium currents and thus reduce cell-spiking. Currents were recorded under voltage-clamp with a Warner PC-501A patch clamp amplifier (Warner Instrument Corp., Hamden, CT). Critieria for cell acceptability were the same as previously detailed (Ling and Benardo, 1995). Cells accepted for study had resting input resistances of ≥100 MΩ (typically ≥150 MΩ) and access resistances ≤20 MΩ (typically <15 MΩ). Cells were discarded if access resistance increased significantly (>20%) during the experiment. Membrane holding potentials were not corrected for the junction potential between bath and pipette solutions (∼9 mV). Signals were digitized at 47 kHz via a 14-bit PCM interface (VR-10B Digital Data Recorder, Instrutech Corp., Elmont, NY) and stored on VHS videotape for posthoc analysis. Recorded data were filtered off-line at 1–5 kHz (–3 dB, four pole Bessel) and digitally sampled at 5–20 kHz with pCLAMP 8.0 software (Axon Instruments, Foster City, CA) running on a PC/AT-compatible Pentium-based microcomputer.
Synaptic currents were evoked by extracellular stimulation with tungsten electrodes placed in layer VI, lateral to the recording electrode. Cathodal shocks (2–10 V; 200 μs) were delivered through a stimulus isolation unit (World Precision Instruments, Sarasota, FL) at a low frequency of 0.1 Hz. Total charge flux, defined as the integral of the current trace, was calculated from the point of current onset to its decay back to baseline using piecewise approximations of IPSC integrals with 0.5 ms bins. Because of the variability in IPSC values between neurons, input–output plots were constructed using averaged normalized values and ‘relative’ stimulus intensities (i.e. adjusted such that 0 V represents threshold stimulus) (Ling and Benardo, 1995).
Spontaneous miniature synaptic currents were detected and measured using Mini Analysis software (Synaptosoft, Inc., Decatur, GA), which identifies spontaneous events on the basis of several criteria including threshold amplitude and the area under each event (Ling and Benardo, 1999). As a routine check, we visually inspected all spontaneous events detected by the software and rejected any which did not exhibit the general shape expected for synaptic currents. Background peak-to-peak noise was measured from quiescent sections of records (i.e., devoid of spontaneous events) and ranged from 2 to 5 pA.
All data throughout this report are expressed as means ± SE.
Results
Effect of Aniracetam on Evoked IPSCs
Whole-cell recordings of isolated IPSCs were obtained from layer V neocortical pyramidal cells by clamping cells at the empirically determined EPSC reversal potential (∼0 mV), while EPSCs were likewise recorded at the IPSC reversal (∼−75 mV) (Ling and Benardo, 1995). The addition of aniracetam (2.5–5.0 mM) to the perfusion solution induced an increase in the peak magnitude and duration of evoked EPSCs (Fig. 1A), consistent with the drug's previously reported effects on excitation (Ito et al., 1990; Tang et al., 1991). Aniracetam also enhanced the strength of evoked IPSCs (Fig. 1A), raising the amplitude of the maximal IPSC by 24.9 ± 5.7% (n = 6 cells). This was reflected in the IPSC input–output relationship, which revealed that aniracetam enhanced IPSC amplitude over the entire range of stimulus intensities applied (Fig. 1C). At the lowest strength stimulus that evoked maximal IPSCs, the EPSC/IPSC ratio of peak amplitude was increased by 22.4 ± 3.2% (Fig. 1B; P < 0.05, Student's paired t-test), suggesting an augmentation of excitation over inhibition. However, peak amplitude is only one measure of synaptic strength, as changes in IPSC kinetics and, in turn, total charge surely influence the efficacy of inhibition. We calculated the effects of aniracetam on the total charge carried by synaptic currents, taken as the area under each event. In the presence of aniracetam, the total charge carried by evoked IPSCs was increased at all stimulus intensities (Fig. 1C), with a mean increase of 216.4 ± 15.3% (from 24.4 ± 4.0 to 70.8 ± 6.7 pC). At the same time, the total charge flux of EPSCs was raised by 52.5 ± 13.5% (from 43.2 ± 13.6 to 61.2 ± 14.3 pC). Consequently, the ratio of EPSC/IPSC charge transfer was decreased by 47.8 ± 7.7% (Fig. 1B; P < 0.05) at the lowest strength stimulus that evoked maximal IPSCs. At the highest stimulus intensity applied (i.e. 10 V), the charge transfer ratio was likewise reduced (by 28.0 ± 6.2%). However, it is important to note that all IPSCs were recorded at ∼0 mV holding potentials and thus likely overestimate the values that would be observed at normal cell resting potentials. Nonetheless, these findings suggest that aniracetam induces significant changes in inhibitory synaptic strength.
