





Successful implantation is the result of reciprocal interactions between the implantation-competent blastocyst and receptive uterus. Although various cellular aspects and molecular pathways of this dialogue have been identified, a comprehensive understanding of the implantation process is still missing. The receptive state of the uterus, which lasts for a limited period, is defined as the time when the uterine environment is conducive to blastocyst acceptance and implantation. A better understanding of the molecular signals that regulate uterine receptivity and implantation competency of the blastocyst is of clinical relevance because unraveling the nature of these signals may lead to strategies to correct implantation failure and improve pregnancy rates. Gene expression studies and genetically engineered mouse models have provided valuable clues to the implantation process with respect to specific growth factors, cytokines, lipid mediators, adhesion molecules, and transcription factors. However, a staggering amount of information from microarray experiments is also being generated at a rapid pace. If properly annotated and explored, this information will expand our knowledge regarding yet-to-be-identified unique, complementary, and/or redundant molecular pathways in implantation. It is hoped that the forthcoming information will generate new ideas and concepts for a process that is essential for maintaining procreation and solving major reproductive health issues in women.
I. Introduction
II. Preimplantation Embryo Development and Genomic Activation
III. Species-Specific Morphological Blueprint and Timing of Implantation
IV. Delayed Implantation
V. Window of Implantation: A Transient and Unique Moment
VI. Embryo-Uterine Signaling Pathways in Implantation
A. Steroid hormone signaling
B. Signaling via adhesion molecules: cell-cell interactions
C. Signaling by vasoactive factors
D. Signaling by growth factors
E. Signaling by cytokines
F. Homeobox genes in implantation
G. Ligand-dependent nuclear receptors and coactivators in implantation
H. Cell cycle regulation and signaling in implantation and decidualization
I. Matrix remodeling and angiogenesis during implantation and decidualization
VII. Emerging Concepts
A. Endocannabinoid signaling in implantation
B. Developmental genes in implantation
C. Discovery of novel implantation-related genes
VIII. Perspectives and Future Directions
I. Introduction
PROCREATION, BOTH SEXUAL and asexual, is a fundamental evolutionary process necessary to sustain life. Viviparity is a landmark in the process of evolution. Sexual procreation in higher eukaryotes, especially in mammals, is often inferior to asexual procreation in prokaryotes and in some eukaryotes with respect to shear number of progeny. Thus, mammalian reproduction is more complex and highly regulated for the propagation of superior offspring to carry on the task of procreation. The nurturing of an offspring within the body and producing a live birth is an enduring task. This process demands safeguard regulatory systems at various critical steps. The assembly of a new life first depends on the union between a sperm and an egg (ovum) culminating in fertilization; failure to achieve such a union leads to their demise. The one-cell fertilized egg, termed embryo, undergoes several mitotic cell divisions, eventually forming a differentiated tissue called the blastocyst with two distinct cell populations, the inner cell mass (ICM) and a layer of trophectoderm cells surrounding the ICM (1). The embryo proper is derived exclusively from the ICM, whereas the placenta and extraembryonic membranes are produced from cells contributed mainly by the trophectoderm. A two-way interaction between the blastocyst and maternal uterine luminal epithelium initiates the process of implantation, a process by which blood vessels of the embryo are brought into functional communication with the maternal circulation leading to the establishment of a functional placenta and pregnancy. Maternal resources filtered across the selective barrier of the placenta protect and nourish the conceptus. Placental types have been classified into three categories: hemochorial (rodents, humans, and nonhuman primates), epitheliochorial (horses, cows, sheep, and pigs), and endotheliochorial (most carnivores) (reviewed in Ref. 2).
A significant pregnancy loss resulting from preimplantation embryonic death is common to many mammals and is considered to be a selection process leading to the survival of superior embryos for implantation. However, dysregulation of the events before, during, or immediately after implantation also may often be a cause for poor pregnancy rates in eutherian mammals. Understanding the mechanism of preimplantation embryonic development and implantation in the uterus has been a challenge to reproductive and developmental biologists with the goal of alleviating the problems of human infertility and ensuring the birth of quality offspring. Such knowledge is also necessary for developing novel contraceptive approaches to restrict world population growth.
The current state of our knowledge of preimplantation and implantation physiology is the result of the accumulation of scientific observations gathered over many years. Implantation is a complex process involving spatiotemporally regulated endocrine, paracrine, autocrine, and juxtacrine modulators that span cell-cell and cell-matrix interactions. However, the precise sequence and details of the molecular interactions involved have not yet been defined. Furthermore, the implantation process varies among species, thus precluding the formulation of a unified theme. In addition, ethical restrictions and experimental difficulties prevent direct analysis of embryo-uterine interactions during human implantation. Thus, it is an onerous task to write a review on the molecular basis of embryo-uterine interactions during implantation that could be relevant to mammals in general. This review focuses primarily on the physiological and molecular basis of implantation in mice because more mechanistic information is now available for this species. However, an attempt has been made to indicate comparative analyses based on limited work in other species.
Despite experimental success in initiating embryonic development outside the womb and in identifying numerous molecules involved in the embryo-uterine dialogue (3–7), there is a yet-to-be-filled significant knowledge gap in understanding the in vivo events of implantation. The successful implantation of an embryo is contingent upon cellular and molecular cross-talk between the uterus and the embryo. The coordination of the endocrine, cellular, and molecular events via paracrine, autocrine, and/or juxtacrine factors in a dynamic manner produces within the uterus a favorable environment, the receptive state, to support implantation. The embryo also functions as an active unit with its own molecular program of cell growth and differentiation. Thus, deficiencies in uterine receptivity, embryo development, or the embryo-uterine dialogue will compromise fertility. This review of the implantation process focuses on the molecular basis of embryo homing and attachment, on elucidating the reciprocal signaling networks between the embryo and uterus, and on determining genetic causes of implantation failure.
II. Preimplantation Embryo Development and Genomic Activation
Preimplantation embryo development and differentiation, which culminate in the formation of a blastocyst, require the activation of the embryonic genome, a process that is essential to implantation. The maternal-zygotic transition occurs at the two-cell stage in mice and other rodents, between the eight- and 16-cell stages in cows and sheep, and between the four- and eight-cell stages in humans (reviewed in Ref. 8). Upon activation of the embryonic genome, the embryo grows rapidly to form a blastocyst. At the blastocyst stage, embryos mature and escape from their zona pellucidae to gain implantation competency. The differentiated and expanded blastocyst is composed of three cell types: the outer polarized epithelial trophectoderm, the primitive endoderm, and the pluripotent ICM. The ICM provides the future cell lineages for the embryo proper (9, 10), and the trophectoderm, the very first epithelial cell type in the developmental process, makes the initial physical and physiological connection with the uterine luminal epithelium. The ICM is not identifiable in marsupial blastocysts that appear as a hollow ball of cells with similar morphological characteristics. It is also not known which cells are programmed to form the embryo proper (reviewed in Ref. 10). The formation of the trophectoderm and its subsequent development into trophoblast tissue are crucial steps for the initiation of implantation and the establishment of pregnancy. Trophoblast cells produce a variety of growth factors, cytokines, and hormones that influence the conceptus and maternal physiology in an autocrine, paracrine, and/or juxtacrine manner (11, 12).
Preimplantation embryo development normally occurs within the zona pellucida. However, zona removal by various experimental manipulations does not deter embryonic development in vitro (13), suggesting that this glycoprotein barrier is not essential for development to progress. The nonadhesive nature of the zona pellucida is thought to facilitate the journey of embryos through the oviduct and from the oviduct to the uterus. In mice and rats, normal embryonic development to the blastocyst stage within the reproductive tract requires the presence of ovarian estrogen and progesterone (14). There is a reduced number of embryos and a reduced number of cells per embryo in the absence of these hormones (15), but treatment with estrogen and progesterone reverses these defects (16). Because there is no convincing evidence that estrogen and/or progesterone act directly on the preimplantation embryo (17), embryonic development is considered to depend on growth-promoting factors originating from the reproductive tract under the influence of these hormones. However, apparently normal development in simple defined media in culture suggests that preimplantation embryos are capable of producing their own growth-promoting factors (reviewed in Ref. 18). In fact, several growth factors, cytokines, and their receptors are expressed in the embryo, and the proliferative and differentiating effects of these factors on embryonic development and functions have been observed (reviewed in Refs. 3 and 18–20).
III. Species-Specific Morphological Blueprint and Timing of Implantation
Implantation is the process by which the blastocyst comes into intimate physical and physiological contact with the uterine endometrium. Enders and Schlafke (21, 22) have classified the process of implantation into three stages: apposition, adhesion, and penetration. Apposition is the stage when embryonic trophectoderm cells become closely apposed to the uterine luminal epithelium. This is followed by the adhesion stage in which the association of the trophectoderm and the luminal epithelium is sufficiently intimate as to resist dislocation of the blastocyst by flushing the uterine lumen. The stage of penetration involves the invasion of the luminal epithelium by the trophectoderm. Stromal cell differentiation into decidual cells (decidualization) is more extensive, and the loss of the luminal epithelium is evident at this stage. These three stages of implantation form a continuum.
In mammals, especially rodents, a generalized stromal edema occurs before the beginning of apposition. This event leads to the closure of the uterine lumen, which results in interdigitation of the microvilli of the trophectoderm and the luminal epithelia (apposition), followed by closer contact between them (the adhesion or attachment reaction). Luminal closure occurs throughout the entire uterus during pregnancy or pseudopregnancy and thus does not require the presence of blastocysts. Priming of the uterus with progesterone alone appears to be sufficient for this event to occur; luminal closure does not occur in the absence of progesterone. Although luminal closure and apposition occur in progesterone-treated delayed implanting mice, the attachment reaction does not occur. Estrogen treatment is required for attachment to occur.
Bonnet (23), on the basis of different types of blastocyst-uterine cell-cell interactions, classified implantation into three categories: central, eccentric, and interstitial. Central implantation occurs in mammals such as rabbits, ferrets, and some marsupials. In these animals, blastocysts grow and expand extensively before implantation. In contrast, the blastocysts of mice, rats, and hamsters are small and show modest expansion. In these species, an implantation chamber is formed by the invagination of the uterine epithelium, which is a characteristic of eccentric implantation. In guinea pigs, chimpanzees, and humans, the implantation process is of the interstitial type, i.e., blastocysts are embedded within the subepithelial stroma. The results of ultrastructural studies led Schlafke and Enders (24) to classify implantation into intrusive, displacement, and fusion types. In the intrusive type of implantation, which occurs in humans and guinea pigs, the trophoblasts penetrate through the luminal epithelium, reaching and extending through the basal lamina. The displacement type of implantation occurs in rodents; the luminal epithelium is freed from the underlying basal lamina, facilitating the spread of trophoblasts through the epithelium. The fusion type of implantation, in which trophoblasts make a connection with the luminal epithelium by forming symplasma, occurs in the rabbit. In many rodents, including mice and rats, implantation always occurs at the antimesometrial side of the uterus, whereas in some bats, implantation is mesometrial. In other animals, the embryos elongate and either attach over the entire endometrium (horse, pig, and wallaby) or only at specialized areas known as caruncles (cow and sheep) (2). Schematic diagrams for different types of implantation have been illustrated previously (2, 3).
The attachment reaction coincides with a localized increase in stromal vascular permeability at the site of the blastocyst, as can be demonstrated by iv injection of a macromolecular blue dye (uterine blue reaction) (25). The first sign of the attachment reaction (apposition stage) in the process of implantation occurs in the mouse and rat on the evenings of d 4 and d 5, respectively, and on d 6.5 in the rabbit (25–27). In the primates, the attachment reaction occurs approximately on d 8 in humans and baboons, on d 9 in macaques, and on d 11 in marmoset monkeys (28, 29). In large domestic animals, the first signs of attachment occur on d 13 in pigs, on d 20 in cows, on d 16 in sheep, and on d 19 in goats (30).
In both mice and humans, stromal cells surrounding the implanting blastocyst undergo decidualization, eventually embedding the embryo into the antimesometrial stromal bed. In mice, blastocysts are oriented with their ICMs directed mesometrially, whereas in humans the ICM is directed antimesometrially. The mechanism by which the blastocyst is directed to the antimesometrial luminal epithelium or by which the orientation of the blastocyst is achieved at the time of implantation remains elusive. There is evidence that in progesterone-treated delayed implanting mice, blastocysts are placed antimesometrially, and interdigitation (apposition) of luminal epithelial cell microvilli occurs with those of the abembryonic or lateral trophectoderm cells of the blastocyst with its ICM oriented toward the uterine lumen. This observation led to the suggestion that upon initiation of the attachment reaction and subsequently the implantation process by estrogen, blastocysts retain the orientation they adopted during delay. During normal implantation in mice with the onset of luminal closure, blastocysts are placed at the antimesometrial side of the lumen along the uterine axis. Shortly after the luminal closure, zona-encased blastocysts are located in implantation chambers with random orientation of the ICMs. However, by the beginning of the attachment reaction, blastocysts are correctly oriented with their ICMs directed at the mesometrial pole. This observation suggested that the trophectoderm of the entire blastocyst surface has the potential for attachment to the luminal epithelium, and that attachment occurs randomly immediately after the loss of the zona pellucida. Evidence was presented to suggest that the correct orientation of the blastocyst is achieved by free movement of the ICM. However, further investigation is necessary to resolve this issue. All of these events, from the luminal closure to the attachment reaction, occur between about 86 and 92 h after coitum in mice (reviewed in Refs. 31 and 32).
