





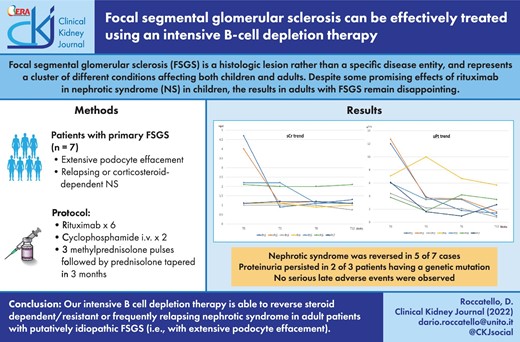
ABSTRACT
Focal segmental glomerular sclerosis (FSGS) is a histologic lesion rather than a specific disease entity and represents a cluster of different conditions affecting both children and adults that includes primary, secondary and genetically mediated forms. These forms can be distinguished by electron microscopy and genetic assessment and show different responsiveness to steroids and immunosuppressants. Despite some promising effects of rituximab in nephrotic syndrome in children, the results in adults with FSGS are disappointing. Our group previously explored the effectiveness of rituximab in eight adult patients with unselected forms of FSGS and achieved a consistent reduction in proteinuria in one case. Following this experience, we developed an alternative therapeutic option intended to enhance the potential of rituximab with the support of other synergic drugs. We herein report the results of this therapeutic protocol (six administrations of rituximab plus two of intravenous cyclophosphamide plus glucocorticoids) in seven prospectively enrolled patients with extensive podocyte effacement and recurrent relapses or steroid dependence.
Patients had a median baseline serum creatinine level of 2.2 mg/dl (range 1–4.7) that decreased to 1.1 mg/dl (range 0.9–2.2) and 1.1 mg/dl (range 0.75–2.21) after 3 and 6 months, respectively, and remained unchanged at 12 months. Three of five patients with renal failure turned to normal function while the other two patients maintained a stable impairment after 18 and 52 months. The median proteinuria decreased from 6.1 g/24 h to 3.5, 3.5 and 1.9 g/24 h at 3, 6 and 12 months, respectively. Specifically, five of seven patients had a partial response at 12 months and became non-nephrotic. One of them had a complete response at 18 months and was still in complete remission at the last follow-up visit at 36 months. Proteinuria persisted unchanged in two of seven patients with a genetic-related disease. No serious late adverse events were observed.
Our results show that intensive B-cell depletion therapy is able to reverse the nephrotic syndrome of steroid-dependent or frequently relapsing adult patients with putatively idiopathic FSGS (i.e. with extensive podocyte effacement).
Lay Summary
Focal segmental glomerular sclerosis (FSGS) is a histologic lesion shared by a cluster of idiopathic, secondary (so-called maladaptive) and genetically mediated glomerular diseases that can be distinguished by combining histologic, clinical and genetic features. Most of the clinical studies published were not based on these combined criteria and mixed cases of putatively different aetiologies, especially in adults. As a result, response to standard therapy (steroids and immunosuppressants), and even to newer approaches (including rituximab), have widely differed. Consequently, patients have often been exposed to prolonged ineffective treatments. We herein report the results of a protocol intended to enhance the potential of rituximab with the synergic support of small doses of cyclophosphamide, combined with three bolus doses of methylprednisolone followed by prednisone tapered until discontinuation by month 3. In the patients with no genetic mutations, this protocol was able to reverse the nephrotic syndrome in severe FSGS adults despite previous recurrent relapses or steroid dependence/resistance.