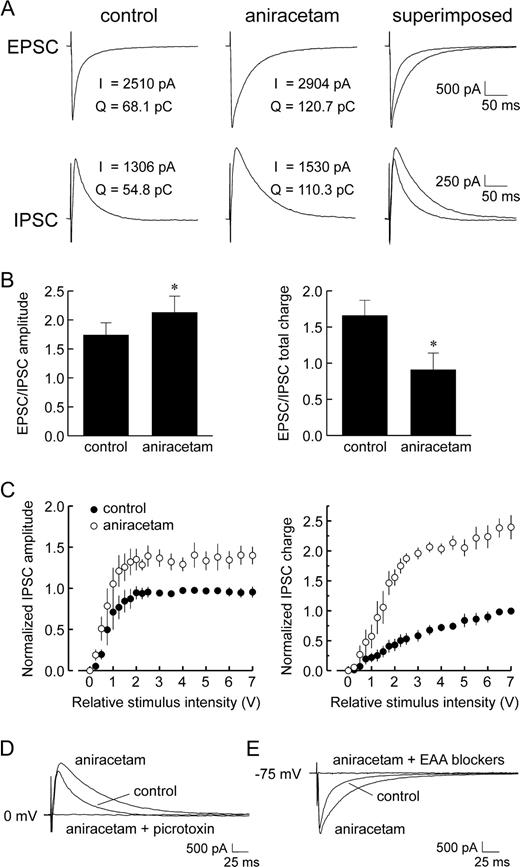
{kind=link}
Effect of aniracetam on the recruitment of IPSCs in layer V pyramidal cells. (A) Bath application of aniracetam (2.5 mM) increased the magnitude and time course of evoked EPSCs (top traces), as expected. Aniracetam also increased the magnitude and time course of maximal amplitude IPSCs (bottom traces), leading to an augmentation of the total charge carried. Shown are responses to 10 V stimuli. (B) The enhancement of postsynaptic currents resulted in an increase in the EPSC/IPSC ratio of peak amplitudes (left panel), but a decrease in the ratio of total charge transfer (right panel). (C) Input–output relationships for IPSC peak amplitude (left panel) and total charge (right panel) were plotted for fast inhibitory events evoked with graded stimulation in control media (filled circles) and following addition of aniracetam (open circles). Over the entire range of stimuli applied, aniracetam increased the peak amplitude and total charge flux of fast IPSCs. Due to cell-to-cell variability in IPSCs, values for amplitude and total charge were normalized to the maximum control values for each cell (Ling and Benardo, 1995). Data points represent the means ± SE of normalized IPSC measurements (n = 5). (D) Addition of picrotoxin (50 μM) to the perfusate completely blocked aniracetam-enhanced IPSCs evoked at 0 mV, confirming that aniracetam did not lead to the activation of non-GABAA receptor-mediated events. (E) In separate experiments, addition of the EAA receptor blockers, CNQX (5 μM) and CPP (10 μM), to the perfusate abolished aniracetam-enhanced EPSCs evoked at −75 mV (i.e. approximate chloride reversal), indicating that there was no alteration in the fast IPSC reversal potential.
Aniracetam-enhanced IPSCs were completely abolished by the subsequent addition of picrotoxin (50 μM; Fig. 1D), indicating that no non-GABAA receptor-mediated currents were recruited as a result of bath application of aniracetam. In separate experiments (n = 3), exposure to the excitatory amino acid (EAA) receptor antagonists CNQX (5 μM) and CPP (10 μM) completely blocked aniracetam-enhanced EPSCs recorded at −75 mV (Fig. 1E), indicating that there was no alteration in the IPSC reversal, as evidenced by the absence of evoked synaptic currents. These initial findings suggested that aniracetam's enhancement of synaptically activated IPSCs resulted from the positive modulation of AMPA/kainate receptors, most likely located on inhibitory interneurons.