IV. Delayed Implantation
Delayed implantation is a process by which implantation is postponed for a certain period. The uterus remains in a quiescent state, and embryos at the blastocyst stage become dormant. Delayed implantation occurs in many vertebrate species, but the underlying mechanisms that direct this process are different in various species that have adapted to this reproductive strategy (33). In mice and rats, ovariectomy before the presumed estrogen surge in the morning of d 4 of pregnancy results in the failure of implantation and initiates a state of dormancy of the blastocyst within the uterine lumen (25, 34). This condition is referred to as delayed implantation and can be maintained for many days by continued treatment with progesterone. The process of implantation with blastocyst activation can be rapidly initiated by a single injection of estrogen in the progesterone-primed uterus (25, 34). The mechanisms by which estrogen mediates the processes of blastocyst activation and implantation are poorly understood. Delayed implantation also occurs naturally (facultative) during lactation after postpartum ovulation and fertilization of the egg in mice and rats (35, 36). However, implantation ensues rapidly after termination of the suckling stimulus. The occurrence of lactational delay in these species is due to the secretion of an insufficient amount of ovarian estrogen. Mustelids, marsupials, and many other species also exhibit obligatory seasonal delayed implantation (37–40). Delayed implantation does not occur in some species such as the hamster, guinea pig, rabbit, and pig. Whether this phenomenon occurs in humans is not known. The delayed implantation models in mice and in other species could be exploited more extensively to better understand the molecular signaling that emanates from the embryo and influences uterine biology and vice versa.
V. Window of Implantation: A Transient and Unique Moment
In all eutherian mammals thus far studied, the uterus differentiates into an altered state when blastocysts are capable of effective two-way communication to initiate the process of implantation. This state is termed uterine receptivity for implantation and lasts for a limited period. At this stage, the uterine environment is able to support blastocyst growth, attachment, and the subsequent events of implantation (30, 41–43). The major factors that specify uterine receptivity are the ovarian steroids, progesterone and/or estrogens. Ovarian progesterone and estrogen are crucial for implantation in mice and rats, but ovarian estrogen is not essential for implantation in pigs, guinea pigs, rabbits, and hamsters (41, 44–48). Estrogen-synthesizing capacity has been demonstrated in rabbit and pig embryos, but whether embryonic estrogen plays a role in implantation in these species is still debatable. Recent evidence suggests that hamster blastocysts express the aromatase gene (Paria, B. C., unpublished results). The mouse embryo lacks aromatase activity necessary for estrogen synthesis (reviewed in Ref. 49). Whether preimplantation estrogen secretion by the ovary or embryo plays a role in human implantation is unknown.
In mice and rats, the coordinated actions of progesterone and estrogen regulating proliferation and/or differentiation of uterine cells in a spatiotemporal manner establish the window of implantation (50). For example, on the first day of pregnancy (vaginal plug) in mice, uterine epithelial cells undergo proliferation under the influence of the preovulatory estrogen secretion. Rising levels of progesterone secreted from freshly formed corpora lutea initiate stromal cell proliferation from d 3 onward. The stromal cell proliferation is further stimulated by a small amount of ovarian estrogen secreted on the morning of d 4 of pregnancy. These coordinated effects of progesterone and estrogen result in the cessation of uterine epithelial cell proliferation, initiating differentiation (50). During normal pregnancy, the presence of an active blastocyst in the uterus is the stimulus for the implantation reaction. After the attachment reaction is initiated on d 4 at 2400 h, stromal cells surrounding the implanting blastocyst begin to proliferate extensively and differentiate into decidual cells (decidualization) (30). In pseudopregnant mice, the steroid hormonal milieu within the uterus is similarly maintained due to the presence of newly formed corpora lutea. Thus, the sensitivity of the pseudopregnant uterus to implantation on d 1–4 is quite similar to normal pregnancy, and blastocyst transfer into the uterine lumen during the receptive phase provokes normal implantation reactions and subsequent decidualization. Although blastocysts are the normal inducers of these events, various nonspecific stimuli, such as intraluminal infusion of oil, air, and mechanical stimuli can also initiate certain aspects of the decidual cell reaction (deciduoma) in pseudopregnant or steroid hormonally prepared uteri (30). However, there is evidence that the initial uterine reactions induced by nonspecific stimuli are different from those induced by blastocysts (51, 52).
Uterine sensitivity with respect to steroid hormonal requirements and implantation has been classified as prereceptive, receptive, and nonreceptive (refractory) phases (30, 41). These phases have been defined by employing embryo transfer experiments in pseudopregnant mice. In the mouse, whereas the uterus is fully receptive on d 4, it is considered prereceptive on d 1–3 of pregnancy or pseudopregnancy. The mouse uterus can be rendered receptive to blastocyst implantation only if exposed to a small amount of estrogen after 24–48 h of progesterone priming (53). Evidence suggests that the uterus is most receptive to implantation on d 4 (43), and the efficiency of implantation decreases with time (54). By d 6, the uterus becomes completely refractory to blastocyst implantation. Recent evidence suggests that concentration of estrogen within a very narrow range determines the duration of the window of uterine receptivity in mice; uterine receptivity remains open for an extended period at lower estrogen levels but rapidly closes at higher levels. Uterine nonreceptivity induced at high estrogen levels is accompanied by aberrant uterine expression of implantation-related genes. These results suggest that careful regulation of estrogen levels could improve female fertility in in vitro fertilization programs (55). Another critical factor determining the window of implantation is the state of activity of the blastocyst, as described below.
In mice and rats, ovariectomy before the preimplantation estrogen secretion on the morning of d 4 of pregnancy induces delayed implantation (34, 43). This status can be maintained for many days if progesterone treatment is continuously provided. Under this condition, blastocysts undergo zona hatching, albeit at a slower pace, but they become dormant without initiating the attachment reaction, and the progesterone-primed uterus remains in the neutral stage. However, a single injection of estrogen promptly induces blastocyst activation with the initiation of implantation in the progesterone-primed uterus. Active and dormant blastocysts are molecularly and physiologically distinguishable. Epidermal growth factor (EGF) receptor (EGF-R), cyclooxygenase-2 (COX-2), and histamine type 2 receptor (H2), the factors that are associated with blastocyst attachment reaction, are expressed in normal or active blastocysts but are down-regulated in dormant blastocysts (43, 56–59). In contrast, the G protein-coupled cannabinoid receptor CB1, which is activated by natural and endocannabinoids, is down-regulated in active blastocysts but remains up-regulated in dormant blastocysts (60). Collectively, these findings suggest that a complex array of molecular networks regulates blastocyst activation and dormancy.
Although estrogen is essential for blastocyst activation and implantation in the progesterone-primed mouse uterus, the mechanisms by which estrogen initiates these responses remain elusive. We speculated that estrogen actions in uterine preparation and blastocyst activation for implantation are two distinct events. Embryo transfer experiments in delayed implanting recipient mice provide evidence that whereas the primary estrogen, 17β-estradiol, initiates uterine events for implantation, its catechol metabolite, 4-hydroxy-17β-estradiol (4-OH-E2), participates in activation of dormant blastocysts (17). Blastocyst activation by 4-OH-E2 involves COX-2-derived prostaglandins (PGs) and cAMP (17). The use of an estrogen receptor (ER) antagonist ICI-182,780 showed that, whereas estradiol via its interaction with the nuclear ERs participates in the preparation of the progesterone-primed uterus to the receptive state in an endocrine manner, 4-OH-E2 produced from estradiol in the uterus mediates blastocyst activation in a paracrine manner that does not involve nuclear ERs. These results provide evidence that both primary and catecholestrogens are required for embryo-uterine interactions to ensure successful implantation and that implantation occurs only when uterine receptivity coincides with the activated state of the blastocyst. Molecular pathways that are potentially involved in uterine receptivity and blastocyst activation are discussed below in more detail.
VI. Embryo-Uterine Signaling Pathways in Implantation
Recent advances in molecular and genetic approaches have led to the discovery of numerous molecules involved in embryo-uterine interactions; however, the precise sequence and details of the signaling cascades for many of these molecules have not yet been defined. This review attempts to focus on a select number of signaling molecules and pathways that are implicated in embryo-uterine interactions in relation to implantation (Fig. 1). A large number of other growth factors, cytokines, lipid mediators, and vasoactive agents that could well be involved in implantation are not addressed here due to space limitations.
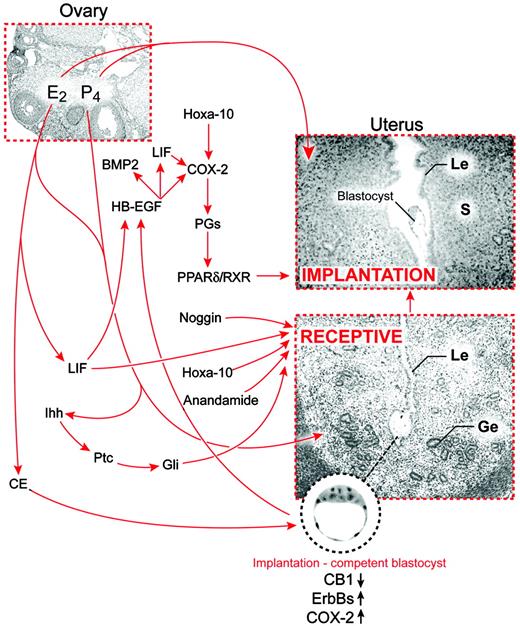
A scheme of signaling networks in embryo-uterine communication during implantation in mice. Implantation in mammals absolutely depends upon synchronized development of the blastocyst to the stage when it is competent to implant and the uterus to the stage when it is receptive to blastocyst growth and implantation. Ovarian estrogen (E2) and progesterone (P4) are the primary effectors that direct the prereceptive uterus to a receptive state via a number of locally expressed growth factors, cytokines, transcription factors, and vasoactive mediators in the uterus, whereas uterine-derived catecholestrogen and regulated levels of endocannabinoids activate the blastocyst to an implantation-competent state. During the attachment phase, signaling and adhesive events embracing the uterus and the blastocyst lead to implantation. Le, Luminal epithelium; Ge, glandular epithelium; S, stroma; CE, catechol estrogen.
A. Steroid hormone signaling
As described earlier, ovarian estrogen is essential for blastocyst implantation in the progesterone-primed uterus in mice and rats. Despite considerable progress regarding the molecular mechanism of estrogen action, many fundamental questions remain unanswered. Estrogen action is normally considered to involve its interaction with a nuclear receptor (ER), a ligand-dependent transcription factor (61, 62). The nuclear ER exists primarily in two isoforms, ERα and ERβ (63, 64). The estrogen-ER complex forms a homodimer that binds to cis-acting estrogen response elements (EREs) in the regulatory regions of the target genes. The EREs for transcription activators are usually present in the 5′-flanking region of specific genes (65). Although a perfect palindromic consensus was identified as AGGTCA(nnn)TGACCT (66), most estrogen-responsive genes have imperfect palindromes or do not have recognizable EREs (65, 67–70). Thus, the contention that steroids or their mimics function only by interacting with nuclear receptors to serve as transcription activators for specific DNA response elements is no longer tenable. Several recent reports highlight the complexities in gene regulation by estrogens, including many of the protein-protein interactions mediated via the ER and coregulators (71–73). For example, cholera toxin, which has no ER-binding capacity, mimics the mitogenic action of estrogen in the uterus presumably by increasing cAMP levels and protein kinase A activity (74, 75). Protein kinase C can also modulate uterine ER levels, and protein kinase C inhibitors can reduce estrogen-induced mitogenic action (76). These results suggest that membrane-bound receptors acting via protein kinases can increase the expression of the same genes activated by steroid hormone nuclear receptors. This forms the basis for the view that other nonnuclear receptors also interact with steroids or their mimics. Although the presence of a membrane ER was postulated more than two decades ago (77), the subject remains controversial because of the lack of molecular identity. However, signaling by plasma membrane ERs is an emerging concept and requires special attention (78).
A large variety of natural and synthetic compounds that mimic natural estrogens and bind to the nuclear ERα are present in the environment (79, 80). A few of these xenobiotics possess an even higher affinity for ERβ than for ERα (81, 82). The interaction of xenoestrogens with ER is considered as the basis for their reproductive toxicity, although their affinity for the receptor is quite low (83, 84). However, there is evidence that responses to an estrogenic compound in target tissues are not necessarily related to its affinity for the receptor (85–88), suggesting the presence of other signaling pathways. Thus, xenoestrogens may interact with ER or other binding proteins that may not result in a similar kind of transactivation that normally occurs with natural ligands. Their estrogen-like effects in the uterus include an increase in water imbibition and ornithine decarboxylase activity, as well as increased DNA and protein synthesis. Xenoestrogens can also induce implantation in progesterone-primed rodents (89). Although the mechanisms of action of these xenoestrogens are not clear, these compounds have received a great deal of attention as a possible cause of certain cancers and impaired reproductive functions (reviewed in Refs. 80 and 90).
Emerging evidence indicates that many of the rapid uterine effects of estrogenic compounds do not involve the classical genomic effects (91–95). We and others have observed that ERα mutant female mice, which show negligible classical responses to estrogen (96), nevertheless display estrogenic responses to catecholestrogens or xenoestrogens (91–95). For example, early uterine estrogenic responses, including gene expression and macromolecular uptake, are elicited by catecholestrogen or kepone independent of ERα and/or ERβ (91). Furthermore, genetic evidence suggests that several other early responsive target genes are induced in the mouse uterus by primary and catechol estrogens independent of nuclear ERs, suggesting nongenomic downstream events. These downstream signaling pathways represent Wnt signaling, protein processing, and calcium homeostasis (92). In contrast, estrogen, which induces the expression of many growth factors and their receptors and other secretory proteins, may facilitate their transport to the membrane and/or secretion by activating the membrane-trafficking pathway via nuclear ER (97). These results suggest that estrogen regulates diverse but interdependent signaling pathways via ER-dependent and -independent processes.