INTRODUCTION
Focal segmental glomerulosclerosis (FSGS) is a glomerular disorder characterized by the presence of sclerotic lesions at a segmental part of the glomerulus with a focal distribution (more than one glomerulus in the whole biopsy specimen). Rather than a specific disease entity, FSGS is a histologic lesion and represents a cluster of different conditions affecting both children and adults [1] that collectively represent the top causes of end-stage kidney disease due to glomerular diseases [2]. The lesion of FSGS can be classified as primary, secondary, genetic and unknown [3]. The main pathogenic mechanism in primary FSGS, probably promoted by an unknown circulating factor, leads to a widespread foot process effacement of podocytes visible on electron microscopy (EM), hence the term ‘podocytopathy’. Clinically, primary FSGS typically erupts with nephrotic syndrome. Secondary FSGS generally occurs as an adaptive phenomenon that results from a reduction in nephron mass or direct toxicity from drugs or viral infections. In this case, podocyte effacement is not diffuse, and the proteinuria is usually <3.5 g/day or, at most, in the nephrotic range without overt nephrotic syndrome. FSGS can be also caused by a number of genetic mutations in genes that code for proteins expressed in podocytes and at the slit diaphragm. Finally, there are cases of typical FSGS with clinical presentation and EM findings that are similar to those in patients with secondary FSGS but with an unknown aetiology in spite of an extensive clinical and genetic evaluation. These cases are classified as FSGS of unknown cause [4].
Distinguishing between the different forms of FSGS has important implications for treatment and prognosis. Remission of primary FSGS can be achieved by treating patients with steroids, but sometimes the response is either partial or not long lasting. In patients with recurrent relapses or steroid dependence, steroid-sparing immunosuppressive drugs, such as cyclosporine A (CyA), mycophenolate mofetil, azathioprine, tacrolimus, levamisole, cyclophosphamide (CYC) and chlorambucil are usually administered [5]. In an effort to avoid the adverse events and toxicity of long-term treatment with these second-line agents, the role of rituximab (RTX) has been also investigated.
RTX, a chimeric monoclonal antibody targeting CD20-positive B cells, is an essential option in several immune-mediated diseases, including rheumatoid arthritis [6] and anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis [7]. In the context of nephrotic syndrome, RTX has been primarily proposed as an alternative therapy in children with frequently relapsing, steroid-dependent or resistant nephrotic disease [8]. Although not proven, the anti-CD20, B-cell-depleting antibody RTX might reduce the production of putative permeability factors or have direct beneficial effects on podocytes [5].
However, adult patients with steroid-resistant FSGS usually fail to respond.
In a case series of eight adult patients treated with eight weekly doses of 375 mg/m2 of RTX as a monotherapy, a sustained positive and lasting response with a reduction of 80% in the proteinuria value from baseline was shown in one case, while the other seven did not show any consistent benefit [9]. A limitation of that study was that the EM examination was not available. That study concluded that a beneficial role of RTX in the specific sample of patients with widespread foot process effacement could not be ruled out.
Recently the efficacy of the so-called rheumatoid arthritis protocol of RTX (1000 mg, given on days 1 and 15) was evaluated in nine adults with FSGS and diffuse podocyte effacement carefully selected for a high level of soluble urokinase plasminogen activator receptor (suPAR) and evidence of β3 integrin activation. A transient response at 6 months was noted in two patients without a parallel change in the suPAR level. At 12 months there was no statistically significant improvement in the proteinuria level and all participants remained nephrotic [10].
Following our previous experience [9] we developed a therapeutic option intended to enhance the potential of RTX with the support of other synergic drugs to be applied in patients with FSGS with extensive podocyte effacement on the EM examination. This newly designed regimen [intensive B-cell depletion therapy (IBCDT)], which is based on the combination of RTX, CYC and methylprednisolone with subsequent oral prednisone tapering at 3 months, proved to be effective in lupus nephritis [11], severe systemic scleroderma [12] and ANCA-associated vasculitis [13].
MATERIALS AND METHODS
Seven patients with complex cases of primary FSGS were prospectively enrolled in our study, meeting the following criteria: biopsy-proven primary FSGS with extensive podocyte effacement, relapsing or corticosteroid-dependent nephrotic syndrome (defined as proteinuria >3.5 g/day and serum albumin <3 g/dl) and an absence of concomitant infections, comorbid conditions or systemic diseases that could have a pathogenic relationship with nephrotic syndrome. Patients who showed a worsening of the renal parameters (serum creatinine and/or proteinuria) while tapering steroids were defined as steroid dependent.
