





Abstract
Acute interstitial nephritis (AIN) is a common cause of acute kidney injury (AKI), particularly in hospitalized patients. It can be difficult for clinicians to differentiate between AIN and other common causes of AKI, most notably acute tubular necrosis (ATN) and prerenal injury. Clinicians often struggle with the clinical history and laboratory data available to definitively diagnose AIN. Sometimes they diagnose ATN or AIN based on these flawed data. Thus it is important that clinicians be familiar with the utility of commonly ordered tests used to aid in the diagnosis. Unfortunately, no single test performs particularly well on its own, and until a biomarker is rigorously shown to be diagnostic of AIN, most patients require a kidney biopsy to definitively establish the diagnosis and direct further management.
Acute interstitial nephritis (AIN) is a common cause of acute kidney injury (AKI), particularly among hospitalized patients. In fact, AIN is estimated to be the cause of AKI in 10–27% of hospitalized patients, making it the third most common etiology of hospital-acquired AKI, behind acute tubular necrosis (ATN) and prerenal AKI [1–6] (Figure 1). Differentiating between these various etiologies of AKI can be challenging using noninvasive tests. Ultimately, kidney biopsy may be the only means of obtaining a definitive diagnosis, especially when AIN is the underlying cause. Therefore it is important for clinicians to be familiar with the utility of the many clinical tests that are often obtained to assess the possibility of AIN as the cause of AKI.
![Pie chart demonstrating common causes of in-hospital AKI. ATN accounts for ∼50% of in-hospital AKI [5], followed by prerenal injury (20–30%) [6] and AIN [1–4]. The remainder is accounted for by obstructive uropathy, glomerular diseases/vasculitides and atheroembolic diseases, as well as other less common etiologies [5, 6].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ckj/12/6/10.1093_ckj_sfz080/2/m_sfz080f1.jpeg?Expires=1716396751&Signature=vpp9OssTf30O3jFFY1OEfHib-txg~jPhyT1-UZOEkgRve19uhdp5sMQrknKyb3mtd5rFEI-3mSWAgYBM9QiDhAhAbMV9XGHF75fOh03TuhC~dYxieVRL7EiopaD8-L-796dy1qJnV~H0rpa3tqQSOaU4vJj7hVIJUsC-Io1c8Vc0IiqUdFnAJYHbWP7joYddIAWY529G8FEackzP0weUKoQ~0roICyZmnu60meg1FmcYMf~7SkOweNOmYgRiuKAmffsiQQbVnTSM0VhIHHqwjLjsJ~2HvYDZjdXU1p2RRu2j0DFPamDVVOFseVrj5kiseK-5OxDqVe99XlwmPJCEpQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Pie chart demonstrating common causes of in-hospital AKI. ATN accounts for ∼50% of in-hospital AKI [5], followed by prerenal injury (20–30%) [6] and AIN [1–4]. The remainder is accounted for by obstructive uropathy, glomerular diseases/vasculitides and atheroembolic diseases, as well as other less common etiologies [5, 6].
ETIOLOGY
In developed countries, exposure to an offending medication is by far the most common cause of AIN, accounting for between 70% and 90% of cases [4, 7, 8]. A 2014 case series from the Mayo Clinic, which is fairly representative of the published literature, noted that drugs were the cause of biopsy-proven AIN in 71% of 133 patients, followed by autoimmune diseases, which accounted for ∼20% [9]. The most commonly implicated drugs (Table 1) in the study were antibiotics (∼50%), proton pump inhibitors (∼14%) and nonsteroidal anti-inflammatory drugs (NSAIDs) (11%). Among antibiotics, amoxicillin and ciprofloxacin were the most common offending agents [9]. In addition, newer anticancer agents, in particular the immune checkpoint inhibitors, are associated with AIN [10, 11]. An incidence of ∼2–4% is observed with these drugs, which are monoclonal antibodies used to treat certain malignancies. Immune checkpoint inhibitors activate the quiescent T cell by blocking inhibitory receptor cytotoxic T lymphocyte-antigen-4 (CTLA-4) binding to the antigen-presenting cell CD80/86 ligand [10, 11]. They also blunt tumor-induced T-cell deactivation by blocking the immunosuppressive receptor programmed death-1 (PD-1) ligand from binding the tumor-expressed ligands [10, 11]. However, activating T cells can lead to widespread activation of the immune system that affects both target (tumor) and nontarget (healthy) tissue, including the kidneys. In this case, AIN complicates therapy and is the most common kidney lesion described with these agents [10, 11]. Additionally, while uncommon, AIN has been observed in kidney transplant recipients in spite of immunosuppressive therapy [12–15]. The etiologies in this patient population appear similar to those of antibiotics, in particular trimethoprim–sulfamethoxazole is frequently cited [12, 13]. NSAID-induced AIN has also been reported in allograft recipients [14], as has infectious AIN, which can present with granulomatous lesions [15].