Effect of Aniracetam on Monosynaptic IPSCs
However, other effects by aniracetam at postsynaptic or presynaptic loci could not be ruled out. For example, aniracetam-induced augmentation of maximal IPSCs could partly arise from direct modulation of GABA receptor-mediated synaptic transmission. Accordingly, we examined the effects of aniracetam on evoked monosynaptic IPSCs (mono-IPSCs), which represent pharmacologically isolated GABA receptor-mediated postsynaptic events recruited via direct electrical activation of inhibitory interneurons in the absence of excitatory synaptic drive. MonoIPSCs were evoked during blockade of excitatory transmission with saline containing the EAA receptor blockers, CNQX and CPP. Subsequent exposure to aniracetam (2.5–5.0 mM) failed to change the peak amplitude of mono-IPSCs (Fig. 2A) over the entire range of stimuli applied (Fig. 2B; n = 4). However the decay time was prolonged at all stimulus intensities, resulting in a 68.8 ± 8.2% increase in the total charge carried by mono-IPSCs (Fig. 2B, right panel). This alteration in mono-IPSC decay kinetics suggested that aniracetam also directly enhances GABAergic transmission. However, the magnitude of this facilitation was much less than that observed with polysynaptic IPSCs (i.e. in the absence of EAA blockers), suggesting that aniracetam-induced enhancement of fast inhibition is predominantly mediated through actions on excitatory drive to interneurons.
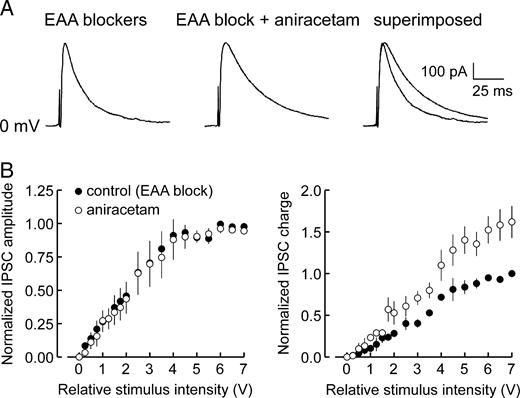
{kind=link}
Effect of aniracetam on monosynaptic IPSCs (mono-IPSCs). (A) Aniracetam did not affect the peak amplitude of mono-IPSCs recorded during blockade of EAA receptors, but did prolong mono-IPSC decay time, resulting in an increase in total charge transfer (example shown, 10 V stimulus). (B) Input–output relationships of IPSCs recorded in control saline (filled circles) and saline containing aniracetam (open circles) revealed that at all stimulus intensities applied, aniracetam had no effect on mono-IPSC peak amplitude (left panel), but did enhance the total charge transfer (right panel). Data points represent mean normalized values ± SE (n = 4; see Fig. 1C).
Effect of Aniracetam on Spontaneous Miniature IPSCs
To further assess possible direct effects of aniracetam on GABA-mediated transmission, we examined miniature IPSCs (mIPSCs) recorded in the presence of tetrodotoxin (TTX, 1–10 μM, Fig. 3A), as mIPSCs are thought to arise from the spontaneous release of single vesicles of transmitter and thus report responses at individual synaptic terminals. Exposure of slices to aniracetam caused a 72.3 ± 10.8% increase in the mean decay time of mIPSCs (n = 6, P < 0.5, from 6.8 ± 0.9 to 12.1 ± 2.2 ms), but had no effect on the mean peak amplitude or rise time (Fig. 3B). The average mIPSC frequency was also unchanged, suggesting that the prolongation of mIPSCs by aniracetam resulted from direct actions on postsynaptic GABAA receptors.