The main demographic and clinical characteristics were collected for each patient, also searching for possible genetic mutations (Table 1). Indications for starting IBCDT are detailed in Table 1. The extension of foot process effacement, presence (+) or absence (−) of diffuse interstitial fibrosis and arteriolosclerosis were also reported. Laboratory parameters were recorded at baseline and at 3, 6 and 12 months after the IBCDT and at the last visit (duration of follow-up is presented in Table 2). Complete response was defined as a reduction of proteinuria to <0.3 g/day with normalization of albuminemia. Partial response was defined as achievement of halving proteinuria into a subnephrotic range and increasing albuminemia up to 3.5 g/dl.
Demographic, genetic and histological characteristics.
| Renal biopsy | ||||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Age (years) | Gender | Genetic mutation | Relapses or no response, n | Previous treatment | Effacement (%) | Interstitial fibrosis | Arteriosclerosis |
| 1 | 54 | M | No | Relapses (7) | GC, CYC, CyA | 80 | + | + |
| 2 | 62 | M | No | Relapses (3) | GC | 88 | + | + |
| 3 | 51 | F | No | Relapse (1) | GC | 85 | + | − |
| 4 | 32 | M | Yes | No response | GC | 90 | − | − |
| 5 | 39 | M | No | Relapse (1) | GC | 70 | + | − |
| 6 | 26 | F | Yes | No response | GC | 75 | − | − |
| 7 | 53 | F | Yes | No response | GC | 80 | − | + |
| Renal biopsy | ||||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Age (years) | Gender | Genetic mutation | Relapses or no response, n | Previous treatment | Effacement (%) | Interstitial fibrosis | Arteriosclerosis |
| 1 | 54 | M | No | Relapses (7) | GC, CYC, CyA | 80 | + | + |
| 2 | 62 | M | No | Relapses (3) | GC | 88 | + | + |
| 3 | 51 | F | No | Relapse (1) | GC | 85 | + | − |
| 4 | 32 | M | Yes | No response | GC | 90 | − | − |
| 5 | 39 | M | No | Relapse (1) | GC | 70 | + | − |
| 6 | 26 | F | Yes | No response | GC | 75 | − | − |
| 7 | 53 | F | Yes | No response | GC | 80 | − | + |
Relapse: recurrence when patients are still on GC therapy or within 2 weeks of discontinuation; no response: non-responder.
Demographic, genetic and histological characteristics.
| Renal biopsy | ||||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Age (years) | Gender | Genetic mutation | Relapses or no response, n | Previous treatment | Effacement (%) | Interstitial fibrosis | Arteriosclerosis |
| 1 | 54 | M | No | Relapses (7) | GC, CYC, CyA | 80 | + | + |
| 2 | 62 | M | No | Relapses (3) | GC | 88 | + | + |
| 3 | 51 | F | No | Relapse (1) | GC | 85 | + | − |
| 4 | 32 | M | Yes | No response | GC | 90 | − | − |
| 5 | 39 | M | No | Relapse (1) | GC | 70 | + | − |
| 6 | 26 | F | Yes | No response | GC | 75 | − | − |
| 7 | 53 | F | Yes | No response | GC | 80 | − | + |
| Renal biopsy | ||||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Age (years) | Gender | Genetic mutation | Relapses or no response, n | Previous treatment | Effacement (%) | Interstitial fibrosis | Arteriosclerosis |
| 1 | 54 | M | No | Relapses (7) | GC, CYC, CyA | 80 | + | + |
| 2 | 62 | M | No | Relapses (3) | GC | 88 | + | + |
| 3 | 51 | F | No | Relapse (1) | GC | 85 | + | − |
| 4 | 32 | M | Yes | No response | GC | 90 | − | − |
| 5 | 39 | M | No | Relapse (1) | GC | 70 | + | − |
| 6 | 26 | F | Yes | No response | GC | 75 | − | − |
| 7 | 53 | F | Yes | No response | GC | 80 | − | + |
Relapse: recurrence when patients are still on GC therapy or within 2 weeks of discontinuation; no response: non-responder.