Common causes of drug-induced AIN
| Antibiotics | β-lactam drugs (penicillins, cephalosporins) |
| Fluoroquinolones | |
| Sulfa-based antimicrobials (TMP–SMX) | |
| Rifampin | |
| Antacid medications | Proton pump inhibitors |
| Histamine-2 blockers | |
| Analgesics | NSAIDs, including COX-2 inhibitors |
| Immunotherapeutic agents | PD-1/PD-L1 inhibitors (nivolumab, pembrolizumab, cemiplimab, atezolizumab, durvalumab, avelumab) |
| CTLA-4 inhibitors (ipilimumab, tremelimumab) | |
| Anti-angiogenesis drugs | Bevacizumab, tyrosine kinase inhibitors (sorafenib, sunitanib) |
| Diuretics | Loop diuretics (furosemide, bumetanide) |
| Thiazide diuretics (HCTZ) | |
| Antiviral agents | Acyclovir |
| Abacavir | |
| Indinavir | |
| Atazanavir | |
| Foscarnet | |
| Anticonvulsants | Phenytoin |
| Carbamazepine | |
| Phenobarbirtal | |
| Other agents | Allopurinol |
| Mesalamine | |
| Lithium | |
| Isofamide | |
| Pemetrexed |
| Antibiotics | β-lactam drugs (penicillins, cephalosporins) |
| Fluoroquinolones | |
| Sulfa-based antimicrobials (TMP–SMX) | |
| Rifampin | |
| Antacid medications | Proton pump inhibitors |
| Histamine-2 blockers | |
| Analgesics | NSAIDs, including COX-2 inhibitors |
| Immunotherapeutic agents | PD-1/PD-L1 inhibitors (nivolumab, pembrolizumab, cemiplimab, atezolizumab, durvalumab, avelumab) |
| CTLA-4 inhibitors (ipilimumab, tremelimumab) | |
| Anti-angiogenesis drugs | Bevacizumab, tyrosine kinase inhibitors (sorafenib, sunitanib) |
| Diuretics | Loop diuretics (furosemide, bumetanide) |
| Thiazide diuretics (HCTZ) | |
| Antiviral agents | Acyclovir |
| Abacavir | |
| Indinavir | |
| Atazanavir | |
| Foscarnet | |
| Anticonvulsants | Phenytoin |
| Carbamazepine | |
| Phenobarbirtal | |
| Other agents | Allopurinol |
| Mesalamine | |
| Lithium | |
| Isofamide | |
| Pemetrexed |
TMP–SMX, trimethoprim–sulfamethoxazole; COX-2, cyclooxygenase-2; PD-1, programmed cell death protein-1; PD-L1, programmed death ligand 1; CTLA-4, cytotoxic T-lymphocyte antigen-4; HCTZ, hydrochlorothiazide; NSAIDs, non-steroidal anti-inflammatory drugs.