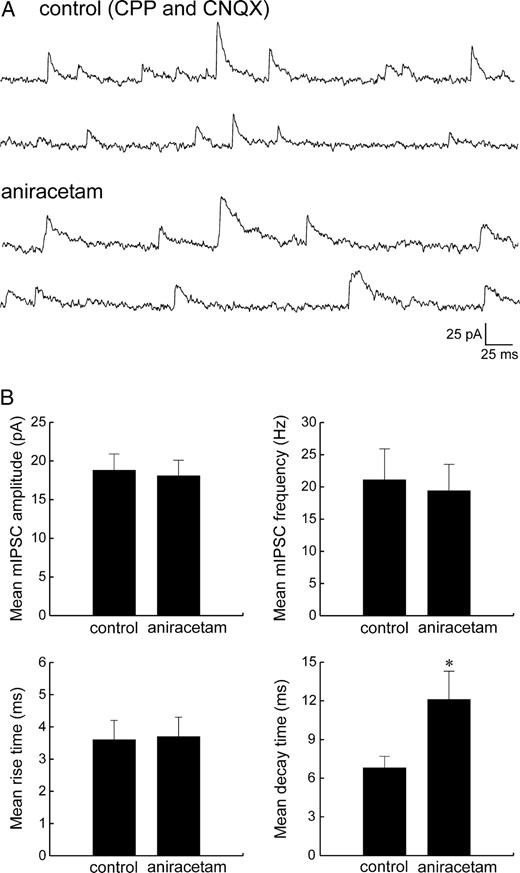
{kind=link}
Effect of aniracetam on miniature IPSCs (mIPSCs). (A) Spontaneously occurring mIPSCs were recorded in the presence of TTX (1–10 μM) at the EPSC reversal potential (∼0 mV), before and after bath application of aniracetam (2.5 mM). (B) Aniracetam had no effect on the mean peak amplitude (top left panel), frequency (top right panel), or 10–90% rise time (bottom left panel) of mIPSCs, but did cause a prolongation of the mean decay time (bottom right panel).
Effect of Cyclothiazide on Evoked IPSCs
Exposure of slices to cyclothiazide (50–100 μM) increased the peak amplitude of evoked EPSCs, confirming its positive modulatory effects on AMPAR-mediated currents (Yamada and Tang, 1993) (Fig. 4A). Cyclothiazide also enhanced evoked inhibitory events in a manner similar to that of aniracetam, increasing the peak amplitude and duration of maximal IPSCs (Fig. 4A) by 25.0 ± 1.7 and 107.1 ± 0.9%, respectively (n = 4 cells). This was likewise reflected in the IPSC input–output relationships, which showed that cyclothiazide enhanced IPSC peak amplitude and total charge over the entire range of stimuli applied (Fig. 4C). At the lowest strength stimulus that evoked maximal IPSCs, the EPSC/IPSC ratio of peak amplitude was increased 27.7 ± 3.1%, but the ratio of total charge transfer was decreased by −34.3 ± 11.8 % (Fig. 4B, P < 0.5). At the highest intensity stimulus applied, the charge transfer ratio was reduced by −16.2 ± 5.9%.
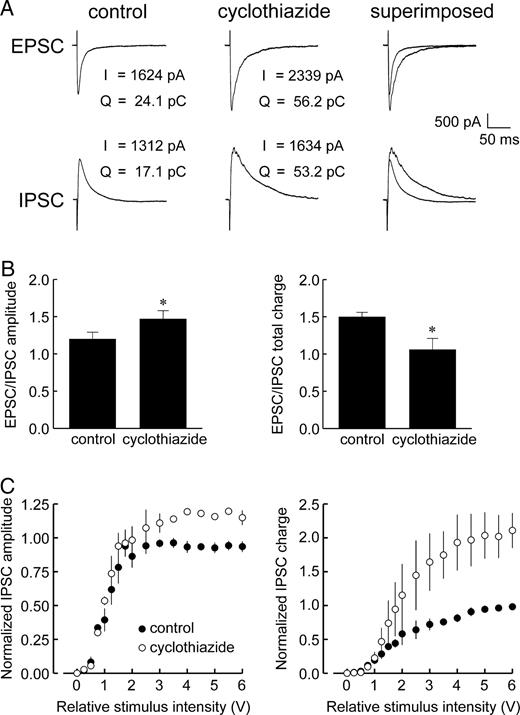
{kind=link}
Effect of cyclothiazide on the recruitment of fast IPSCs. (A) Bath application of cyclothiazide (50 μM) increased the magnitude, time course, and in turn, total charge of evoked EPSCs and IPSCs, thereby mimicking the effects of aniracetam (shown, 10 V stimulus). (B) Similar to aniracetam, cyclothiazide-induced enhancement of postsynaptic currents led to an increase in the EPSC/IPSC ratio of peak amplitude (left panel), but a decrease in the ratio of charge transfer (right panel). (C) Input–output relationships of IPSCs recorded in control saline (filled circles) and saline containing cyclothiazide (open circles) revealed that cyclothiazide increased both IPSC peak amplitude (left panel) and total charge over the entire range of stimuli applied. Data points represent mean normalized values ± SE (n = 4).