Duration of follow-up and laboratory data at baseline and 3, 6, 9 and 12 months and at last visit.
| sCr (mg/dl)/proteinuria (g/24 h) | ||||||
|---|---|---|---|---|---|---|
| Patient | Duration of follow-up (months) | T0 | T3 | T6 | T12 | Last follow-up |
| 1 | 60 | 4.7/12 | 0.9/3.9 | 1.1/1.3 | 1.3/1.4 | 1.3/1.1 |
| 2 | 18 | 4/12.7 | 1.1/3.7 | 1.2/3.6 | 1.1/1.6 | 1/1.8 |
| 3 | 36 | 1/4.4 | 1/2.2 | 1/2.1 | 0.75/0.8 | 0.5/0.3 |
| 4 | 12 | 1.5/7.1 | 1.1/10 | 0.9/6.7 | 1.0/5.7 | – |
| 5 | 52 | 2.2/6 | 2.2/3.5 | 1.9/3.5 | 2.1/1.9 | 2.1/0.8 |
| 6 | 18 | 2.1/3.8 | 2/1.7 | 2/4.2 | 2.1/3.5 | 2.1/3.8 |
| 7a | 14 | 1.1/6.1 | 1.2/1.6 | 1.2/1 | 1.1/2.7 | 1.1/2.6 |
| sCr (mg/dl)/proteinuria (g/24 h) | ||||||
|---|---|---|---|---|---|---|
| Patient | Duration of follow-up (months) | T0 | T3 | T6 | T12 | Last follow-up |
| 1 | 60 | 4.7/12 | 0.9/3.9 | 1.1/1.3 | 1.3/1.4 | 1.3/1.1 |
| 2 | 18 | 4/12.7 | 1.1/3.7 | 1.2/3.6 | 1.1/1.6 | 1/1.8 |
| 3 | 36 | 1/4.4 | 1/2.2 | 1/2.1 | 0.75/0.8 | 0.5/0.3 |
| 4 | 12 | 1.5/7.1 | 1.1/10 | 0.9/6.7 | 1.0/5.7 | – |
| 5 | 52 | 2.2/6 | 2.2/3.5 | 1.9/3.5 | 2.1/1.9 | 2.1/0.8 |
| 6 | 18 | 2.1/3.8 | 2/1.7 | 2/4.2 | 2.1/3.5 | 2.1/3.8 |
| 7a | 14 | 1.1/6.1 | 1.2/1.6 | 1.2/1 | 1.1/2.7 | 1.1/2.6 |
aPatient 7 started maintenance therapy with RTX at month 12.
Duration of follow-up and laboratory data at baseline and 3, 6, 9 and 12 months and at last visit.
| sCr (mg/dl)/proteinuria (g/24 h) | ||||||
|---|---|---|---|---|---|---|
| Patient | Duration of follow-up (months) | T0 | T3 | T6 | T12 | Last follow-up |
| 1 | 60 | 4.7/12 | 0.9/3.9 | 1.1/1.3 | 1.3/1.4 | 1.3/1.1 |
| 2 | 18 | 4/12.7 | 1.1/3.7 | 1.2/3.6 | 1.1/1.6 | 1/1.8 |
| 3 | 36 | 1/4.4 | 1/2.2 | 1/2.1 | 0.75/0.8 | 0.5/0.3 |
| 4 | 12 | 1.5/7.1 | 1.1/10 | 0.9/6.7 | 1.0/5.7 | – |
| 5 | 52 | 2.2/6 | 2.2/3.5 | 1.9/3.5 | 2.1/1.9 | 2.1/0.8 |
| 6 | 18 | 2.1/3.8 | 2/1.7 | 2/4.2 | 2.1/3.5 | 2.1/3.8 |
| 7a | 14 | 1.1/6.1 | 1.2/1.6 | 1.2/1 | 1.1/2.7 | 1.1/2.6 |
| sCr (mg/dl)/proteinuria (g/24 h) | ||||||
|---|---|---|---|---|---|---|
| Patient | Duration of follow-up (months) | T0 | T3 | T6 | T12 | Last follow-up |
| 1 | 60 | 4.7/12 | 0.9/3.9 | 1.1/1.3 | 1.3/1.4 | 1.3/1.1 |
| 2 | 18 | 4/12.7 | 1.1/3.7 | 1.2/3.6 | 1.1/1.6 | 1/1.8 |
| 3 | 36 | 1/4.4 | 1/2.2 | 1/2.1 | 0.75/0.8 | 0.5/0.3 |
| 4 | 12 | 1.5/7.1 | 1.1/10 | 0.9/6.7 | 1.0/5.7 | – |
| 5 | 52 | 2.2/6 | 2.2/3.5 | 1.9/3.5 | 2.1/1.9 | 2.1/0.8 |
| 6 | 18 | 2.1/3.8 | 2/1.7 | 2/4.2 | 2.1/3.5 | 2.1/3.8 |
| 7a | 14 | 1.1/6.1 | 1.2/1.6 | 1.2/1 | 1.1/2.7 | 1.1/2.6 |
aPatient 7 started maintenance therapy with RTX at month 12.