Common causes of drug-induced AIN
| Antibiotics | β-lactam drugs (penicillins, cephalosporins) |
| Fluoroquinolones | |
| Sulfa-based antimicrobials (TMP–SMX) | |
| Rifampin | |
| Antacid medications | Proton pump inhibitors |
| Histamine-2 blockers | |
| Analgesics | NSAIDs, including COX-2 inhibitors |
| Immunotherapeutic agents | PD-1/PD-L1 inhibitors (nivolumab, pembrolizumab, cemiplimab, atezolizumab, durvalumab, avelumab) |
| CTLA-4 inhibitors (ipilimumab, tremelimumab) | |
| Anti-angiogenesis drugs | Bevacizumab, tyrosine kinase inhibitors (sorafenib, sunitanib) |
| Diuretics | Loop diuretics (furosemide, bumetanide) |
| Thiazide diuretics (HCTZ) | |
| Antiviral agents | Acyclovir |
| Abacavir | |
| Indinavir | |
| Atazanavir | |
| Foscarnet | |
| Anticonvulsants | Phenytoin |
| Carbamazepine | |
| Phenobarbirtal | |
| Other agents | Allopurinol |
| Mesalamine | |
| Lithium | |
| Isofamide | |
| Pemetrexed |
| Antibiotics | β-lactam drugs (penicillins, cephalosporins) |
| Fluoroquinolones | |
| Sulfa-based antimicrobials (TMP–SMX) | |
| Rifampin | |
| Antacid medications | Proton pump inhibitors |
| Histamine-2 blockers | |
| Analgesics | NSAIDs, including COX-2 inhibitors |
| Immunotherapeutic agents | PD-1/PD-L1 inhibitors (nivolumab, pembrolizumab, cemiplimab, atezolizumab, durvalumab, avelumab) |
| CTLA-4 inhibitors (ipilimumab, tremelimumab) | |
| Anti-angiogenesis drugs | Bevacizumab, tyrosine kinase inhibitors (sorafenib, sunitanib) |
| Diuretics | Loop diuretics (furosemide, bumetanide) |
| Thiazide diuretics (HCTZ) | |
| Antiviral agents | Acyclovir |
| Abacavir | |
| Indinavir | |
| Atazanavir | |
| Foscarnet | |
| Anticonvulsants | Phenytoin |
| Carbamazepine | |
| Phenobarbirtal | |
| Other agents | Allopurinol |
| Mesalamine | |
| Lithium | |
| Isofamide | |
| Pemetrexed |
TMP–SMX, trimethoprim–sulfamethoxazole; COX-2, cyclooxygenase-2; PD-1, programmed cell death protein-1; PD-L1, programmed death ligand 1; CTLA-4, cytotoxic T-lymphocyte antigen-4; HCTZ, hydrochlorothiazide; NSAIDs, non-steroidal anti-inflammatory drugs.
CLINICAL HISTORY AND PHYSICAL EXAMINATION
A careful history should be elicited searching for possible exposure to new medications, particularly those known to be associated with AIN, as well as other causes such as underlying autoimmune conditions. However, it is important to recognize that the time course with drug-induced AIN can be quite variable. For example, AKI can present days, weeks or even months after exposure to an offending medication, depending on the class of drug [7]. Making matters more challenging, symptoms tend to be vague and nonspecific (malaise, arthralgias, nausea) and many patients may be asymptomatic. Commonly cited physical exam findings include fever and rash (Table 2). However, a 2004 review of 128 patients with AIN found that fever was present in only 15% and rash in only 27% [16]. Furthermore, two retrospective series yielded relatively similar findings, with fever present in 36% and rash in 22% of cases [17, 18]. Urine volume is also unhelpful, as patients can be either oliguric or nonoliguric. In a 2010 retrospective study of 60 biopsy-proven cases, oliguria was present in 51% of cases [19].
Clinical features of AIN
| Fever | Rash | Eosinophilia (blood) | Triad of fever, rash and eosinophilia | Oliguria |
|---|---|---|---|---|
| Present in 15–36% of cases | Present in 22–27% of cases | Present in 23–36% of patients | Present in up to 10% of cases | Present in ∼50% of cases |
| Fever | Rash | Eosinophilia (blood) | Triad of fever, rash and eosinophilia | Oliguria |
|---|---|---|---|---|
| Present in 15–36% of cases | Present in 22–27% of cases | Present in 23–36% of patients | Present in up to 10% of cases | Present in ∼50% of cases |
Clinical features of AIN
| Fever | Rash | Eosinophilia (blood) | Triad of fever, rash and eosinophilia | Oliguria |
|---|---|---|---|---|
| Present in 15–36% of cases | Present in 22–27% of cases | Present in 23–36% of patients | Present in up to 10% of cases | Present in ∼50% of cases |
| Fever | Rash | Eosinophilia (blood) | Triad of fever, rash and eosinophilia | Oliguria |
|---|---|---|---|---|
| Present in 15–36% of cases | Present in 22–27% of cases | Present in 23–36% of patients | Present in up to 10% of cases | Present in ∼50% of cases |
COMMONLY ORDERED TESTS
A battery of urine tests are often ordered to evaluate and differentiate the various causes of AKI that develops in hospitalized patients (Table 3). The search for novel biomarkers of acute kidney disease is a prominent research topic in nephrology. New information on biomarkers for AIN is an active area of research.