Despite the evidence for nongenomic steroid hormone actions, differential uterine cell-specific expression of nuclear ERα and ERβ and the progesterone receptor (PR) during the periimplantation period in mice suggests that the coordinated effects of estrogen and progesterone in uterine events for implantation are primarily mediated via these nuclear receptors (98). Furthermore, gene-targeting experiments in mice lacking ER or PR provide valuable information regarding their roles in uterine biology that were not previously recognized. ERα(−/−) mice exhibit infertility due to hyperstimulated ovaries and hypoplastic uteri (99). Furthermore, blastocyst transfers in estrogen- and progesterone-treated uteri of ERα(−/−) mice have established that functional ERα is required for implantation (100). In contrast, ERα(−/−) mice can induce and support decidualization (deciduoma) in response to artificial stimuli if primed appropriately with progesterone alone (101, 102). These results suggest that ERα(−/−) mice fail to initiate and support implantation, perhaps due to the failure of the attachment reaction rather than the failure of decidualization events (91). Indeed, the genes and signaling pathways involved in decidualization are induced in ERα(−/−) after application of an artificial stimulus (100). PR(−/−) mice show pleiotropic reproductive defects including impaired ovulation, uterine hyperplasia, and failure in decidualization (103). Selective deletion of the PR-A isoform also showed infertility but with a milder phenotype, suggesting that PR-A and PR-B serve as functionally distinct mediators of progesterone signaling in vivo (104). The results of experiments that used both PR(−/−) and PR-A(−/−) mice further reinforce a requirement of progesterone in decidualization (103, 104).
Gene-targeting experiments have established the importance of both ER and PR in uterine preparation for implantation in mice (99). However (98, 103), whether the preimplantation embryo is a direct target for steroid hormones remains unclear. Recent evidence suggests that preimplantation estrogen secretion on d 4 of pregnancy in mice has a dual role as primary estrogen and as a catecholestrogen with distinct targets (17). Whereas primary estrogen acts via the uterine ER to prepare the uterus for implantation, catecholestrogens formed locally in the uterus from the primary estrogen participate in blastocyst activation. However, it is still a mystery how catecholestrogens mediate activation of blastocysts (17). Although nuclear ERα is present in both active and dormant blastocysts (105), dormant blastocysts do not respond to estradiol and fail to attain implantation competency in vitro. In contrast, dormant blastocysts do respond to a catecholestrogen 4-OH-E2 and become implantation competent in vitro. The ER antagonist ICI-182,780 fails to reverse this response, suggesting that nuclear ER signaling is not critical to blastocyst activation (17). These observations are surprising in the light of other recent findings that ERα, ERβ, and efp mRNAs are expressed in the preimplantation embryos (106, 107). Examining the direct roles of estrogens and/or progesterone in preimplantation embryo function and how steroid hormone signaling in the embryo and uterus are coordinated for implantation will require further investigation.
Estradiol undergoes hydroxylation to 4-OH-E2 by a P450-linked enzyme CYP1B1 (108). This enzyme is present throughout the mouse uterus on d 4, but disappears from the implantation site on d 5 (17). Activation of dormant blastocysts appears to involve an early response to 4-OH-E2, because dormant blastocysts transferred into delayed implanting recipient uteri within 1 h of estradiol administration of the recipients show implantation, whereas similar blastocysts transferred beyond this 1-h period fail to implant (17). These results suggest that a rapid response that is critical to implantation occurs in utero. In contrast, dormant blastocysts cultured in the presence of 4-OH-E2, but not estradiol, gain implantation competency and, upon transfer, implant in pseudopregnant recipients well beyond the 1-h window of estradiol treatment. Similar results were also obtained by culturing dormant blastocysts in the presence of PGE2 or a permeable analog of cAMP. This effect apparently involves the COX-2 signaling pathway (17). For example, coincubation of dormant blastocysts with a selective COX-2 inhibitor and 4-OH-E2 efficiently blocks their activation and implantation upon transfer to suitable recipients. This effect of the COX-2 inhibitor was partially reversed by addition of PGE2 to the culture media. The results strongly suggest that the action of 4-OH-E2 on dormant blastocysts is mediated via the COX-2 signaling pathway, leading to an increase in intracellular cAMP levels. Further investigation will reveal the types of PGs and receptors involved in this event.
B. Signaling via adhesion molecules: cell-cell interactions
Many glycoproteins and carbohydrate ligands and their receptors are expressed in the uterine luminal epithelium and blastocyst cell surfaces (reviewed in Refs. 109 and 110). Primary adhesion molecules that are implicated in implantation are selectins, galectins, heparan sulfate proteoglycans, Muc-1, integrins, cadherins, and the trophinin-tastin-bystin complex. Muc1 acts as an antiadhesive masking molecule (111). Muc1, a stretch of long carbohydrate moieties, is expressed in the mouse uterine epithelium before implantation. The physical hindrance created by these branches is thought to prevent interaction between the embryo and the luminal epithelium of the uterus before the attachment reaction. This is consistent with timely down-regulation of Muc1 from the luminal epithelium throughout the uterus before the attachment reaction in mice (reviewed in Ref. 3). In contrast, overall Muc1 expression increases in the rabbit and human uterus during the receptive period. However, careful examination revealed that there is indeed a decrease in Muc1 levels at the site of implantation in rabbits (112). In humans, the situation appears to be more complicated. During the apposition phase, the presence of an embryo increases the levels of Muc1 in the epithelium, but at the adhesion phase, the embryo induces a cleavage of Muc1 at the implantation site (113). Collectively, these findings suggest that Muc1 acts as an antiadhesive molecule that must be removed from the site of implantation.
Among the adhesion molecules, integrins have been studied more extensively in the human endometrium because of their cycle-dependent changes and the potential role in uterine receptivity. The members of the integrin family serve as receptors for various extracellular matrix (ECM) ligands and modulate cell-cell adhesion and signal transduction events (114). Each integrin is comprised of two subunits, α and β, and each αβ-combination has its own binding specificity and signaling properties. As membrane-associated receptors, integrins possess short cytoplasmic tails with no enzymatic activity. Signaling by integrins is mediated by associating adaptor proteins that bridge them to the cytoskeleton, cytoplasmic kinases, and transmembrane growth factor receptors (114). Several members of the integrin family, including αvβ3, are known to interact with the RGD (Arg-Gly-Asp) peptide sequence present in many ECM proteins, such as fibronectin, laminin, and entactin. Although many integrin heterodimers show constitutive expression in the uterine epithelium or stroma, α1β1, α3β1, α6β1, αvβ3, and αvβ1 heterodimers exhibit cycle-dependent changes (115–119). Of special interest is the expression of α1β1, which shows implantation-related changes. For example, the expression of α1β1 is restricted to early- and midsecretory phases both in the epithelium and stroma and primarily restricted to the stroma during the predecidual phase (117). Furthermore, unexplained infertility in women is associated with the deficiency of αvβ3 or α4β1 in the uterus during the window of implantation (120). However, the functional definition of these markers in uterine receptivity or under pathological conditions still awaits further investigation. In mice, αvβ3 is expressed in both the uterine luminal epithelium and the blastocyst during implantation. It has been shown that an intrauterine injection of RGD peptide or neutralizing antibody against αvβ3 reduces the number of implantation sites in mice and rabbits (121).
Among many subunits, α5β1, α6β1, and αvβ3 are expressed in the mouse embryo throughout the periimplantation period, whereas several others exhibit stage-specific expression (122). Integrins are also expressed in the differentiating trophoblasts at later stages (122), suggesting their roles in trophoblast differentiation and adhesion. A role for fibronectin via integrin binding in blastocyst outgrowth was further confirmed in vitro using antibodies against the αv, α5, β1, or β3, which inhibited adhesiveness on the outer surface of the trophoblast inducible by fibronectin (123). In addition, a gene-targeting experiment revealed that deletion of the β1 gene results in ICM defects and embryonic lethality (124). However, the mutant embryos form morphologically normal blastocysts and initiate implantation, but trophoblast invasion becomes defective (124). Adhesion-competent, late-blastocyst-stage trophoblasts undergo intracellular signaling initiated upon ligation of α5β1 and αvβ3 by fibronectin (125). Integrin signaling mobilizes cytoplasmic Ca2+ and induces the trafficking of intracellular vesicles, resulting in stronger adhesion to fibronectin at the apical surface. Therefore, blastocyst adhesion to the endometrium during implantation is considered to be regulated by the endogenous developmental program, as well as through interactions with ECM components in the local environment (126, 127). Although there is evidence that the embryo is a site of action for integrin signaling, it is not yet clear whether the uterus is a site of action. Results of gene-targeting experiments of integrin subunits are not very informative in relation to their roles in implantation because of the complex phenotypes and apparent compensation by other subunits (128–131).
Invasive mouse trophoblasts adhere, spread, and migrate on ECM substrates (132–135) and penetrate three-dimensional ECM structures (136, 137). Several ECM components that are up-regulated in the periimplantation endometrium, including fibronectin, laminin, and collagen type IV (138–140), support trophoblast outgrowth in vitro (132, 133). Trophoblast interactions with the ECM are mediated primarily by integrins (122, 123, 133–135, 141). Hexapeptides containing the RGD sequence recognized by integrins (142) block trophoblast outgrowth on fibronectin, collagen type II and IV, entactin, and vitronectin (133, 135, 141). However, trophoblast adhesion to type I laminin is independent of its RGD sequence and is primarily mediated through interaction of α7β1 with the E8 integrin-recognition domain of laminin (143, 144).
Trophinin was identified by cDNA library screening of a human trophoblastic cell line (145). This transmembrane protein can mediate homophilic interactions between two different cell types. For example, it mediates an interaction between a human endometrial cell line and a trophoblastic cell line, but this interaction is complex (145). Trophinin requires the presence of a cytoplasmic protein tastin to sustain adhesion between these two cell types. In addition, the presence of bystin, another cytoplasmic protein, is required for effective interaction between trophinin and tastin. This adhesion complex, which is present in both trophoblastic teratocarcinomas and endometrial adenocarcinomas, mediates adhesion between them. In humans and monkeys, trophinin is specifically expressed in cells involved in implantation. Furthermore, the trophinin complex was detected in both trophoblast and decidual cells at the human fetalmaternal interface as early as the sixth week of pregnancy (146). Although trophinin expression in mice coincides with the timing of implantation, it is expressed in the luminal and glandular epithelium throughout the uterus irrespective of the presence or absence of blastocysts, raising doubts regarding its role specific to implantation (147).
E-cadherin, a calcium-dependent cell-cell adhesion molecule, participates in the formation of the epithelial adherens junctions in cooperation with α- and β-catenins (148, 149). E-cadherin is a critical factor for blastocyst formation, because its targeted deletion leads to defective embryonic development resulting in failure to form the trophectoderm (150, 151). E-cadherin is implicated in uterine-embryo interactions because of its homotypic adhesive activity (152). Whereas the trophectoderm highly expresses E-cadherin, the components of the adherens junctional complex are also expressed in the uterine luminal epithelium at the time of the attachment reaction. The expression subsequently becomes evident in the subepithelial stroma surrounding the implanting blastocysts with apoptosis occurring in the luminal epithelium (152, 153). Therefore, it is speculated that temporal and cell-specific expression of the adherence junction proteins in the uterus results in molecular guidance, which is important for blastocyst attachment and subsequent invasion.
A recent study has highlighted the critical role of the selectin adhesion system in human implantation (154). This adhesion system, which is involved in leukocyte capture from the bloodstream, is also operative during implantation and placentation. On the maternal side, selectin oligosaccharide ligands are expressed in the receptive uterine epithelium, and on the embryonic side, trophoblast cells expressed L-selectin receptors. It was observed that beads coated with specific selectin ligands adhere to the trophoblast, suggesting that the trophoblast cell surface receptors are functional. This investigation suggests that trophoblast L-selectin mediates interactions with the uterus to establish an adhesion mechanism for implantation. This is an exciting finding in several respects. First, this study affirms that specific ligand-receptor signaling pathways between the embryo and uterus are critical for implantation and subsequent pregnancy establishment. Second, it shows that the same mechanism operative during implantation is also operative during the later phases of pregnancy. Third, it shows that trophoblast cells share a system known to be active in the blood-vascular system. Similar adhesion signaling between the transmembrane form of heparin-binding EFG-like growth factor (HB-EFG) expressed on the luminal epithelial surface and ErbB receptors present on the trophectoderm cell surface for implantation in mice and humans has been reported previously (57, 155).
C. Signaling by vasoactive factors
It has long been speculated that vasoactive agents, such as histamine and PGs, are involved in many aspects of reproduction including ovulation, fertilization, implantation, and decidualization. Histamine functions as a ubiquitous mediator of cell-cell signaling and is synthesized from L-histidine by histidine decarboxylase (HDC) both in peripheral tissues and in the nervous system (156). Histamine is a well-known neurotransmitter in the brain (157), but it is also involved in other physiological responses including gastric acid secretion, regulation of allergic reactions, and vascular permeability (reviewed in Ref. 158). Because the process of implantation is considered a proinflammatory reaction and because increased vascular permeability at the site of blastocyst implantation is common to many species, it was suggested that histamine plays a role in implantation and decidualization (158). Earlier observations suggested that uterine mast cells are a possible source of histamine, and its release from mast cells by estrogen is important for implantation (159, 160). This suggestion was based on the observations that local histamine application stimulates uterine hyperemic and edematous responses (161) and that there is a reduction in uterine mast cell numbers and histamine content after estrogen treatment and during implantation (162). Furthermore, a histamine antagonist pyrathiazine or an inhibitor of HDC was shown to interfere with implantation when instilled into the uterine lumens of rats and rabbits (159, 163). Histamine works via at least four histamine receptor subtypes (H1, H2, H3, and H4) (164–166) and blocking both H1 and H2 receptors was shown to interfere with implantation (167). Subsequent studies also showed that histamine induces implantation in delayed implanting rats when injected with a suboptimal dose of estrogen (168). However, successful implantation and birth of live offspring in mast cell-deficient mice and other evidence suggest that uterine mast cell histamine is not essential for implantation (136, 169, 170). Thus, if histamine is involved in implantation, it should be provided either by major uterine cell types or by embryonic cells. A recent study showed that mouse blastocysts do not have the capacity for histamine synthesis (59). However, HDC is expressed in uterine epithelial cells on d 4 of pregnancy in mice before implantation, but not in decidual cells (171). Thus, although histamine may have a role in implantation in mice, its role in decidualization is unlikely. Whereas H1, H2, and H3 receptor subtypes are not detectable in the uterus, H2 receptors are expressed in preimplantation mouse blastocysts. These observations, as well as the inhibition of blastocyst zona-hatching and implantation by H2 antagonists and an HDC inhibitor, suggest that uterine histamine targets the blastocyst for implantation (59). However, apparently normal implantation occurs in mice lacking HDC or H2 type histamine receptor genes, suggesting the possible involvement of other vasoactive agents with overlapping functions in this process (165, 172).