The IBCDT protocol administered to these seven patients consisted of RTX (every 4 weeks followed by doses every 2 months 375 mg/m2), CYC (two pulses of 10 mg/kg on days 4 and 17, with a 30% reduction in patients with an estimated glomerular filtration rate (eGFR) <50 ml/min and methylprednisolone (three bolus doses of 15 mg/kg) followed by oral prednisone (starting dose 50 mg) tapered until complete discontinuation in 3 months. Supportive treatments of nephrotic syndrome (including diuretics, statins, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers) given at the time of the intensive protocol administration were maintained during the follow-up.
All subjects provided written consent prior to treatment, according to the Declaration of Helsinki. This study was performed according to regulations established by the Regional Health Department on the Off-Label Therapy in Rare Diseases in Piedmont (northwest Italy). Statistical analysis for comparison of variables at baseline and follow-up included Student's t-test for normally distributed parameters and the non-parametric Mann–Whitney test for non-normally distributed parameters. Correlations were calculated and significance determined by the Fisher's test. For these analyses, SPSS (IBM, Armonk, NY, USA) software was used and a value of P < .05 was considered significant.
RESULTS
The main demographic, genetic and histological characteristics are summarized in Table 1. Laboratory data at baseline and during follow-up are summarized in Table 2. Three women and four men were recruited with a mean age of 45.3 years (range 26–62). The mean follow-up was 30 months (range 12–60). Three patients (4, 6 and 7) had a genetic mutation (Table 3).
Identified genetic mutations.
| Patient | Gene | Variant identified | Genotype |
|---|---|---|---|
| 1 | No mutation identified | – | |
| 2 | No mutation identified | – | |
| 3 | N/A | – | |
| 4 | FN1, chromosome 2q35 | c.07018+7A>G | Heterozygous |
| 5 | No mutation identified | ||
| 6 | INF2, chromosome 14q32.33 | c.212A>C p.(Gln71Pro) | Heterozygous |
| 7 | CFHR1, chromosome 1q31.3 CFHR3, chromosome 1q31.3 | Deletion chromosome 1: 196794596–196801139 Deletion chromosome 1: 196744006–196762653 | Homozygous |
| Patient | Gene | Variant identified | Genotype |
|---|---|---|---|
| 1 | No mutation identified | – | |
| 2 | No mutation identified | – | |
| 3 | N/A | – | |
| 4 | FN1, chromosome 2q35 | c.07018+7A>G | Heterozygous |
| 5 | No mutation identified | ||
| 6 | INF2, chromosome 14q32.33 | c.212A>C p.(Gln71Pro) | Heterozygous |
| 7 | CFHR1, chromosome 1q31.3 CFHR3, chromosome 1q31.3 | Deletion chromosome 1: 196794596–196801139 Deletion chromosome 1: 196744006–196762653 | Homozygous |
Identified genetic mutations.