Urine diagnostics in AIN
| Basic urinalysis | Urine microscopy | Urine eosinophils | Urine chemistries |
|---|---|---|---|
| Proteinuria: present in ∼ 90% of cases | WBC casts: present in 3–14% of cases | Sensitivity: 31% | FENa: can be >1% or <1% |
| Nephrotic-range proteinuria rare | RBC casts: present in up to 29% cases | Specificity: 68% | FEUrea: can be >35% or <35% |
| Hematuria: present in ∼50% of cases | RTE and granular casts: present in up to 86% cases | Also present in ATN, GN and other renal diseases | |
| Pyuria: present in 50–80% of cases | Bland urine sediment: present in ∼20% cases |
| Basic urinalysis | Urine microscopy | Urine eosinophils | Urine chemistries |
|---|---|---|---|
| Proteinuria: present in ∼ 90% of cases | WBC casts: present in 3–14% of cases | Sensitivity: 31% | FENa: can be >1% or <1% |
| Nephrotic-range proteinuria rare | RBC casts: present in up to 29% cases | Specificity: 68% | FEUrea: can be >35% or <35% |
| Hematuria: present in ∼50% of cases | RTE and granular casts: present in up to 86% cases | Also present in ATN, GN and other renal diseases | |
| Pyuria: present in 50–80% of cases | Bland urine sediment: present in ∼20% cases |
GN, glomerulonephritis.
Urine diagnostics in AIN
| Basic urinalysis | Urine microscopy | Urine eosinophils | Urine chemistries |
|---|---|---|---|
| Proteinuria: present in ∼ 90% of cases | WBC casts: present in 3–14% of cases | Sensitivity: 31% | FENa: can be >1% or <1% |
| Nephrotic-range proteinuria rare | RBC casts: present in up to 29% cases | Specificity: 68% | FEUrea: can be >35% or <35% |
| Hematuria: present in ∼50% of cases | RTE and granular casts: present in up to 86% cases | Also present in ATN, GN and other renal diseases | |
| Pyuria: present in 50–80% of cases | Bland urine sediment: present in ∼20% cases |
| Basic urinalysis | Urine microscopy | Urine eosinophils | Urine chemistries |
|---|---|---|---|
| Proteinuria: present in ∼ 90% of cases | WBC casts: present in 3–14% of cases | Sensitivity: 31% | FENa: can be >1% or <1% |
| Nephrotic-range proteinuria rare | RBC casts: present in up to 29% cases | Specificity: 68% | FEUrea: can be >35% or <35% |
| Hematuria: present in ∼50% of cases | RTE and granular casts: present in up to 86% cases | Also present in ATN, GN and other renal diseases | |
| Pyuria: present in 50–80% of cases | Bland urine sediment: present in ∼20% cases |
GN, glomerulonephritis.
Urinalysis and urine microscopy
Pyuria
In a 2012 case series by Fogazzi et al. [20], pyuria was found to be present in 57% of 21 biopsy-proven cases of AIN. A larger case series of 133 patients with biopsy-proven AIN published in 2014 by Muriithi et al. [9] revealed similar findings, with pyuria present in 47% of cases. A more recent Australian single-center case series of 40 biopsy-confirmed AIN cases revealed >10 white blood cells (WBCs) per high-power field in 73% of cases [21]. In fact, a review of the published literature noted that dipstick pyuria is present in 60–80% of cases, but is more likely to be absent in cases not caused by antibiotics [8]. In summary, sterile pyuria (Figure 2A) is present in one-half to three-fourths of cases; however, the absence of pyuria does not exclude a diagnosis of AIN.