PGs possess vasoactive, mitogenic, and differentiating properties (173) and are implicated in various female reproductive functions. COX, which exists in two isoforms, COX-1 and COX-2, is the rate-limiting enzyme in the biosynthesis of PGs. COX mediates the conversion of arachidonic acid into PGH2, which is then converted to various PGs by specific synthases (173). The COX isoforms are encoded by two separate genes and exhibit distinct cell-specific expression, regulation, and subcellular localization, yet share similar structural and kinetic properties. COX-1 is considered to be a constitutive enzyme that mediates housekeeping functions. In contrast, COX-2 is an inducible enzyme and is induced in a variety of cell types by growth factors, cytokines, and inflammatory stimuli (173). PGs normally exert their function by interacting with cell surface G protein-coupled receptors, but they can also function as ligands for nuclear peroxisome proliferator-activated receptors (PPARs) (174–177). Because COX-2 is primarily responsible for increased PG production during inflammation, this isoform is the target for development of selective antiinflammatory drugs (178, 179). COX-2 overexpression is also associated with tumorigenesis (180, 181).
The processes of ovulation and implantation are considered analogous to proinflammatory responses, and thus participation of PGs in these events has been speculated (182, 183). For example, PGs are considered to participate in follicular rupture during ovulation (reviewed in Ref. 184). This is consistent with gonadotropin-mediated induction of COX-2 in ovarian follicles preceding ovulation (85, 184). PGs are also implicated as important mediators of increased endometrial vascular permeability during implantation and decidualization (185). A unique pattern of expression of Cox-1 and Cox-2 genes in the periimplantation mouse uterus further suggests that PGs play important roles in these processes (185). Cox-1 is expressed in uterine luminal and glandular epithelial cells on the morning of d 4 of pregnancy, but its expression becomes undetectable in the luminal epithelial cells by the time of the attachment reaction. In contrast, Cox-2 is expressed in the luminal epithelium and underlying stromal cells solely at the site of blastocyst attachment. Using the delayed implantation model, this study also established that the expression of Cox-2 in the receptive uterus requires the presence of active blastocysts. The results suggested that Cox-2 expression during the attachment reaction is critical to implantation (185). Indeed, gene-targeting experiments have demonstrated that COX-2-derived PGs are essential for implantation and decidualization (186–188). Experiments with Cox-1(−/−) mice suggest that the loss of COX-1 is compensated by the expression of Cox-2 for implantation (189). Among various PGs, the levels of prostacyclin (PGI2) are highest at the implantation sites of wild-type mice, and implantation defects are partially restored in Cox-2(−/−) mice by administration of a more stable PGI2 agonist, carbaprostacyclin (190).
The role of PGs is further illustrated by the reduced fertility of female mice lacking cytoplasmic phospholipase A2, which is involved in the liberation of arachidonic acid from membrane phospholipids for PG synthesis by the COX system (191–193). The reduced fertility in these females is due to deferral of on-time implantation, leading to subsequent retarded fetoplacental development and reduced litter size (54). Collectively, these results indicate that the cytoplasmic phospholipase A2-COX-2 axis is crucial to implantation. However, one recent study described that wild-type (B6C3H) blastocysts transferred into COX-2-deficient female mice on a mixed (C57Bl6/JX129S7/SvEvBrd) background on d 3 of pseudopregnancy implanted and produced live offspring, although decidual growth was retarded (194). Because this study did not follow experimental protocols similar to other studies, the discrepancy between this study and others cannot be compared scientifically. However, some of the results reported by Cheng and Stewart (194) remain uninterpretable. For example, dissected decidual weights reported by these investigators seem abnormally high on various days of pregnancy, and the sizes of d 7 implantation sites shown appeared larger than d 8 implantation sites and at a more advanced stage of pregnancy. Nonetheless, recent evidence demonstrates that Cox-2 is expressed either in the uterus, blastocyst, or both during implantation in a variety of species with different modes of implantation, including sheep, mink, skunk, baboon, and pig (195–198). COX-2 expression in human endometrium has also been reported (199, 200). These results suggest a conserved function of COX-2 in implantation in various species. However, it has been discovered recently that, depending on the genetic background, COX-1 can rescue female infertility in COX-2-deficient mice (201).
PGs exert diverse functions using both cell surface PG receptors and PPARs. Receptors for PGE2, PGF2α, PGD2, PGI2, and thromboxanes have been named as EP1–EP4, FP, DP, IP, and TP, respectively; they belong to the G protein-coupled family of cell surface receptors (reviewed in Refs. 202 and 203). Although PGE2 synthase is expressed at the implantation sites with the presence of PGE2 and EP receptors (203–205) and although PGE2 has been shown to be associated with implantation and decidualization (206), gene-targeting experiments show that three of the four EP receptor subtypes (EP1–EP3) are not critical for implantation. EP4 deficiency results most frequently in perinatal lethality, and thus its role in implantation has not yet been determined (reviewed in Ref. 203). Furthermore, mice deficient in FP or IP show normal implantation. PGs can also exert their effects by utilizing PPARs that belong to a nuclear hormone receptor superfamily. The evidence that PG-mediated PPAR signaling is involved in implantation is discussed below in more detail. PGs also appear to be important for embryonic functions relevant to preimplantation embryo development and implantation. Preimplantation embryos produce PGs, and inhibitors of PG synthesis have been shown to inhibit embryonic growth, functions, and zona hatching in vitro (207 , 208). Dormant mouse blastocysts can achieve implantation competence if cultured in the presence of PGE2 or a permeable analog of cAMP. This effect apparently involves the COX-2 signaling pathway (17). However, normal development of Cox-1(−/−) /Cox-2(−/−) double-mutant embryos in the uterus suggests that PGs of embryonic origin are not essential for embryo development (208). However, compensation by maternal PGs in embryonic development cannot be ruled out. Vascular endothelial growth factor (VEGF), also known as vascular permeability factor, is highly vasoactive in nature. It is a potent inducer of vasodilation and angiogenesis. Its role in implantation is discussed below.
D. Signaling by growth factors
The expression of various growth factors and their receptors in the uterus in a temporal and cell-specific manner during the periimplantation period suggests that these factors are important for implantation (3–6, 209, 210). The present review highlights primarily the importance of the EGF family of growth factors; however, the roles of other growth factors, such as TGFβs, fibroblast growth factors (FGFs), IGFs, platelet-derived growth factors, and many others should not be ignored. The EGF family of growth factors includes EGF itself, TGFα, HB-EGF, amphiregulin, betacellulin, epiregulin, and neuregulins (58, 211). HB-EGF is the earliest molecular marker found in the uterus exclusively at the sites of active blastocysts appearing several hours before the attachment reaction in mice (26). This induction is followed by the expression of betacellulin, epiregulin, neuregulin-1, and Cox-2 around the time of the attachment reaction (58, 185, 211). In contrast, amphiregulin is expressed throughout the uterine epithelium on the morning of d 4 of pregnancy and is well characterized as a progesterone-responsive gene in the uterus (212). Around the time of the attachment reaction, strong expression of amphiregulin in the luminal epithelium is found only around the implanting blastocysts, and this expression is absent by the morning of d 5. Although these results suggested that amphiregulin has a role in implantation, amphiregulin-deficient mice or compound knockout mice for EGF/TGFα/amphiregulin do not exhibit implantation defects (213, 214). Because HB-EGF, betacellulin, epiregulin, neuregulin, and amphiregulin all show overlapping uterine expression patterns around the implanting blastocyst at the time of attachment reaction (reviewed in Refs. 3 and 211), it is assumed that a compensatory mechanism rescues implantation in the absence of one or more members of the EGF family.
The EGF-like growth factors interact with the receptor subtypes of the erbB gene family, which is comprised of four receptor tyrosine kinases: ErbB1 (EGF-R), ErbB2, ErbB3, and ErbB4. They share common structural features but differ in their ligand specificity and kinase activity (215). The initial dimerization between coexpressed receptors upon ligand binding constitutes the classical mechanism of action of EGF-like ligands. Spatiotemporal expression patterns of EGF gene family members and ErbBs in the uterus during the periimplantation period suggest compartmentalized functions of EGF-like growth factors in implantation (58).
A number of growth factors and their receptors are expressed in preimplantation embryos of several species, suggesting their roles in preimplantation mammalian development (216, 217). In this review, we focus on potential roles of the EGF family of ligands with respect to preimplantation embryo development and implantation. ErbB1 (EGF-R), ErbB2, and ErbB4, the receptor subtypes for the EGF family of growth factors, are expressed in the mouse blastocyst (Refs. 56 and 218 and our unpublished results), and EGF or TGFα has beneficial effects on embryonic development in vitro (18). Using genetic and biochemical approaches, the roles of embryonic ErbB1 and/or ErbB4 in interacting with uterine HB-EGF in blastocyst implantation have recently been highlighted in mice (57, 218). HB-EGF is expressed as soluble and transmembrane forms in the uterine luminal epithelium at the site of the blastocyst before the attachment reaction, suggesting paracrine and/or juxtacrine interactions with embryonic ErbBs, as well as autocrine, paracrine, and/or juxtacrine interactions with uterine ErbBs that are expressed in a spatiotemporal manner during the periimplantation period (26, 58, 219). For example, the expression of both ErbB1 and ErbB4 is down-regulated in dormant blastocysts during delayed implantation but is readily up-regulated with blastocyst activation and initiation of implantation (56, 218). Furthermore, whereas a recombinant soluble HB-EGF can promote blastocyst growth and differentiation (26), cells that express the transmembrane form of HB-EGF can adhere to active, but not dormant, blastocysts in vitro (57), suggesting paracrine and juxtacrine functions of HB-EGF. In addition, by directing an HB-EGF-toxin conjugate toward wild-type and erbB1(−/−) blastocysts, it was shown that HB-EGF could also interact with embryonic ErbB4 and heparan sulfate proteoglycan molecules (218). Collectively, these results suggest that an interaction between uterine HB-EGF and blastocyst ErbBs is important for the attachment reaction. However, the absolute necessity of HB-EGF in implantation requires genetic evidence. A recent report shows that most HB-EGF mutant mice die early in postnatal life due to cardiac defects, precluding critical examination of the implantation phenotype (220). It is also to be noted that early events of implantation do not appear to be affected by blastocysts deficient in either ErbB1 or ErbB4 (221, 222), although the implantation-initiating efficiency of blastocysts deficient in more than one receptor type needs to be tested to delineate the functional redundancy among the receptor family. In conclusion, detailed expression and gene-targeting experiments with all of the ligands and receptors are required to define paracrine, autocrine, and/or juxtacrine roles of specific ligand or its receptors in implantation.
Among many growth factors that have been studied in humans, HB-EGF appears to play a role in implantation and embryonic development. Its expression is maximal during the late secretory phase (cycle d 20–24) when the endometrium becomes receptive for implantation (223, 224) and cells expressing the transmembrane form of HB-EGF adhere to human blastocysts displaying cell surface ErbB4 (155). Furthermore, HB-EGF was shown to be one of the most potent growth factors for enhancing the development of human in vitro fertilization-derived embryos to blastocysts and subsequent zona hatching (225). Thus, cumulative evidence suggests that HB-EGF has a significant role in preimplantation embryo development and implantation as a paracrine and/or juxtacrine factor in various species.
E. Signaling by cytokines
The expression of various cytokines and their receptors in the uterus and embryo during early pregnancy suggests their roles in various aspects of implantation (reviewed in Refs. 3 , 20 , 225 , and 226). However, gene-targeting studies show that mice lacking TNFα, IL-1β, IL-1 receptor antagonist, IL-1 receptor type 1, IL-6, and granulocyte/macrophage-colony stimulating factor apparently do not manifest overt reproductive defects (reviewed in Ref. 20). These observations suggest that either these molecules have minor roles in implantation or the loss of one cytokine is compensated by other cytokines with overlapping functions. In contrast, some cytokines are important for normal female fertility (227–229). For example, female op/op mice with a naturally occurring mutation of the M-CSF gene have markedly impaired fertility (227), and mice with a null mutation of the Lif gene encoding leukemia inhibitory factor (LIF) show complete failure of implantation, and blastocysts in these mutant mice undergo dormancy (228, 230). Studies using IL-11Rα mutant mice have also shown that IL-11 is crucial to decidualization, but not for the attachment reaction (229). Interestingly, both LIF and IL-11 are members of the IL-6 family, which includes IL-6 itself, oncostatin M, ciliary neurotrophic factor, and cardiotrophin (231). LIF and IL-11 bind to ligand-specific receptors, LIFR and IL-11R, respectively, and share gp130 as a signal transduction partner (231), suggesting that gp130 signaling is critically involved in implantation. Although the mechanism underlying implantation and decidualization failures in the absence of LIF still remains to be elucidated, recent evidence shows that there is a loss or an aberrant expression of certain implantation-related genes in pregnant Lif mutant mice (232). For example, uterine expression of HB-EGF and epiregulin is absent, and Cox-2 expression is aberrant at the sites of blastocysts in Lif mutant mice during the anticipated time of implantation.