| Patient | Gene | Variant identified | Genotype |
|---|---|---|---|
| 1 | No mutation identified | – | |
| 2 | No mutation identified | – | |
| 3 | N/A | – | |
| 4 | FN1, chromosome 2q35 | c.07018+7A>G | Heterozygous |
| 5 | No mutation identified | ||
| 6 | INF2, chromosome 14q32.33 | c.212A>C p.(Gln71Pro) | Heterozygous |
| 7 | CFHR1, chromosome 1q31.3 CFHR3, chromosome 1q31.3 | Deletion chromosome 1: 196794596–196801139 Deletion chromosome 1: 196744006–196762653 | Homozygous |
| Patient | Gene | Variant identified | Genotype |
|---|---|---|---|
| 1 | No mutation identified | – | |
| 2 | No mutation identified | – | |
| 3 | N/A | – | |
| 4 | FN1, chromosome 2q35 | c.07018+7A>G | Heterozygous |
| 5 | No mutation identified | ||
| 6 | INF2, chromosome 14q32.33 | c.212A>C p.(Gln71Pro) | Heterozygous |
| 7 | CFHR1, chromosome 1q31.3 CFHR3, chromosome 1q31.3 | Deletion chromosome 1: 196794596–196801139 Deletion chromosome 1: 196744006–196762653 | Homozygous |
The CD19 depletion lasted up to 18 months, except patients 6 and 7 who showed a repopulation at 11 and 12 months, respectively. This figure is consonant with the long-lasting B-cell depletion achieved with the IBCDT in other pathologic disorders [11, 13].
Patient 1 had seven relapses and was previously treated with prolonged and repeated courses of glucocorticosteroids (GCs), oral CYC and CyA. Patients 2 and 5, previously treated with GCs, had three and one relapses, respectively, before receiving the IBCDT. Patients 3, 4, 6 and 7 did not respond to a full 4-month course of oral GCs.
In five cases, foot process effacement on EM had an extent >80%. Patient 5 (with a genetic mutation) and 6 had an estimated effacement of 70% and 75%, respectively.
Patients had a median baseline serum creatinine (sCr) level of 2.2 mg/dl (range 1–4.7). The median sCr value decreased to 114 mg/dl (range 0.9–2.2) and 1.1 mg/dl (range 0.9–2) after 3 and 6 months, respectively, and remained unchanged at 12 months (Fig. 1). Three of five patients had a renal impairment (defined as an eGFR <60 ml/min/1.73 m2) at baseline that became normal after therapy, while two other patients maintained a stable impairment over follow-ups of 18 and 52 months, respectively.
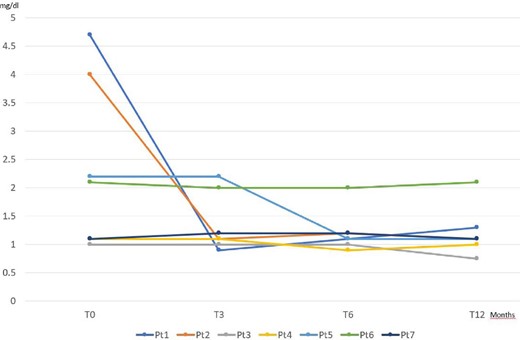
Profile of sCr at baseline and 3, 6 and 12 months.
The median proteinuria decreased from 6.1 g/24 h (range 3.8–12) to 3.5 (range 1.6–10), 3.5 (range 1–6.7) and 1.9 (range 0.8–5.7) at 3, 6 and 12 months, respectively (Fig. 2). Specifically, five of seven patients had a partial response (as defined above) at 12 months. One of them was found to have a complete response (<0.3 g/day proteinuria with >3.5 g/dl albumin) at 18 months and remained in complete remission at the last follow-up visit at 36 months.
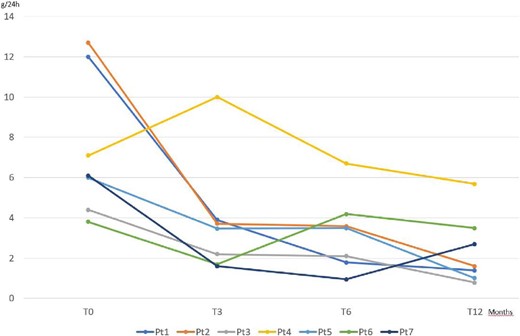
Profile of proteinuria at baseline and 3, 6 and 12 months.