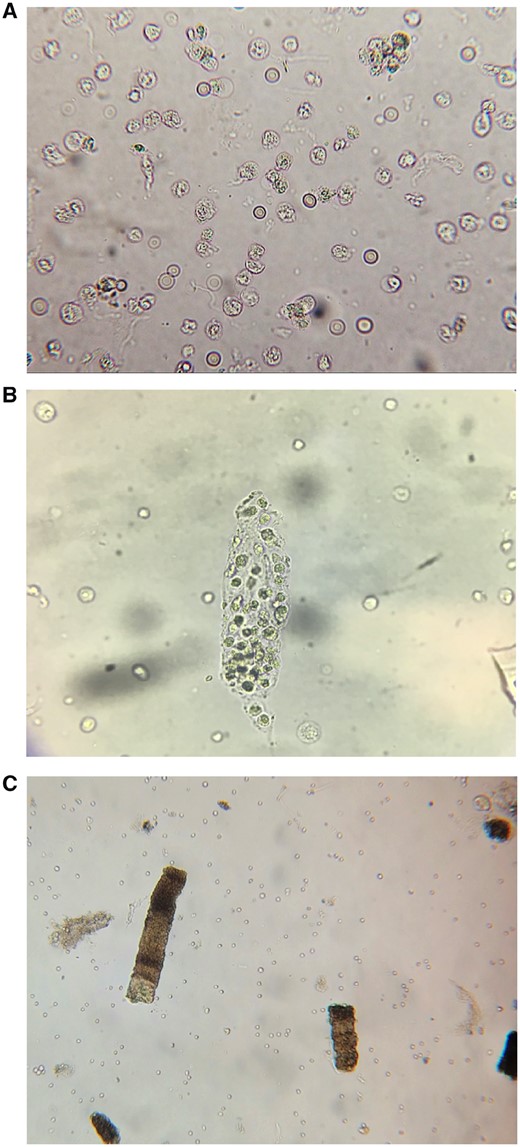
Urine microscopy findings in AIN. (A) WBCs and RBCs. (B) WBC cast. (C) Granular ‘muddy brown’ casts.
Proteinuria
Nonnephrotic range proteinuria or ‘tubular proteinuria’ is common in AIN. In the 133-patient case series by Muriithi et al. [9], 92% of patients had low-grade proteinuria, defined as >150 mg of urinary protein excretion per day. The mean protein excretion in this study was 1.2 g/24 h, with a range of 0.5–3.4 g/day. Higher levels of proteinuria reflected concurrent glomerular disease. Fogazzi et al. [20] found similar results, with 90% of patients having some degree of proteinuria. However, nephrotic-range proteinuria is rare [1, 2, 8, 9, 20, 21]. Indeed, in the study by Fogazzi et al. [20], the average protein excretion was 1.36 g/24 h with a range of 0.29–12.1 g. Only one patient demonstrated urine protein excretion >3.5 g/24 h (12.1 g). One notable exception is in patients who develop interstitial nephritis secondary to NSAID use, which can concurrently cause minimal change disease [2, 8, 22, 23]. Indeed, NSAIDs were the offending agent in the lone case of nephrotic-range proteinuria in the Fogazzi et al. study. Also, patients with diabetic kidney disease and other glomerular diseases may have high-grade proteinuria in the setting of AIN that is due to their underlying glomerulopathy rather than AIN. In summary, mild to moderate proteinuria is typically present in AIN. However, nephrotic-range proteinuria is rare and when observed in the setting of AIN should raise suspicion for NSAID-induced nephrotoxicity or underlying glomerular disease.
Hematuria
Hematuria is a relatively common finding in AIN, however, as with pyuria, it is not universally present (Figure 2A). Several studies note that hematuria is present in a mean of ∼50% (range 20–80%) of AIN cases [8, 20]. Thus hematuria is a nonspecific and insensitive laboratory test for AIN.
White blood cell (WBC) casts
WBC casts often indicate the presence of inflammatory pathology within the kidney parenchyma (Figure 2B). They may be seen in the setting of intrarenal infection, such as pyelonephritis, or with inflammatory kidney lesions, such as proliferative glomerulonephritis and AIN. The 2012 case series published by Fogazzi et al. [20] noted that WBC casts were present in only 3 of 21 (14%) patients with biopsy-proven AIN. Perhaps, surprisingly, in the even larger case series by Muriithi et al. [9] of 133 biopsy-proven cases, only 3% of patients had WBC casts seen by microscopy. Notably, WBC casts were only observed in patients with drug-induced AIN. Thus WBC casts are neither a sensitive nor specific test for diagnosing AIN.