It has been reported that cyclothiazide may directly facilitate neurotransmitter release from presynaptic terminals (Diamond and Jahr, 1995; Bellingham and Walmsley, 1999). Accordingly, the effects of cyclothiazide on mono-IPSCs were assessed to evaluate any contributions from direct effects on GABA-mediated transmission, i.e. independent of feed-forward, glutamatergic inputs to inhibitory cells. With EAA receptors blocked, cyclothiazide had no effect on the amplitude or time course of mono-IPSCs (Fig. 5, n = 4), confirming that it does not directly influence GABAA receptor-mediated responses. Thus, cyclothiazide-induced potentiation of polysynaptic IPSCs appears to stem solely from modulation of inhibitory cell AMPAR activity, serving to further distinguish the actions of cyclothiazide from those of aniracetam.
{kind=link}
Effect of cyclothiazide on evoked monosynaptic IPSCs. (A) MonoIPSCs recruited during perfusion with EAA blockers were unaffected by the addition of cyclothiazide to the bathing medium. (B) Input–output plots of IPSC peak amplitude (left panel) and total charge (right panel) indicated that cyclothiazide had no effect on mono-IPSCs over the entire range of stimuli applied. Data points represent mean normalized values ± SE (n = 4).
Effect of Concanavalin A on Evoked IPSCs
In separate experiments, slices were exposed to bathing solutions containing Con A (0.3 mg/ml) to assess potential contributions from kainate receptor activity. Although it is generally accepted that kainate receptors do not appear to have a primary role in excitatory synaptic transmission, it was important to consider them, as recent studies have implicated kainate receptors as mediators of EPSCs in specific neuronal populations, most notably hippocampal inhibitory interneurons (Cossart et al., 1998; Frerking et al., 1998). Con A has been shown to preferentially inhibit kainate receptor desensitization, with only weak effects on AMPARs. Exposure of slices to Con A had no effect on either the amplitude or duration of evoked EPSCs (Fig. 6), confirming that AMPAR are the prime (perhaps sole) mediators of excitatory transmission to cortical principal cells. Con A also failed to alter recruitment of evoked IPSCs, as reflected in IPSC input–output plots of peak amplitude and total charge (Fig. 6B, n = 4 cells), suggesting that positive modulation of kainate receptors is insufficient to potentiate either the magnitude or time course of fast inhibitory events. This finding reinforces the hypothesis that the primary excitatory drive to inhibitory circuits in the deep layers of neocortex is mediated by AMPARs. These results cannot preclude the existence of kainate receptors on neocortical interneurons, but the actions of synaptically released glutamate on kainate receptors does not appear to be a prominent factor in regulating fast IPSC recruitment.
{kind=link}
Effect of concanavalin A on the recruitment of fast IPSCs. (A) Bath application of Con A did not alter the magnitude or time course of maximal amplitude IPSCs. (B) Input–output plots of IPSC peak amplitude (left panel) and total charge (right panel) indicated that Con A had no significant effect on fast IPSCs at any of the stimulus intensities applied. Data points represent mean normalized values ± SE (n = 4).