Proteinuria persisted unchanged in patients 4 and 6, both of them having a putatively genetic-related disease. Patient 7 showed a partial response (with an improvement of serum albumin to 4.2 g/dl) despite a genetic mutation.
The intravenous infusions were well tolerated. No serious late adverse events were observed. Patient 7 had an asymptomatic urinary tract infection.
Complete peripheral blood B-cell depletion was achieved in all patients who received IBCDT. The CD20+/CD19+ B cells were assessed every 3 months for the first 18 months and every 6 months thereafter. The CD20+/CD19+ B cells were detectable in the circulation after a median of 12 months (range 11–18).
DISCUSSION
A few case series and isolated reports have suggested a promising role of RTX in nephrotic syndrome in children and have created considerable expectations in primary FSGS. However, results of RTX in adult patients with FSGS remain disappointing. We previously explored the effectiveness of infusions of 375 mg/m2 of RTX every 8 weeks in an unselected sample of adult patients with FSGS who were not systematically examined for foot process effacement on EM. This regimen achieved a consistent reduction in proteinuria in one of eight cases [9]. Recently, similar results were obtained in nine carefully selected patients with extensive foot process effacement receiving two standard doses of 1 g RTX every 2 weeks [10].
In the present study on similarly selected patients, we observed a remarkable decrease in proteinuria and a reversal from nephrotic syndrome in five of seven patients receiving an IBCDT consisting of a combination of six RTX infusions of 375 mg/m2, two intravenous doses of CYC and three pulses of methylprednisolone followed by a short course of oral prednisone. This intensive B-cell depletion protocol proved to be highly effective in several immune-mediated disorders, including lupus nephritis [11], severe ANCA-associated vasculitis [13] and systemic scleroderma [12]. This scheme ensures prolonged B cell depletion because of a relatively high cumulative dose of RTX (usually >3000 mg in 3 months), which is further potentiated by the synergic effects of two intravenous doses of CYC. This probably explains the difference between our results and those reported by Hladunewich et al. [10]. While not supported by solid evidence, we feel that patients with severe/relapsing putative primary FSGS (i.e. with extensive foot process effacement and nephrotic proteinuria) could benefit from a more intensive B-cell depletion regimen than standard protocols.
One could not exclude that steroid-dependent/frequently relapsing patients could have responded to standard doses of RTX, as shown in some available reports [14–17]. However, our previous experience using doses of RTX, even augmented, when compared with standard protocols [9], makes this possibility uncertain. It is also worth noting that, in agreement with previous reports [10, 18], two of three patients treated with IBCDT because of resistant disease (patients 4, 6 and 7) did not respond even to an intensified approach.
Genetic analysis is desirable. Indeed, the two unresponsive patients both had a genetic mutation. Nevertheless, it is worth noting that the impact of the genetic mutation on the immunomodulatory effects of IBCDT can be different. The effect of IBCDT in patient 7 suggested that in the presence of a mutation, especially if not surely pathogenic, clinicians should not be discouraged to attempt a trial of IBCDT, which was found to achieve at least a partial response. Future studies are required to assess the role of the identified mutations in the context of FSGF.
Finally, while a synergic role of RTX in combination with CYC has been associated with prolonged B-cell depletions [19–21], other putative reasons for improvement cannot be ruled out.
CONCLUSIONS
Our results showed promising effects of an intensified B-cell depletion regimen in adult patients with primary FSGS. Among seven patients who had massive nephrotic proteinuria at baseline, five showed a sustained response with a remarkable reduction of proteinuria without side effects, while both the unresponsive patients had a genetic mutation.
AUTHORS’ CONTRIBUTIONS
All authors participated in the data collection, data analysis, data interpretation, manuscript drafting and editing. All authors approved the final version.
DATA AVAILABILITY STATEMENT
The data underlying this research are available in the article.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
Author notes
Contributed equally to this study.


{kind=link}
{kind=link}
{kind=link}
Comments