LIF and its receptors, LIFR and gp130, exist in both soluble and membrane-bound forms, and soluble forms of these two receptors antagonize the actions of their ligands, implying the complexity of the LIF signaling pathway (233–235). Lif is transiently expressed in uterine glands on d 4 of pregnancy in mice, suggesting its role in implantation (236). However, our recent studies show that uterine Lif expression is biphasic on d 4. Not only is Lif expressed in glands, but it is also expressed in stromal cells surrounding the blastocyst at the time of the attachment reaction (232). This suggests that LIF has dual roles: first in the preparation of the uterus, and later in the attachment reaction. However, the molecular mechanism by which LIF executes its effects on implantation is not yet known. In this regard, it would be useful to establish the complicated ligand-receptor interactions and detailed expression patterns of LIF receptors that occur during the periimplantation period. However, a recent report shows that inactivation of gp130 by deleting all signal transducers and activators of transcription binding (STAT) sites results in implantation failure (237), reinforcing the importance of LIF signaling in implantation.
The uterine milieu in Lif mutant mice fails to induce implantation irrespective of the blastocyst genotypes, since Lif(−/−) blastocysts can implant after transfer to wild-type pseudopregnant recipients (228, 230). These reciprocal embryo transfer experiments suggest that maternal LIF is essential for blastocyst implantation. However, a role for this cytokine in embryonic functions cannot be ignored, because LifR and gp130 are expressed at the blastocyst stage, and administration of exogenous LIF improves embryo viability and hatching in several species (238–240). Taken together, these data suggest that both the preimplantation embryo and the uterus are sites of LIF action. However, embryos lacking either LIFR or gp130 develop to the blastocyst stage and implant normally but die during the perinatal period (241, 242). These results raise questions about the role of LIF signaling in preimplantation embryo development.
Lif expression in the uterus is maximal around the time of implantation in most species examined, although the steroid hormonal requirements for the preparation of uterine receptivity and implantation differ depending on the species. Whereas uterine Lif expression in several species appears to be regulated by P4 (reviewed in Ref. 242), estrogen regulates Lif expression in the mouse uterus. This is evident from Lif expression on d 1 of pregnancy and during the estrous stage of the cycle when the uterus is under the influence of estrogen stimulation (236, 243, 244). In addition, Lif is not expressed in the uterus during experimentally induced delayed implantation but is rapidly induced by an injection of estrogen (232, 236). However, it has yet to be learned how estrogen induces Lif expression in the mouse uterus and the mechanism by which it is regulated by progesterone in other species. In humans, Lif is expressed in the endometrium and at higher levels in the glandular epithelium of the secretory endometrium (245). Furthermore, LIF deficiency has been associated with unexplained recurrent abortions and infertility in women (246).
F. Homeobox genes in implantation
Hox genes are transcription factors that belong to a multigene family. They are developmentally regulated and share a common highly conserved sequence element called the homeobox that encodes a 61-amino acid helix-turn-helix DNA-binding domain (247). Hox genes are organized in four clusters (A, B, C, and D) on four different chromosomes in mice and humans and follow a stringent pattern of spatial and temporal colinearity during embryogenesis (247). Several Hox genes at the 5′-end of each cluster are classified as AbdB-like Hox genes, because of their homology with the Drosophila AbdB gene. In vertebrates, AbdB-like Hox genes, similar to their Drosophila ortholog, are expressed in developing genitourinary systems (248). For example, Hoxa-10 and Hoxa-11 are highly expressed in developing genitourinary tracts and the adult female reproductive tract, suggesting roles in reproductive events (248–250). Hoxa-10 mutant mice exhibit oviductal transformation of the proximal one third of the uterus. Furthermore, adult female mice deficient in Hoxa-10 show failures in blastocyst implantation and decidualization unrelated to the oviductal transformation (248). Subsequent studies revealed that uterine stromal cells in Hoxa-10-deficient female mice show reduced proliferation in response to progesterone, leading to decidualization defects (248, 251). The uterus in Hoxa-11-deficient mice is hypoplastic and devoid of uterine glands due to developmental defects. Defective proliferation of stromal cells in Hoxa-10(−/−) female mice suggests that Hoxa-10 is involved in the local events of cellular proliferation by regulating cell cycle molecules. Indeed, cyclin D3 is aberrantly expressed in Hoxa-10 mutant uteri in response to a decidualizing stimulus (252). Furthermore, because several progesterone-responsive genes are dysregulated in the uterine stroma of Hoxa-10 mutant mice (251), Hoxa-10, as a transcriptional factor, may convey progesterone responsiveness in the uterine stroma by regulating gene expression. A similar, but more severe, phenotype was also noted in Hoxa-11-deficient female mice (250).
A recent study using microarray analysis further revealed that the absence of Hoxa-10 in the uterus in response to progesterone is associated with two cellular disturbances (253). First, among many genes that were up-regulated in the Hoxa-10-deficient uteri, two cell cycle molecules, p15 and p57, were notable. These two genes are both cyclin-dependent kinase inhibitors (CKIs), suggesting that the previously observed defect in stromal cell proliferation in Hoxa-10 mutant mice could be associated with this up-regulation of CKIs. Second, the microarray experiments and follow-up fluorescence activated cell sorter analyses demonstrated that there was hyperproliferation of T lymphocytes in the Hoxa-10-deficient uterine stroma in response to progesterone. These results suggest that an aberrant lymphoproliferation has adverse effects on implantation in the Hoxa-10-deficient mice. In humans, both Hoxa-10 and Hoxa-11 genes are markedly up-regulated in the uterus during the midsecretory phase in steroid hormone-dependent manner (254), suggesting their roles in human implantation.
There is a homeobox gene family, unrelated to other larger classes of homeobox genes, called the Hmx family of transcription factors. These genes show overlapping expression during development, but gene-targeting experiments have revealed a unique role for Hmx3 in female reproduction (255). Hmx3 mutant female mice show normal fertilization and preimplantation embryo development to blastocysts. However, the blastocysts fail to implant in the uterus and subsequently die. Because Hmx3 is primarily expressed in the myometrium during early pregnancy, the mechanism of infertility in these mice is different from that of Hoxa-10 or Hoxa-11 mutant mice and has yet to be explored.
G. Ligand-dependent nuclear receptors and coactivators in implantation
The nuclear receptor superfamily of transcription factors modulates expression of target genes by binding to specific DNA elements. The members of this superfamily span from well-characterized steroid hormone receptors to orphan nuclear receptors with no known ligands. Steroid hormone receptors aside, the PPAR family of nuclear receptors has been implicated in female reproductive events. Three members of the PPAR family are PPARα, PPARγ, and PPARδ. To act as a transcriptional activator, PPARs must form a heterodimer with a member of the retinoid X receptor (RXR) subfamily (176, 190). As described below, whereas developmental defects are common in PPARδ and PPARγ mutant mice, no apparent reproductive phenotype is evident in PPARα mutant mice.
1. PPARδ(β)
PGs can act via dual receptor signaling systems, either via classical cell surface receptors or through nuclear receptor systems. PPARs can respond to a wide variety of ligands including natural and synthetic eicosanoids, fatty acids, and hypolipidemic and hypoglycemic drugs (256). There is evidence that COX-2-derived PGI2 participates in implantation via activation of PPARδ (190), because the implantation defects in Cox-2(−/−) mice are reversible by a PGI2 agonist or a combination of PPARδ and RXR agonists. PGI2 is the most abundant PG in the early pregnant mouse uterus and is higher at implantation sites than in interimplantation sites (190). Consistent with the finding that COX-2-driven uterine PG production is crucial to implantation, Cox-2 and prostacyclin synthase (Pgis) are coexpressed at the implantation site, suggesting the availability of PGI2 directly to uterine cells. In searching for a target receptor for PGI2 in the uterus, the expression of known PGI2 receptors, such as IP, PPARα, and PPARδ, was examined. Among these, PPARδ was colocalized at similar regions of the implantation sites with Cox-2 and Pgis; the expression of IP and PPARα was very low to undetectable. The functionality of PPARδ as a PGI2 receptor was further examined in vivo, using COX-2-deficient mice as an in vivo model. Administration of cPGI or L-165041 (a selective PPARδ agonist) to these mice improved implantation and decidualization (190). In conjunction with other in vitro evidence, this work suggests that PPARδ expressed in the uterine stroma responds to a PGI2 agonist to mediate embryo implantation (190, 257, 258). Three independent groups have reported diverse phenotypes of PPARδ knockout mice (259–261). Because of severe early developmental defects of PPARδ mutant embryos, it is very difficult to utilize this model to directly address whether the absence of maternal PPARδ affects implantation as in COX-2-deficient mice. Therefore, the conditional knockout mouse model with uterine-specific deletion of PPARδ is necessary to address this issue.
2. PPARγ
PPARγ is well known as a key metabolic regulator participating in obesity control, diabetes, atherosclerosis, and other processes (262). PPARγ-activating ligands include naturally occurring compounds, such as 15-deoxy-Δ (12, 14)-PGJ2, and the thiazolidinedione class of insulin-sensitizing synthetic compounds (262). Whereas the roles of PPARγ in several metabolic processes have been well documented, gene-targeting experiments reveal that PPARγ is also required for normal development of placental, cardiac, and adipose tissues. PPARγ mutant mice die around embryonic d 10 due to defective terminal differentiation of the trophoblast and placental vascularization (263). Furthermore, in a study using a tissue-specific PPARγ knockout mouse model generated by cross-breeding of mouse mammary tumor virus-cAMP response element and conditional PPARγ-null mice, it was shown that deletion of ovarian PPARγ in female mice led to impaired implantation (264). However, the cause of uterine dysfunction is not due to the absence of uterine PPARγ, because PPARγ was not deleted in the uterus by this cross-breeding. Further study is required to elucidate the underlying mechanism of impaired uterine function in the absence of ovarian PPARγ.
3. Transcriptional cofactors
Many transcription factors including nuclear receptors modulate transcription by direct binding to sequence-specific DNA response elements in promoters of target genes, resulting in activation or repression of transcription in a promoter-specific manner (265, 266). Although biological functions and profiles of activating ligands are extremely diverse, ligand-activated nuclear receptors utilize a converging point of transcriptional cofactors to activate/repress downstream target genes (267). Thus, although many cofactors were cloned based on their interaction with a specific nuclear receptor, various studies show a wide range of sharing of cofactors among the family of nuclear receptors (268), supporting the notion of a functional convergence in vivo. Because of the promiscuous nature of transcriptional cofactors, they have been extensively studied in relation to various nuclear receptors. Recently, several gene-targeted mouse models demonstrate that many of these cofactors are involved in developmental and reproductive processes.
a. cAMP response element binding protein (CREB)-binding protein (CBP)/p300
CBP and the related p300 are called cointegrators because of their many-sided interactions with nuclear receptors, cofactors, and basal transcriptional machinery (268). Thus, as a common limiting cofactor for diverse transcriptional activators and coactivators, CBP apparently organizes multiple signals into an integrated response at promoters containing multiple cis-acting elements (268). CBP generally exhibits constant levels of expression in various cell lines and in the developing embryo (269). Furthermore, the gene dosage-dependent role of CBP and p300 is well demonstrated in experiments with knockout mice in that CBP(−/−), p300(−/−), and CBP(+/−)xp300(+/−) double heterozygous mice all die in utero during midgestation due to multiple developmental defects (269). This suggests an essential role of cointegrators in embryonic development.
b. Steroid receptor coactivator (SRC)
The SRC family consists of three members: SRC-1, transcriptional intermediary factor 1 (TIF1) (SRC-2/glucocorticoid receptor interacting protein 1), and p/CIP(SRC-3/AlB1/RAC3/ACTR). The SRC family members interact with a variety of nuclear receptors including ER, PR, and PPAR, indicating their roles in various reproductive functions (270, 271). The three SRC family members share similar properties with respect to interacting with nuclear receptors and transcriptional activity, but it is suggested that these factors exhibit diverse expression patterns and functions (268). Whereas SRC-1-deficient mice are viable and fertile, they show reduced hormone responsiveness in several target organs, including the uterus, prostate, and testis (272). In contrast, fertility of both male and female TIF1(−/−) mice is impaired (273). Female TIF1(−/−) mice exhibit increases in embryonic resorption between d 12.5 and 18.5 gestation, possibly resulting from placental hypoplasia. The placental hypoplasia in TIF1(−/−) female mice seems to be due to the maternal deficiency of TIF1 in decidual stromal cells. Lastly, SRC-3(−/−) mice also exhibit reduced fertility primarily due to decreased ovulation (274). These results suggest that three members of SRC family cofactors play diverse roles in reproductive processes.
c. PPAR-binding protein (PBP)
PBP was first identified in a yeast two-hybrid screening system as a PPARγ cofactor. This 165-kDa protein was also independently cloned by other groups as vitamin D receptor-interacting protein 205 or thyroid hormone receptor (TR)-associated protein 220, each being a part of a multiunit complex for transcriptional activation by vitamin D receptor or TR, respectively (275, 276). PBP is widely expressed in various mouse tissues and can interact with ER and PPAR (277, 278). Gene-targeting experiments showed that PBP(−/−) embryos die in utero during midgestation due to poor placental development (279, 280).
d. PPAR-interacting protein (PRIP)/nuclear reaceptor-activating protein 250
PRIP or nuclear receptor-activating protein 250 was identified as a PPARγ or PPARα cofactor independently (281, 282). PRIP interacts with members of the PPAR, retinoic acid receptor and RXR families, as well as ER and TR. This gene is widely expressed in various tissues including reproductive organs (281, 282), and PRIP(−/−) mice exhibit severe developmental defects showing placental and cardiac hypoplasia (283, 284).
e. Receptor-interacting protein 140 (RIP140)
Two independent groups cloned RIP140 as a cofactor for PPARα or TR2 (an orphan receptor) (285, 286). This factor actively competes with SRC-1 in transcriptional assays and therefore works as a true transcriptional corepressor (285). RIP140 is involved in female fertility, as demonstrated in gene-targeting experiments (287). RIP140 mutant female mice show reduced ovulation that leads to small litter size, and a follow-up study showed that uterine function in RIP140-deficient mice is normal (288).