Discussion
In this study, we examined the effects of nootropic drugs, potent modulators of AMPA/kainate receptor function, on the recruitment of fast inhibitory synaptic events in rat neocortical layer V pyramidal cells to determine whether positive AMPA/kainate receptor modulators can significantly enhance cortical inhibition. We also considered the corollary postulate that nootropic modulation of inhibition provides an overriding influence on dynamic interactions of excitatory and inhibitory networks. Indeed, in addition to their expected actions on synaptic excitation, both aniracetam and cyclothiazide augmented the strength of fast population IPSCs, increasing both the peak amplitude and duration of these events. Con A had no effect on evoked EPSCs or IPSCs, suggesting that modulation of IPSCs by aniracetam and cyclothiazide stems primarily from actions on AMPARs, presumably on interneurons. Aniracetam also extended the duration, but not the amplitude, of mono-IPSCs and mIPSCs, probably indicative of a direct modulatory effect on postsynaptic GABAA receptors that further enhances inhibitory output beyond that of cyclothiazide.
AMPAR Desensitization and Deactivation and Modulation of Evoked GABA Inhibition
We previously found that the magnitude of synaptically driven fast IPSCs plateaus at levels of stimulation where glutamatergic excitation continues to increase, and that this limit was unaltered by changes in NMDA receptor-mediated excitatory drive (e.g. by magnesium-free medium) (Ling and Benardo, 1995). However, if interneurons do not possess significant functional NMDA receptors, as we have suggested (Ling and Benardo, 1995), then changes in the efficacy of NMDA receptor-mediated transmission would have little or no effect on synaptic excitation of interneurons. On the other hand, enhancement of AMPAR-driven currents could lead to increased synaptic excitation in interneurons and, in turn, augmented IPSCs in synaptically connected pyramidal cells, as was observed in the present study.
Previous investigations have shown that increased synchronous firing among inhibitory interneurons can lead to enhancements in the size of population inhibitory events (Michelson and Wong, 1994; Benardo, 1997). Thus, alterations in the kinetics of AMPAR-mediated excitation could result in an extension of the IPSC maximum. Theoretical studies of hippocampal circuits suggests that synchronous firing of cell aggregates is determined, in part, by the kinetics of synaptic events in individual cells (Wang and Buzsáki, 1996; Baker et al., 2002). Rapid desensitization or deactivation of AMPARs would promote fast decay of excitatory synaptic inputs to cells (Trussell and Fischbach, 1989), resulting in less coordinated firing within cell aggregates as individual cells are activated within narrow time windows, leading to states in which some neurons are turning ‘off’ just as others are turning ‘on’. This limits the number of cells that are able to fire in precise synchrony. However, if the time course of excitatory inputs to interneurons was prolonged, the window of probability for coordinated firing among interneurons would increase. In addition, due to significant electrotonic coupling (Benardo, 1997; Deans et al., 2001), the inhibitory neuronal network can function as a high-fidelity throughput system, further fostering synchronous interneuronal firing. Thus, we postulate that blockade of interneuronal AMPAR desensitization or deactivation induces more synchronized firing of the coupled interneuronal network. This would lead to more simultaneous release of GABA and, in turn, larger ensemble GABA IPSCs in target pyramidal cells, increasing the amplitude of fast GABA-mediated events.
An alternative, but not mutually exclusive, possibility is that AMPAR modulators uncover ‘silent’ interneurons (Ling and Benardo, 1999). We previously reported that blockade of GABA uptake had no effect on the maximal IPSC amplitude and suggested, as have others (Segal and Barker, 1984; Dingledine and Korn, 1985), that this was because peak IPSC conductance is dependent only on the number of interneurons that are activated and not on GABA clearance (Ling and Benardo, 1998). If allosteric modulation of AMPA receptor–ionophore complexes by nootropics enhances excitation to levels sufficient to fire subthreshold, putatively ‘silent’ interneurons, then the observed alteration of the IPSC input–output relationship (see Fig. 1C) could result.