Red blood cell (RBC) casts
RBC casts are often considered quite specific for glomerular pathology and indicative of a primarily glomerular process. In general, these casts have not been considered a common finding in AIN and their presence was once thought to be sufficient to exclude a diagnosis of AIN. However, one study found that RBC casts were present in 29% of biopsy-proven cases of AIN [9]. How is this possible? One possible explanation for the formation of RBC casts in AIN is disruption of interstitial blood vessels with RBC extravasation and entry into tubular lumens where they admix with uromodulin and form the cellular cast [9].
Renal tubular epithelial cell and granular casts
Renal tubular epithelial (RTE) cells and granular casts (Figure 2C) are most often seen with ischemic or nephrotoxic acute tubular injury. However, since AIN also causes tubulitis and tubular injury, they are commonly seen with this inflammatory lesion. In fact, these urinary sediment findings have been observed in up to 86% of cases [2, 9]. Therefore their presence should not exclude AIN as a possible cause of AKI and elicit the classic ‘this is ATN’ response.
Bland urine sediment
Since AIN is an inflammatory kidney lesion, many clinicians expect to see an active urine sediment. In fact, a bland urine sediment is mistakenly considered diagnostic of prerenal AKI. However, 20% of patients with AIN may have a bland urine sediment (despite an aggressive inflammatory infiltrate within the interstitium on kidney biopsy). Thus the absence of the above urinary findings does not exclude a diagnosis of AIN [8].
Urine chemistries
Urine sodium/fractional excretion of sodium
Clinicians frequently order these urine chemistries to help diagnose various causes of AKI, in particular ATN, prerenal AKI and AIN. However, urine sodium concentration and fractional excretion of sodium (FENa) are unhelpful in the diagnosis of AIN, as both can be either elevated or depressed in these patients [1, 2, 24, 25]. The same holds true for fractional excretion of urea (FEUrea). Accordingly, a FENa value <1% and FEUrea <35%, while classically used to support a diagnosis of prerenal AKI, do not rule out a diagnosis of AIN.
Urine and serum and urine eosinophils
Serum eosinophils
Clinicians are classically taught that hypereosinophilia is a reasonably good diagnostic test for allergic reactions, including AIN. However, this is not true, even with drug-induced AIN. In a 2004 study of 128 patients with AIN, eosinophilia was present in only 23% of patients [16]. Two subsequent studies found that eosinophilia was present in 36% [17] and 34% [18] of biopsy-proven cases of AIN. Notably, the combination of fever, rash and eosinophilia is present in only 5–10% patients [1, 7, 16]. Thus the absence of eosinophilia does not exclude AIN.
Urine eosinophils
Measuring urine eosinophils to evaluate for possible AIN is a favorite topic of many clinicians [1, 26]. While it is true that eosinophiluria may be observed in patients with AIN, the test is neither sensitive nor specific for the diagnosis. Perhaps the best study to illustrate this was conducted by Muriithi et al. [27], which was published in 2013. This study identified 566 patients with both a urine eosinophil test and a kidney biopsy performed within 1 week of each other. There were 91 patients in this study with biopsy-proven AIN. Using a urinary eosinophil cutoff of 1%, the sensitivity of a positive urinary eosinophil test was only 31% and specificity 68%. In addition, the use of urinary eosinophils proved unreliable in distinguishing AIN from ATN, a common final differential for AKI. Indeed, much of what makes the test particularly unhelpful is that eosinophiluria can be seen in many renal and nonrenal diseases other than AIN [1, 2, 27]. Examples include pyelonephritis, cystitis, prostatitis, atheroembolic disease, ATN, rapidly progressive glomerulonephritis and bladder malignancies, among others [1, 2, 27, 28].
Novel biomarkers
Recently, several novel biomarkers have been evaluated as potential diagnostic and prognostic tools. Urinary monocyte chemotactic peptide-1 (MCP-1), neutrophil gelatinase-associated lipocalin, α1-microglobulin, β2-microglobulin and N-acetyl-β-d-glucosamidase have been shown to be associated with tubulointerstitial damage [8, 29, 30]. α1-microglobulin and β2-microglobulin, for example, are low molecular weight proteins that are freely filtered through the glomerulus and reabsorbed in the proximal tubule. With tubulointerstitial injury, tubular reabsorption is decreased, thus increasing the urinary concentration of these proteins. MCP-1 appears to be closely associated with the degree of interstitial edema and inflammation and may have a role in prognosticating the severity of the injury, as well as serving a diagnostic role in differentiating AIN from ATN [29, 30]. Unfortunately this study utilized controls without kidney disease (normal volunteers), thereby not examining the utility of these urinary biomarkers in differentiating AIN from ATN and other kidney lesions.