H. Cell cycle regulation and signaling in implantation and decidualization
As stated above, the differentiation of the uterus to support embryo development and implantation is primarily directed by progesterone and estrogen (25, 43). Decidualization is first initiated at the antimesometrial site where blastocysts implant. This process, characterized by stromal cell proliferation and differentiation into specialized type of cells (decidual cells) with polyploidy, is critical to the establishment of pregnancy in many species. The mechanisms by which the cell cycle events govern decidualization are poorly understood. The cell cycle is tightly regulated at two checkpoints, the G1-S and G2-M phases. Normal operation of these phases involves a complex interplay of cyclins, cyclin-dependent kinases (cdks), and CKIs. At the beginning of decidualization, the stromal cells immediately surrounding the implanting blastocyst proliferate (50). In mice, stromal cells close to the embryo cease to proliferate, initiating the formation of the primary decidual zone (PDZ) later on d 5 of pregnancy; the PDZ is fully established by d 6. However, stromal cell proliferation outside the PDZ continues, eventually forming the secondary decidual zone (SDZ) (30). Under normal conditions, the stimulus for decidualization is the implanting blastocyst. However, a similar process (deciduoma) can be experimentally induced in the pseudopregnant or hormonally prepared rodent uterus by intraluminal infusion of various agents including oil (30). The development of decidua or deciduoma in rodents is associated with the formation of multinucleate and giant cells (289–291). In mice, the decidual cells in the antimesometrial zone are characterized by polyploidy and endoreduplication, and most cells in this zone eventually enlarge, containing nuclei with as much as 64n DNA.
The well-known regulators of mammalian cell proliferation are the three D-type cyclins (D1, D2, and D3), also known as G1 cyclins (292). The D-type cyclins accumulate during the G1 phase. Their association with cdk4 or cdk6 is important for forming holoenzymes that facilitate cell entry into the S phase. The retinoblastoma protein (Rb) and its family members, p107 and p130, are negative regulators of the D-type cyclins. Inactivation of these regulators by phosphorylation is dependent on the cyclin/cdk complex activity and allows the cell cycle to progress through the G1 phase (293). Overexpression of D-type cyclins shortens the G1 phase and allows rapid entry into S phase (294). In contrast, cyclins A and B are involved in the progression from the S through G2-M phase. Binding of cyclin A or cyclin B to cdk1 induces phosphorylation and activation of the complex that is essential to the G2-M phase transition, whereas the cyclin A/cdk2 complex participates during progression in the S phase. In general, the action of cdks is constrained by at least two families of CKI, p16 and p21. The p16 family includes p15, p16, p18, and p19, and they inhibit the catalytic partners of D-type cyclins, cdk4 and cdk6. The p21 family consists of p21, p27, and p57, and they inhibit cdks with a broader specificity. CKIs accumulate in quiescent cells, but are down-regulated with the onset of proliferation. Thus, a critical balance between the positive and negative cell cycle regulators is a key decision maker for cell division (295).
The uterus is a unique and dynamic physiological model in which cellular proliferation, differentiation, polyploidization, and apoptosis occur in a spatiotemporal manner during the reproductive cycle and pregnancy. In the human uterus, various cyclins (A, B1, D1, and E) and cyclin-dependent kinases (cdk1, cdk2, and cdk4) and CKI (p27) are regulated during the menstrual cycle or after hormone treatments (57, 58). These molecules are expressed primarily in epithelial and stromal cells during the proliferative phase, suggesting their involvement in rhythmic proliferation of these cells (296). During the secretory phase or after progesterone administration, p27 expression correlates with progesterone-induced growth suppression in endometrial glands and stromal basalis (297). In rodents, however, the uterine expression of D- and E-type cyclins and cdks is regulated by estrogen and/or progesterone in a temporal manner (298–300). Progesterone-dependent growth suppression of the endometrium is considered to be mediated by decreased cdk activity, which presumably occurs via decreased levels of cyclins and increased association of CKI (p27) with cdks (301). There is also evidence that progesterone inhibition of uterine epithelial cell proliferation is mediated by the inhibition of nuclear translocation of cyclin D1 and cdk4 in association with the activation of cyclins A- and E-dependent cdk2 activity (302). Cell cycle molecules are also involved in endoreduplication in trophoblast differentiation during placentation (303, 304).
In mice, the expression of cyclin D3 is up-regulated in decidualizing stromal cells at the implantation site and is associated with cell proliferation (252) (305, 306). Furthermore, cyclin D3 is associated with the large polyploid cells that are defined as terminally differentiated stroma. A schematic model, as shown in Fig. 2, illustrates a possible role for cyclin D3 together with other cell cycle molecules in the developmental regulation of stromal cell decidualization and polyploidy. Cyclin D3 generally associates with cdk4 and/or cdk6 for cell proliferation. The coordinate expression of cdk4 and cyclin D3 at the site of the embryo after the onset of implantation in mice on d 5 of pregnancy suggests that these regulators play roles in proliferation of stromal cells undergoing decidualization. However, the expression of p21 with concomitant down-regulation of cyclin D3 and cdk4 in the PDZ at the implantation site in the afternoon of d 5 supports the view that cell proliferation activity of cdk4/cyclin D3 ceases with the development of the PDZ. Their expression in the decidualizing stroma outside the PDZ is again consistent with their role in proliferation of the stroma at the SDZ. In contrast, down-regulation of cdk4 in the SDZ with persistent expression of p21 on d 6 of pregnancy perhaps directs differentiation of stromal cells in this zone. On this day of pregnancy, a switch from cdk4 to cdk6 with continued expression of cyclin D3 and p21 in stromal cells within the SDZ is noted in polyploid decidual cells. The presence of cyclin E, cyclin A, and cdk2 with concomitant down-regulation of cyclin B and cdk1 in these cells supports the view that these cells are following the endocycle pathway.
A proposed model of stromal cell polyploidy and decidualization. The phase-specific cell cycle regulators in the G1 phase (cyclin D3, p21, cdk4, cdk6, cyclin E, and cdk2) or in the S-G2-M phases (cyclin A, cyclin B, cdk1, and cdk2) are shown with respect to their association in mitotic cell cycle vs. endocycle. The coordinate regulation of cyclin D3 and cdk4 in decidualizing stromal cells suggests that these regulators play roles in proliferation. However, the expression of p21 with concomitant down-regulation of cyclin D3 and cdk4 supports the view that cell proliferation ceases with the development of the PDZ. Furthermore, a switch from cdk4 to cdk6 with sustained expression of cyclin D3 and p21 in cells within the SDZ is consistent with the progression through the G phase for the onset of endocycle. This is further supported by the expression of cyclin E, cyclin A, and cdk2 in the polyploid cells for successful progression in G and S phases of the endocycle pathway. The absence of cyclin B and cdk1 probably plays a role to initiate the first endocycle.
The physiological significance of stromal cell polyploidy during decidualization is still unclear. The life span of decidual cells during pregnancy is limited, and their demise makes room for the rapidly growing embryo. Because most decidual cells become polyploid during their lifetime, it is speculated that polyploidy limits the life span of decidual cells. Furthermore, one of many uterine functions is to support embryonic growth that requires increased protein synthesis by decidual cells. The developing polyploidy may ensure increased synthetic capacity by increasing the number of gene copies for transcription. In conclusion, a tight coordination of the cell cycle molecules appears to be critical for uterine cell proliferation and differentiation during implantation and decidualization.
I. Matrix remodeling and angiogenesis during implantation and decidualization
Tissue remodeling and angiogenesis are two hallmark events during implantation and decidualization. The changing endocrine state of the female during the reproductive cycle and pregnancy results in extensive remodeling in the uterine tissue (139, 307). For example, various basement membrane components, such as type IV collagen, laminin, fibronectin, and proteoglycans, in the human uterus undergo changes throughout the menstrual cycle and pregnancy (307). Likewise, the ECM components undergo remodeling during mouse uterine stromal cell decidualization (139). Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs) are thought to be key mediators for matrix degradation during implantation and decidualization (reviewed in Refs. 307–309). There is evidence that a balance between a select set of MMPs and TIMPs is important for implantation. Mechanisms regulating the MMP and TIMP genes during the periimplantation period are not clear, although growth factors and cytokines including the EGF and TGFβ family members and LIF have been shown to modulate MMPs and TIMPs (reviewed in Ref. 309).
Under physiological conditions, angiogenesis, the process by which new blood vessels develop from preexisting vessels, primarily occurs in the uterus and ovary of the adult during the reproductive cycle and pregnancy (310). Indeed, increased vascular permeability and angiogenesis are crucial to successful implantation, decidualization, and placentation (Fig. 3). A number of studies provided indirect and descriptive evidence for the potential roles of estrogen and progesterone in these processes in various species (reviewed in Refs. 310–312). These studies primarily examined the changes in the whole uterus of the expression of a number of gene products known to regulate vascular permeability and angiogenesis, including VEGF and its receptors, without investigating its angiogenic status. Thus, in vivo roles for estrogen and progesterone in uterine angiogenesis are not fully appreciated. VEGF, originally discovered as a vascular permeability factor (reviewed in Ref. 313), is also a potent mitogen for endothelial cells and a key regulatory growth factor for vasculogenesis and angiogenesis (314). Targeted disruption of even one allele of the Vegf gene results in embryonic death in utero during midgestation with aberrant blood vessel formation (315, 316). Differential splicing of the Vegf gene generates several VEGF isoforms in both humans and mice; VEGF121 and VEGF165 are the predominant isoforms in humans, whereas VEGF120 and VEGF164 are the most abundant isoforms in mice (311, 317).
A scheme showing angiogenic signaling in the uterus during implantation. Increased vascular permeability and angiogenesis at the site of the blastocyst are two hallmarks of the implantation process. The proangiogenic factor VEGF and its receptor Flt1 (VEGFR1) and Flk1 (VEGFR2) are primarily important for uterine vascular permeability and angiogenesis before and during the attachment phase of the implantation process, whereas VEGF in complementation with the angiopoietins (Ang1 and Ang2) and their receptor Tie-2 directs angiogenesis during decidualization. Furthermore, HIFs and COX-2-derived prostaglandins PGs are important for uterine angiogenesis during implantation and decidualization and primarily target the VEGF, but not the angiopoietin, system. Ang1 in collaboration with VEGF induces vessel maturation and maintains vessel leakiness, whereas Ang2 induces vessel destabilization required for further sprouting in the presence of VEGF. Flk1lacZ mice were used to study angiogenesis during implantation. LacZ-stained (blue) blood vessels are shown at the implantation site on d 8 of pregnancy. EC, Endothelial cells; NO, nitric oxide; bFGF, basic FGF; IFN-α, interferon α.
VEGF effects are primarily mediated by two tyrosine kinase receptors: VEGFR1 [fms-like tyrosine kinase 1(FLT1)] and VEGFR2 [fetal liver kinase 1 (FLK1)/kinase insert domain-containing receptor (KDR)] (318–321). Although FLT1 activation does not stimulate endothelial cell mitosis, targeted disruption of the Flt1 gene produces impaired endothelial cell assembly into blood vessels and embryonic lethality (322). FLK1 is the major transducer of VEGF signals that induce chemotaxis, actin reorganization, and proliferation of endothelial cells (314, 323, 324). Targeted deletion of the Flk1 gene in mice produces defects in hematopoietic and endothelial cell development leading to embryonic death by d 9.5 (325). Recently, another multifunctional VEGF receptor has been identified as Neuropilin-1 (NRP1). NRP1 was originally described as a neuronal transmembrane receptor that participates in axonal guidance in the developing nervous system (326, 327) and is a receptor for the collapsin/semaphorin family of proteins (328, 329). It is now known that NRP1 functions as a receptor for at least five different ligands, collapsin-1/semaphorin-IIII/D, semaphorin-E, semaphorin-IV, VEGF165, and placental growth factor (PlGF), which are involved in different biological processes, such as nervous system development, vasculogenesis, and angiogenesis (329, 330). NRP1 is expressed in human endothelial cells as a VEGF165-specific receptor. When coexpressed in endothelial cells with FLK1, NRP1 enhances the binding of VEGF165 to FLK1 and VEGF165-mediated chemotaxis severalfold higher than that of FLK1 alone (331). Conversely, inhibition of VEGF165 binding to NRP1 inhibits its binding to FLK1 and its mitogenic activity in endothelial cells. NRP1-deficient mice show peripheral nervous system abnormalities and die in midgestation due to yolk sac vascular insufficiency and developmental anomalies of the cardiovascular system (332). Mice overexpressing NRP1 also show cardiovascular abnormalities including increased number of blood vessels and abnormal hearts (333). Several structurally similar VEGF family members including PlGF, VEGF-B, VEGF-C, and other VEGF-related proteins have recently been identified, and they are distinct gene products. VEGF-C and PlGF can bind to FLK1 and FLT1, respectively. VEGF-C can also interact with a third structurally related tyrosine kinase receptor, FLT4 (reviewed in Ref. 312). Definitive physiological significance of these newly identified VEGF-related growth factors in uterine angiogenesis during implantation warrants further investigation.
We have recently shown that the genes encoding murine VEGF isoforms and their receptors, FLT1, FLK1, and NRP1, are differentially expressed in the mouse uterus in a spatiotemporal manner during implantation and that the predominant VEGF164 isoform interacts with FLK1 and NRP1 (311, 312). These results suggest that the VEGF system is important for uterine vascular permeability and angiogenesis during implantation. Others have also shown the expression of VEGF and its receptors in the uterus as a whole during pregnancy and in response to steroid hormones (313). For example, estrogen rapidly induces uterine vascular permeability and Vegf expression transcriptionally via nuclear ER (313), and the Vegf gene contains EREs (334). Progesterone also up-regulates uterine Vegf expression via activation of the nuclear PR but at a slower rate (334). Because estrogen rapidly stimulates uterine vascular permeability and Vegf expression, and because vascular permeability is considered a prerequisite for angiogenesis, it is widely believed that estrogen is a potent stimulator of uterine angiogenesis during normal reproductive processes in vivo. However, recent evidence using molecular, genetic, physiological, and pharmacological approaches shows that estrogen and progesterone have different effects in vivo; estrogen promotes uterine vascular permeability but profoundly inhibits angiogenesis, whereas progesterone stimulates angiogenesis with little effect on vascular permeability. These effects of estrogen and progesterone are mediated by differential spatiotemporal expression of proangiogenic factors in the uterus (335).