The prolongation of mono-IPSC decay time by aniracetam suggests that it also exerts direct effects on GABAA receptor-mediated transmission, independent of AMPAR modulation. Since monosynaptic IPSCs arise from the direct electrical activation of interneurons (i.e. independent of feed-forward synaptic drive), there are at least two possible explanations for aniracetam-induced prolongation of mono-IPSCs: (i) increased release of GABA from inhibitory cells; and (ii) direct modulation of postsynaptic GABAA receptors on target pyramidal cells. The piracetam-related agents, including aniracetam, enhance the activity of L-type, low threshold, voltage-gated calcium channels (Yoshii and Watabe, 1994); this could promote interneuron bursting, leading to extended depolarization of synaptic terminals, increased GABA release and, in turn, prolonged pyramidal cell IPSCs (Ling and Benardo, 1998). Direct facilitation of GABA release by aniracetam represents another possibility, as aniracetam has been reported to enhance neurotransmitter release in other cortical subregions (Shirane and Nakamura, 2001; Togashi et al., 2002; Yu and Cai, 2003). However, our finding that aniracetam did not alter the frequency of TTX-insensitive mIPSCs is evidence against a presynaptic site of action (Hill et al., 1998; Perrais and Ropert, 1999). Previous studies using cultured neurons have shown that some pyrrolidinone nootropic agents interact directly with GABAA receptors, leading to alterations in receptor desensitization and GABA-mediated currents (Huang et al., 1996). Although aniracetam reportedly exhibits low activity at this locus (Nabeshima et al., 1990), altered desensitization of pyramidal cell GABAA receptors by aniracetam could lead to prolongation of fast IPSCs (Vicini et al., 1986; Bai et al., 1999) without necessarily affecting mIPSC frequency, amplitude or rise time.
Implications of AMPAR Modulation of GABAergic Inhibition
Inhibition is critical to the regulation of synaptic activity, serving to check the spread of excitation (Connors and Amitai, 1993) and isolate synaptic potentiation to particular dendritic domains (Holmes and Levy, 1997). As such, GABAergic systems are pivotal in regulating the level of neuronal excitability within the cortex (Miles and Wong, 1987; Alger, 1991) and, in turn, normal brain physiology. The importance of this kind of control is clear from studies showing that blockade of fast inhibition releases excitatory transmission through recurrent pathways (Traub and Wong, 1982; Connors, 1984), often inducing synchronous paroxysmal firing and excitotoxicity. Deficits in inhibition are not limited to seizure disorders, as there is increasing evidence that the genesis of Alzheimer's disease, schizophrenia, and bipolar disorder may, in part, be a consequence of diminished GABA function, due to either the loss of cortical interneurons (Solodkin et al., 1996; Benes and Beretta, 2001) or reduced excitatory synaptic drive to inhibitory circuits (Deakin and Simpson, 1997; Zhong et al., 2003). Further augmentation of GABAergic transmission allows intrinsic inhibitory mechanisms to keep pace with escalating excitation and to compensate for declines in inhibitory function (Yang and Benardo, 1998). Well-known modulators of inhibition include agents which prolong or enhance the actions of GABA on pyramidal cells; these have found applications as effective anticonvulsants, mood stabilizers, sedatives and tranquilizers, examples of which are valproate, phenobarbital and clonazepam (Vicini et al., 1986; Hashimoto et al., 2003).
However, GABA-enhancing drugs can also produce cognitive impairments, such as amnesia, that presumably result from amplified GABAergic inhibition impeding normal cortical function (Costa and Guidotti, 1996). As cognition-enhancers, nootropic agents clearly avoid these deleterious effects. Our data suggest that nootropic-induced amplification of inhibition is counterbalanced by the concurrent augmentation of glutamatergic synaptic excitation, and may serve to promote cognitive performance by providing the inhibitory levels needed to efficiently regulate heightened excitatory drive. This dual enhancement of excitation and inhibition may also give rise to additional therapeutic benefits, as evidenced by studies showing that nootropic agents potentiate the anticonvulsant actions of antiepileptic agents (Hawkins and Mellanby, 1986; Mondadori and Schmutz, 1986; Paulus et al., 1991) while countering memory disturbances caused by epilepsy and antiepileptic drugs (Mondadori and Schmutz, 1986; Pohle et al., 1997).