More recently, in a study of 155 patients published by Moledina et al. [31] demonstrated that urinary cytokines tumor necrosis factor-alpha (TNF-α) and interleukin-9 (IL-9) were higher in patients with biopsy-proven AIN compared with controls with other forms of kidney disease (ATN, diabetic nephropathy, glomerulonephritis, etc.). Additionally, these urinary cytokines were higher with increasing histologic severity of disease. Use of these urinary biomarkers was further shown to improve diagnostic accuracy compared with other available tests. A notable strength of this study is that the control patients also had clinically and histologically significant kidney disease, most commonly ATN, thus providing an additional layer of clinical applicability.
PERSPECTIVES ON DIAGNOSTIC TESTS FOR AIN
AIN is a common cause of AKI, particularly among hospitalized patients, where up to 27% of patients have this lesion. As such, it is important that clinicians be familiar with the utility of commonly ordered tests used to aid in the diagnosis. Unfortunately, as detailed above, no single test performs particularly well on its own. Furthermore, there are still several common misconceptions that exist within the medical community regarding the value of these tests. For example, urinary eosinophils are still commonly ordered as part of the standard evaluation for AIN despite recent studies showing that urinary eosinophils are neither sensitive nor specific for AIN. Anecdotally, we encounter this frequently, and in our institution alone, urinary eosinophils were ordered 170 times from December 2017 to 2018. Indeed, given the strength of the evidence, the nephrology community recommends against testing for urinary eosinophils as part of the evaluation of possible AIN or other causes of AKI. Hypereosinophilia is also a nonspecific finding and cannot be relied upon to rule in or rule out AIN.
Another common misconception is the notion that urine microscopy typically reveals characteristic findings unique to AIN that can be considered diagnostic. Unfortunately this is not the case. For example, while WBC casts may signal AIN, it is an infrequent finding, as these casts are observed in only 3–14% of patients with AIN. Thus it is important to keep in mind that the absence of WBC casts by no means rules out a diagnosis of AIN. Furthermore, WBC casts can also be seen with proliferative glomerular diseases, which are often in the differential of hospital-acquired AKI. Notably, RBC casts—which are typically considered a marker solely of glomerular injury—may be seen and their presence by themselves also does not rule out a diagnosis of AIN. Similarly, the urine sediment in AIN can look identical to that seen in ATN with RTE cells, and RTE cells and granular casts can be observed in up to 86% of AIN cases. Lastly, an entirely bland urine sediment may be observed in up to 20% of cases and accordingly should not be considered definitive evidence against AIN. In essence, the take-home point from the available evidence is clear, that is, urine microscopy may be helpful in the diagnostic evaluation of AIN in the right clinical setting. However, urine microscopy by itself cannot be used to confirm or rule out the diagnosis, particularly given the overlap with other common causes of hospital-acquired AKI. The role of new urinary biomarkers, particularly the T-cell-derived cytokine biomarkers, remains to be determined. However, new data on these novel tests are quite promising.
Given the poor sensitivity and specificity of the aforementioned tests, kidney biopsy remains the gold standard to establish the diagnosis of AIN. There is no literature regarding the utility of clinical findings and laboratory tests (including urine studies) in diagnosing AIN in patients with kidney transplants. The sediment in patients with AIN and transplant rejection would look very similar (WBCs, tubular cells, etc.). Thus histologic evaluation is particularly important in kidney transplant patients given the paucity of literature about AIN in this population and the expanded differential for AKI among this subgroup. This is not to say that biopsy is necessary in all patients with suspected AIN. For example, a patient with AKI in the setting of recent antibiotic exposure who develops fever, rash, eosinophilia and pyuria without any other clear cause of kidney injury likely has AIN. In this setting with a high pretest probability, the presence of urinary WBC casts may indeed clinch the diagnosis. However, this constellation of clinical findings is quite rare (<10%) and is certainly the exception. More commonly, hospitalized patients have several possible reasons for AKI without the classic constellation of clinical findings seldom seen in AIN. Until a biomarker is rigorously shown to be diagnostic of AIN, most patients require a kidney biopsy to definitively establish the diagnosis and direct further management.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES


{kind=link}
{kind=link}
Comments