VEGF effects are complemented and coordinated by another class of angiogenic factors, the angiopoietins (336). VEGF acts during the early stages of vessel development (315, 316, 325), whereas angiopoietin-1 (Ang1) acts later to promote angiogenic remodeling, including vessel maturation, stabilization, and leakiness (337–339). In contrast to agonistic functions of Ang1, Ang2 behaves as an antagonist. Thus, Ang1 and Ang2 are naturally occurring positive and negative regulators of angiogenesis, respectively. They interact with an endothelial cell-specific tyrosine kinase receptor Tie2 (340). Two additional members of the angiopoietin family have been identified recently. Ang3, which is expressed in mice, appears to function as an antagonist to Ang1 activation of Tie2 in a fashion similar to Ang2 (341). In contrast, Ang4, the human counterpart of Ang3, functions as an agonist to Tie2 (341). However, definitive biological functions of Ang3 and Ang4 remain unclear. Our recent investigation shows that whereas VEGF and its receptor Flk1 are primarily important for uterine vascular permeability and angiogenesis before and during the attachment phase of the implantation process, VEGF together with the angiopoietins and their receptor Tie2 directs angiogenesis during decidualization after implantation (342). A recent report has shown that Ang2 is required for postnatal angiogenic remodeling, and Ang2 in collaboration with VEGF participates in the development of lymphatic vasculature (343).
PGs, because of their roles in angiogenesis, cell proliferation, and differentiation in other systems, are also likely to participate in uterine vascular permeability and angiogenesis during implantation and decidualization. Indeed, there is now genetic and molecular evidence that COX-2-derived PGs participate in uterine angiogenesis during implantation and decidualization (342). Thus, one cause of failure of implantation and decidualization in Cox-2(−/−) mice is the deregulated vascular events in the absence of COX-2. The attenuation of uterine angiogenesis in these mice is primarily due to defective VEGF signaling rather than the angiopoietin system. Collectively, the results provide evidence that whereas ovarian steroid hormones influence uterine vascular permeability and angiogenesis during the preimplantation period, COX-2-derived PGs direct these events during implantation and decidualization by differentially regulating VEGF and angiopoietin signaling (335, 342).
Oxygen homeostasis is essential for cell survival and is primarily mediated by hypoxia-inducible factors (HIFs). These factors are intimately associated with vascular events and induce Vegf expression by binding to the hypoxia response element in the Vegf promoter. HIFα isoforms function by forming heterodimers with the aryl hydrocarbon nuclear translocator (ARNT) (HIF-β) family members. There is very limited information on the relationship among HIFs, ARNTs, and VEGF in the uterus during early pregnancy, although roles of HIFs in regulating VEGF and angiogenesis in cancers and vasculogenesis during embryogenesis are well documented (344). Using molecular and physiological approaches, we have recently shown that uterine expression of HIFs and ARNTs does not correlate with Vegf expression during the preimplantation period (d 1–4) in mice. In contrast, their expression follows the localization of uterine Vegf expression with increasing angiogenesis during the postimplantation period (d 5–8). This disparate pattern of uterine HIFs, ARNTs, and Vegf expression on d 1–4 of pregnancy suggests that HIFs have multiple roles in addition to the regulation of angiogenesis during the periimplantation period. Using pharmacological, molecular, and genetic approaches, we also observed a novel finding that whereas progesterone primarily up-regulates uterine HIF-1α expression, estrogen transiently stimulates that of HIF-2α (345). The definitive role of hypoxia in uterine angiogenesis warrants further investigation.
VII. Emerging Concepts
A. Endocannabinoid signaling in implantation
Psychoactive cannabinoids are active components of marijuana that work via activation of the G protein-coupled cell surface receptors, CB1 and CB2 (346, 347). The discovery of cannabinoid receptors led to the identification of endogenous cannabinoid ligands, arachidonoylethanolamide (anandamide) and 2-arachidonoylglycerol (2-AG) (348, 349). The mouse uterus synthesizes anandamide, and the levels fluctuate in the uterus during early pregnancy coincident with the window of uterine receptivity for implantation (350). Thus, anandamide levels were found to be lower in the receptive uterus and at the implantation sites, but were higher in the nonreceptive uterus and at interimplantation sites (350). The progesterone-treated delayed implanting uterus also showed elevated levels of anandamide, but the levels were down-regulated with the termination of the delayed implantation by estrogen (60). Experiments using Lif(−/−) pregnant mice also supported this finding, because these mice with implantation failure had higher uterine levels of anandamide than those of wild-type mice (60). Therefore, a correlation between the levels of anandamide and phases of uterine receptivity suggests that endocannabinoid ligand-receptor signaling is an important aspect of defining the window of uterine receptivity for implantation (350). There is experimental evidence that ligand-receptor signaling with cannabinoids directs preimplantation embryo development and implantation. This is consistent with the observation that mouse blastocysts express CB1 (351). CB1 is expressed from the two-cell stage at the time of zygotic gene expression through the blastocyst stage (351). Embryonic CB1 is functional, because two-cell embryos cultured in the presence of natural, synthetic, and endocannabinoids fail to develop to the blastocyst stage, and this failure occurs between the eight-cell and blastocyst stages. This effect is reversed by a CB1-selective antagonist (352). Furthermore, the endocannabinoid anandamide at a low concentration stimulates blastocyst differentiation and trophoblast outgrowth, whereas at higher concentrations it inhibits these events via differential regulation of MAPK and Ca2+ signaling (353, 354). These results suggest that a narrow range of cannabinoid concentrations regulate the embryonic developmental program.
We speculate that a tightly regulated level of uterine anandamide and embryonic CB1 during early pregnancy is important for preimplantation embryonic development and implantation. This speculation is consistent with our recent observations of asynchronous preimplantation embryo development in CB mutant mice, and inhibition of implantation in wild-type mice, but not in CB1(−/−) × CB2(−/−) double-mutant mice, with experimentally induced sustained levels of exogenously administered cannabinoids (60). Collectively, the expression of cannabinoid receptors in the preimplantation mouse embryo, synthesis of anandamide in the uterus, and the dose- and stage-specific effects of anandamide on embryo development and implantation suggest that ligand-receptor signaling with endocannabinoids or cannabinoid agonists is important for these events. The observation that heightened levels of cannabinoids inhibit implantation in mice subsequently led to the discovery that elevated levels of anandamide induce spontaneous pregnancy losses in women (355, 356). Thus, regulated cannabinoid signaling perhaps functions as a physiological surveillance system that assures implantation of healthy, but not abnormal, embryos resulting from aberrant expression of CB1 or their exposure to aberrant levels of endogenous or exogenous cannabinoids (Fig. 4). The present observation identifies unique biological functions of cannabinoid signaling for a very fundamental process that determines the survival or demise of a developing embryo.
A schematic diagram showing potential endocannabinoid signaling in blastocyst activation and implantation. Anandamide and 2-AG are the major endocannabinoids that interact with G protein-coupled cannabinoid receptors, CB1 and CB2. Regulated levels of endocannabinoids in the receptive uterus and CB1 in activated blastocysts at the time of implantation are beneficial for implantation, whereas higher levels are detrimental to this process. Because COX-2 is expressed in the uterus at the site of implantation and because anandamide and 2-AG can serve as substrates for either COX-2 or FAAH, the proposal suggests that uterine endocannabinoids are tightly regulated by the coordinated activity of FAAH and COX-2 in the uterus during early pregnancy. Evidence suggests that regulated uterine levels of endocannabinoids and blastocyst CB1 play a physiological role in synchronizing blastocyst competency with uterine receptivity for implantation. Tr, Trophectoderm; ANA, anandamide; CB1, brain-type cannabinoid receptor; FAAH, fatty acid amide hydrolase; IS, implantation site; Inter-IS, interimplantation site.
B. Developmental genes in implantation
The intricate cross-talk between the blastocyst and uterus during implantation has several features of the reciprocal epithelial-mesenchymal interactions during embryogenesis and involves evolutionarily conserved signaling pathways. An emerging concept is that many of these evolutionary conserved genes, including those encoding FGFs, IGFs, bone morphogenetic proteins (BMPs), Wnts, Noggin, Indian hedgehog (IHH) proteins, and their receptors, are potential players in the process of implantation and embryo spacing in the uterus (52) (Fig. 1). These genes are expressed in the uterus in a spatiotemporal manner during the periimplantation period in mice. For example, the attachment reaction is associated with a localized stromal induction of genes encoding BMP-2, FGF-2, and WNT-4. A simple in vitro model of implantation is not yet available to examine either the hierarchy of the events elicited in the uterus by the embryo or the function of individual signaling proteins. These questions were addressed by selectively delivering factors via blastocyst-sized gelatin beads in the uterine lumen to provoke implantation-like reactions with correct gene expression similar to what is generated by living embryos during normal implantation. We observed that beads soaked in HB-EGF or IGF-I, but not other proteins, induce many of the same discrete local responses elicited by the blastocyst, including increased localized vascular permeability, decidualization, and expression of Bmp-2 and COX-2 at the sites of the beads (4, 52). Furthermore, beads containing BMP-2 do not themselves produce an implantation-like response but alter the spacing of implantation sites induced by blastocysts cotransferred with the beads (52).
Genes encoding the components of the hedgehog-signaling pathway, namely IHH, the multipass transmembrane HH-binding protein/receptor, PATCHED (PTC), and the transcription factors, GLI 1, -2, and -3 (357–359) are expressed in a dynamic temporal and spatial pattern during the preparation of the uterus for implantation (360). The expression of Ihh increases in the luminal epithelium and glands from d 3, reaching high levels on d 4. During the same time, the expression of Ptc, Gli1, and Gli2 is up-regulated in the underlying mesenchymal stroma. Transcription of Ihh in ovariectomized mice is induced by progesterone but not by estrogen. Lower induction of Ihh, Ptc, and Hoxa-10 is also observed in response to progesterone treatment in the uteri of PR mutant mice lacking the progesterone nuclear steroid receptor. This finding suggests that this hormone regulates Ihh via both nuclear receptor-dependent and -independent pathways. Furthermore, in uterine explant cultures, recombinant N-SHH (Sonic hedgehog with N-terminal acylation) protein stimulates the proliferation of mesenchymal cells and the expression of noggin. These findings suggest that IHH generated by the epithelium functions as a paracrine growth factor for stromal cells during the early stages of pregnancy (360). Progesterone regulation of Ihh expression in the mouse uterus has recently been confirmed by another group of investigators (361). Research involving the genes known to be involved in developmental processes in the field of implantation biology is in its infancy and needs to be pursued more vigorously.
C. Discovery of novel implantation-related genes
The identification and characterization of molecules involved in embryo implantation have relied historically on candidate gene approaches with a requirement for knowledge of gene structure or sequence. Exceptions to this include the use of subtractive hybridization, representational difference analysis, and similar techniques (362–366). These approaches have identified a number of implantation-related genes in uteri of rodents and primates including humans, but these studies did not provide information on the expression profile of large gene families. Another approach is to modify unknown genes by insertion of transgenic markers or by chemical inactivation. These large-scale mutagenesis screens offer the ability to randomly inactivate genes throughout the genome and detect sought-after phenotypic changes, such as infertility. Although this approach does not address the expression of multiple genes, it does lead to recognition of important molecules without prior information on gene identity or structure. Unfortunately, there are no mutagenic studies that have identified genes implicated in implantation. Differential display (DDRT-PCR) and serial analysis of gene expression (SAGE) are RT-PCR-based methods that do not require advance knowledge of genetic sequence information. Although there are no published reports that used the SAGE technique to identify implantation-related genes, expression studies using DDRT-PCR have identified a small number of interesting genes that are differentially expressed in the uterus during the periimplantation period (252, 362, 367–372). The DDRT-PCR technique identified a large number of differentially expressed RT-PCR products that are candidates for further evaluation. To date, none of the study designs using DDRT-PCR to examine the implantation process overlap, so the comparison of their results shows little similarities.
The advent of cDNA microarray technology and sequencing of the mouse and human genomes have led to approaches for global analysis of implantation-related genes. Gene array and SAGE results are comparable (373, 374). Neither method requires prior knowledge of gene sequence, and both provide a read-out on the expression profile of a significant segment of the genome at any given time point or under any experimental condition. A series of uterine gene array studies have produced a large number of implantation-specific candidate genes. In animal models, gene arrays have been used to identify genes that show differential expression at implantation vs. interimplantation sites (375), implantation vs. postimplantation periods (376), or genes that are differentially regulated by changes in estrogen and progesterone signaling (253, 361, 377–381). We compared the results of implantation vs. interimplantation arrays to the results of progesterone-treated (delayed implantation) uteri vs. estrogen-stimulated (termination of delayed implantation) uteri to find genes that may be important for implantation. The combined strategy identified a number of genes with previously recognized roles in implantation, confirming the validity of this approach. Among genes that had decreased expression at the implantation site and during progesterone-induced delay, we found a marked shift in the expression of immune-related genes, suggesting active modification of the host immune response to the implanting blastocyst at an early stage. A recent microarray analysis examining the uterine response to progesterone identified a similar subset of Ig-family genes (253). This study identified 1675 progesterone-induced genes that were grouped into 18 unique patterns of progesterone responsiveness. About one half (750 genes) of these genes were also detected by our combined array analyses. Other microarray studies have used PR-deficient mice (361) and RU-486-treated mice (377) to identify novel progesterone-mediated pathways during implantation. After comparing and analyzing the results available for each of these disparate approaches, we selected a subset of genes that are commonly expressed in the mouse uterus under varying periimplantation-like conditions (Table 1). Closer examination of these genes and comparison of results from similar microarray experiments may provide clues to the identity of important genes or gene families in the implantation process.