AMPAR modulators increase inhibitory efficacy in neocortical layer V pyramidal cells by extending the operational limit on GABAAergic IPSC amplitude and prolonging IPSC duration. Given such effects, perhaps we should include these nootropic agents amongst the list of drugs that modulate inhibition. Moreover, these findings suggest that their therapeutic value may well be mediated through alterations of downstream GABAergic inhibition, and not solely through the direct modification of excitation, as previously thought. The possibility of endogenous modulation of desensitization states is also interesting to consider, allowing for another level of dynamic control of local circuit activity.
Although it is possible that nootropic-enhanced AMPAR-mediated events might be capable of overcoming the concomitant augmentation of inhibition, the temporal extension of IPSCs and resulting increases in total charge may promote more efficient filtering of excitatory events, especially those mediated by NMDA receptors. However, it remains unknown whether a key function provided by the GABAergic system is control of NMDA activity, which is somehow lost in diseases targeting cognitive function, and which may be replaced by nootropic agents.
This work was supported by a grant from the New York City Speaker's Fund for Biomedical Research (to D.S.F.L.) and National Institute of Mental Health grants MH-51677 and MH-01431 (to L.S.B.).
References
Alger BE (
Arai A, Kessler M, Rogers G, Lynch G (
Arai A, Guidotti A, Costa E, Lynch G (
Bai D, Pennefather PS, MacDonald JF, Orser BA (
Baker P, Pennefather PS, Orser BA, Skinner FK (
Bellingham MC, Walmsley B (
Benardo LS (
Benes FM, Berretta S (
Connors BW, Amitai Y (
Constantinidis C, Williams GV, Goldman-Rakic PS (
Cossart R, Esclapez M, Hirsch JC, Bernard C, Ben-Ari Y (
Costa E, Guidotti A (
Deakin JF, Simpson MD (
Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL (
Diamond JS, Jahr CE (
Dingledine R, Korn SJ (
Frerking M, Malenka RC, Nicoll RA (
Granger R, Deadwyler S, Davis M, Moskovitz B, Kessler M, Rogers G, Lynch G (
Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA (
Hawkins CA, Mellanby JH (
Hestrin S (
Hill MW, Reddy PA, Covey DF, Rothman SM (
Holmes WR, Levy WB (
Huang CS, Ma JY, Marszalec W, Narahashi T (
Isaacson JS, Nicoll RA (
Ito I, Tanabe S, Kohda A, Sugiyama H (
Leventhal AG, Wang Y, Pu M, Zhou Y, Ma Y (
Ling DSF, Benardo LS (
Ling DSF, Benardo LS (
Ling DSF, Benardo LS (
McBain CJ, Freund TF, Mody I (
Michelson HB, Wong RKS (
Miles R, Wong RKS (
Mondadori C, Schmutz M (
Nabeshima T, Noda Y, Tohyama K, Itoh J, Kameyama T (
Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A (
Paulus W, Ried S, Stodieck SRG, Schimdt D (
Perrais D, Ropert N (
Pohle W, Becker A, Grecksch G, Juhre A, Willenberg A (
Rao SG, Williams GV, Goldman-Rakic PS (
Segal M, Barker JL (
Shirane M, Nakamura K (
Shors TJ, Servatius RJ, Thompson RF, Rogers G, Lynch G (
Solodkin A, Veldhuizen SD, Van Hoesen GW (
Stäubli U, Rogers G, Lynch G (
Tang C-M, Shi Q-Y, Katchman A, Lynch G (
Togashi H, Nakamura K, Matsumoto M, Ueno K, Ohashi S, Saito H, Yoshioka M (
Traub RD, Wong RKS (
Trussell LO, Fischbach GD (
Vicini S, Alho H, Costa E, Mienville JM, Santi MR, Vaccarino FM (
Vyklicky L, Jr., Patneau DK, Mayer ML (
Wang X-J, Buzsáki G (
Yamada KA, Tang C-M (
Yang L, Benardo LS (
Yoshii M, Watabe S. (
Yu S, Cai J (
Author notes
1Department of Physiology and Pharmacology, State University of New York Downstate Medical Center, Brooklyn, NY 11203, USA and 2Department of Neurology, State University of New York Downstate Medical Center, Brooklyn, NY 11203, USA