Identification of implantation-related genes in the mouse
| Progesterone-responsive | Estrogen-responsive | |
|---|---|---|
| Up-regulated | Down-regulated | |
| Alkaline phosphatase | Arachidonate 15-lipoxygenase | BM-90/fibulin |
| Amphiregulin | ATFx | EIG 180 (ethanol-induced gene) |
| Apg-2 (chaperone) | Cytosolic adenylate kinase | Glutathione S-transferase, θ-2 |
| Carbonic anhydrase II | GADD45 protein | Hereditary hemochromatosis-like protein |
| Cathepsin F | Glutamyl-tRNA synthetase | Hoxd4 |
| CCAAT/enhancer binding protein β | Guanine nucleotide regulatory protein | HS1-binding protein 3 |
| Chondroitin sulfate proteoglycan 2 | Heat shock protein, 105 kDa | Intracisternal A particles |
| Claudin-7 | Hexokinase II, exon 1 | Leptin receptor |
| Complement C1q B chain | IL-1 receptor, type 11 | Norrie disease homolog |
| Cyclin-dependent kinase inhibitor 1C | Mitochondrial stress-70 protein | P glycoprotein 3 |
| Dickkopf-3 | NAD-dependent methylenetetrahydrofolate dimethyl cyclohydrolase | Ras-related protein (DEXRAS1) |
| Follistatin | Thioether S-methyltransferase | |
| Glutathione-S-transferase | NM23 metastatic-associated protein | Xeroderma pigmentosum, complementation group C |
| Histidine decarboxylase | Nuclear autoantigenic sperm protein | |
| Hoxa 11 | p45 MAPK kinase | |
| IGF binding protein-3 | Procollagen, type VI, α 2 | |
| IL-13 receptor, 2 | Protein kinase inhibitor p58 | |
| Keratin complex 1 | RAB geranylgeranyl transferase | |
| Lactotransferrin | RAMP3 | |
| Leukocyte 12/15 lipoxygenase | Ran GTPase | |
| LRG-21 | RAN GTPase-activating protein 1 | |
| Membrane metalloendopeptidase | RNAse L inhibitor (Mu-RLI) | |
| Metallothionein 1 | Small proline-rich protein 2F | |
| Norrie disease homolog | Splicing factor, arginine/serine-rich 10 | |
| Osteoblast-specific factor 2 | Squalene epoxidase | |
| Peptidylarginine deiminase | Squalene synthase | |
| Procollagen type V 2 | Type VI collagen, α 3 | |
| Procollagen type XV | ||
| Ras-like GTP-binding protein Rem | ||
| Small proline-rich protein 2F | ||
| Snail homolog | ||
| Spermidine synthase | ||
| Squalene synthase | ||
| Tissue factor pathway inhibitor |
| Progesterone-responsive | Estrogen-responsive | |
|---|---|---|
| Up-regulated | Down-regulated | |
| Alkaline phosphatase | Arachidonate 15-lipoxygenase | BM-90/fibulin |
| Amphiregulin | ATFx | EIG 180 (ethanol-induced gene) |
| Apg-2 (chaperone) | Cytosolic adenylate kinase | Glutathione S-transferase, θ-2 |
| Carbonic anhydrase II | GADD45 protein | Hereditary hemochromatosis-like protein |
| Cathepsin F | Glutamyl-tRNA synthetase | Hoxd4 |
| CCAAT/enhancer binding protein β | Guanine nucleotide regulatory protein | HS1-binding protein 3 |
| Chondroitin sulfate proteoglycan 2 | Heat shock protein, 105 kDa | Intracisternal A particles |
| Claudin-7 | Hexokinase II, exon 1 | Leptin receptor |
| Complement C1q B chain | IL-1 receptor, type 11 | Norrie disease homolog |
| Cyclin-dependent kinase inhibitor 1C | Mitochondrial stress-70 protein | P glycoprotein 3 |
| Dickkopf-3 | NAD-dependent methylenetetrahydrofolate dimethyl cyclohydrolase | Ras-related protein (DEXRAS1) |
| Follistatin | Thioether S-methyltransferase | |
| Glutathione-S-transferase | NM23 metastatic-associated protein | Xeroderma pigmentosum, complementation group C |
| Histidine decarboxylase | Nuclear autoantigenic sperm protein | |
| Hoxa 11 | p45 MAPK kinase | |
| IGF binding protein-3 | Procollagen, type VI, α 2 | |
| IL-13 receptor, 2 | Protein kinase inhibitor p58 | |
| Keratin complex 1 | RAB geranylgeranyl transferase | |
| Lactotransferrin | RAMP3 | |
| Leukocyte 12/15 lipoxygenase | Ran GTPase | |
| LRG-21 | RAN GTPase-activating protein 1 | |
| Membrane metalloendopeptidase | RNAse L inhibitor (Mu-RLI) | |
| Metallothionein 1 | Small proline-rich protein 2F | |
| Norrie disease homolog | Splicing factor, arginine/serine-rich 10 | |
| Osteoblast-specific factor 2 | Squalene epoxidase | |
| Peptidylarginine deiminase | Squalene synthase | |
| Procollagen type V 2 | Type VI collagen, α 3 | |
| Procollagen type XV | ||
| Ras-like GTP-binding protein Rem | ||
| Small proline-rich protein 2F | ||
| Snail homolog | ||
| Spermidine synthase | ||
| Squalene synthase | ||
| Tissue factor pathway inhibitor |
Identification of implantation-related genes in the mouse
| Progesterone-responsive | Estrogen-responsive | |
|---|---|---|
| Up-regulated | Down-regulated | |
| Alkaline phosphatase | Arachidonate 15-lipoxygenase | BM-90/fibulin |
| Amphiregulin | ATFx | EIG 180 (ethanol-induced gene) |
| Apg-2 (chaperone) | Cytosolic adenylate kinase | Glutathione S-transferase, θ-2 |
| Carbonic anhydrase II | GADD45 protein | Hereditary hemochromatosis-like protein |
| Cathepsin F | Glutamyl-tRNA synthetase | Hoxd4 |
| CCAAT/enhancer binding protein β | Guanine nucleotide regulatory protein | HS1-binding protein 3 |
| Chondroitin sulfate proteoglycan 2 | Heat shock protein, 105 kDa | Intracisternal A particles |
| Claudin-7 | Hexokinase II, exon 1 | Leptin receptor |
| Complement C1q B chain | IL-1 receptor, type 11 | Norrie disease homolog |
| Cyclin-dependent kinase inhibitor 1C | Mitochondrial stress-70 protein | P glycoprotein 3 |
| Dickkopf-3 | NAD-dependent methylenetetrahydrofolate dimethyl cyclohydrolase | Ras-related protein (DEXRAS1) |
| Follistatin | Thioether S-methyltransferase | |
| Glutathione-S-transferase | NM23 metastatic-associated protein | Xeroderma pigmentosum, complementation group C |
| Histidine decarboxylase | Nuclear autoantigenic sperm protein | |
| Hoxa 11 | p45 MAPK kinase | |
| IGF binding protein-3 | Procollagen, type VI, α 2 | |
| IL-13 receptor, 2 | Protein kinase inhibitor p58 | |
| Keratin complex 1 | RAB geranylgeranyl transferase | |
| Lactotransferrin | RAMP3 | |
| Leukocyte 12/15 lipoxygenase | Ran GTPase | |
| LRG-21 | RAN GTPase-activating protein 1 | |
| Membrane metalloendopeptidase | RNAse L inhibitor (Mu-RLI) | |
| Metallothionein 1 | Small proline-rich protein 2F | |
| Norrie disease homolog | Splicing factor, arginine/serine-rich 10 | |
| Osteoblast-specific factor 2 | Squalene epoxidase | |
| Peptidylarginine deiminase | Squalene synthase | |
| Procollagen type V 2 | Type VI collagen, α 3 | |
| Procollagen type XV | ||
| Ras-like GTP-binding protein Rem | ||
| Small proline-rich protein 2F | ||
| Snail homolog | ||
| Spermidine synthase | ||
| Squalene synthase | ||
| Tissue factor pathway inhibitor |
| Progesterone-responsive | Estrogen-responsive | |
|---|---|---|
| Up-regulated | Down-regulated | |
| Alkaline phosphatase | Arachidonate 15-lipoxygenase | BM-90/fibulin |
| Amphiregulin | ATFx | EIG 180 (ethanol-induced gene) |
| Apg-2 (chaperone) | Cytosolic adenylate kinase | Glutathione S-transferase, θ-2 |
| Carbonic anhydrase II | GADD45 protein | Hereditary hemochromatosis-like protein |
| Cathepsin F | Glutamyl-tRNA synthetase | Hoxd4 |
| CCAAT/enhancer binding protein β | Guanine nucleotide regulatory protein | HS1-binding protein 3 |
| Chondroitin sulfate proteoglycan 2 | Heat shock protein, 105 kDa | Intracisternal A particles |
| Claudin-7 | Hexokinase II, exon 1 | Leptin receptor |
| Complement C1q B chain | IL-1 receptor, type 11 | Norrie disease homolog |
| Cyclin-dependent kinase inhibitor 1C | Mitochondrial stress-70 protein | P glycoprotein 3 |
| Dickkopf-3 | NAD-dependent methylenetetrahydrofolate dimethyl cyclohydrolase | Ras-related protein (DEXRAS1) |
| Follistatin | Thioether S-methyltransferase | |
| Glutathione-S-transferase | NM23 metastatic-associated protein | Xeroderma pigmentosum, complementation group C |
| Histidine decarboxylase | Nuclear autoantigenic sperm protein | |
| Hoxa 11 | p45 MAPK kinase | |
| IGF binding protein-3 | Procollagen, type VI, α 2 | |
| IL-13 receptor, 2 | Protein kinase inhibitor p58 | |
| Keratin complex 1 | RAB geranylgeranyl transferase | |
| Lactotransferrin | RAMP3 | |
| Leukocyte 12/15 lipoxygenase | Ran GTPase | |
| LRG-21 | RAN GTPase-activating protein 1 | |
| Membrane metalloendopeptidase | RNAse L inhibitor (Mu-RLI) | |
| Metallothionein 1 | Small proline-rich protein 2F | |
| Norrie disease homolog | Splicing factor, arginine/serine-rich 10 | |
| Osteoblast-specific factor 2 | Squalene epoxidase | |
| Peptidylarginine deiminase | Squalene synthase | |
| Procollagen type V 2 | Type VI collagen, α 3 | |
| Procollagen type XV | ||
| Ras-like GTP-binding protein Rem | ||
| Small proline-rich protein 2F | ||
| Snail homolog | ||
| Spermidine synthase | ||
| Squalene synthase | ||
| Tissue factor pathway inhibitor |
In humans, microarray technology has been used to examine uterine cell lines and explant cultures under conditions that simulate the periimplantation period (371, 382, 383). A more physiological application is based on estimates of the putative window of receptivity in women. Endometrial biopsies taken before and during this period have identified significant changes in a modest number of genes that may serve as markers for uterine receptivity in humans. Although some variation exists in their methods, two independent studies used the same microarray platforms for their analysis (384, 385). A comparison of the results shows differential regulation of a small number of genes around the time of uterine receptivity for implantation (Table 2). Interestingly, there are a number of these genes that are also differentially expressed in mouse models of implantation and uterine receptivity.
Identification of implantation-related genes in women
| Up-regulated genes | Down-regulated genes |
|---|---|
| Annexin II | AF1q |
| ApoD | Autotaxin |
| Dickkopf-1 (hdkk-1) | CAP2 adenylyl cyclase-associated protein |
| H2B/g | Centromere protein-A (CENP-A) |
| hCPE-R | Chromosome 1-specific transcript |
| ID4 DNA-binding protein | Cyclin B |
| IL-15 | Dipeptidyl aminopeptidase-like protein |
| IL-15 precursor | Endothelin 3 (EDN3) |
| Igλ gene locus | Fibroblast activation protein |
| Mammaglobin | Frizzled-related protein frpHE |
| Metallothionein-IG (MTIG) | GLI protein |
| NKG5 NK and T cell-specific gene | hFEN1 |
| Osteopontin | Matrilysin |
| Pyruvate carboxylase | Midline 1 fetal kidney isoform 3 (MID1) |
| TGFβ superfamily protein | MSX-2 |
| Neuronal olfactomedin-related ER localized protein | |
| RA-binding protein II (CRABP-II) | |
| Serine/threonine kinase (STK-1) | |
| TRAIL receptor 2 |
| Up-regulated genes | Down-regulated genes |
|---|---|
| Annexin II | AF1q |
| ApoD | Autotaxin |
| Dickkopf-1 (hdkk-1) | CAP2 adenylyl cyclase-associated protein |
| H2B/g | Centromere protein-A (CENP-A) |
| hCPE-R | Chromosome 1-specific transcript |
| ID4 DNA-binding protein | Cyclin B |
| IL-15 | Dipeptidyl aminopeptidase-like protein |
| IL-15 precursor | Endothelin 3 (EDN3) |
| Igλ gene locus | Fibroblast activation protein |
| Mammaglobin | Frizzled-related protein frpHE |
| Metallothionein-IG (MTIG) | GLI protein |
| NKG5 NK and T cell-specific gene | hFEN1 |
| Osteopontin | Matrilysin |
| Pyruvate carboxylase | Midline 1 fetal kidney isoform 3 (MID1) |
| TGFβ superfamily protein | MSX-2 |
| Neuronal olfactomedin-related ER localized protein | |
| RA-binding protein II (CRABP-II) | |
| Serine/threonine kinase (STK-1) | |
| TRAIL receptor 2 |
The results of two different human uterine microarray experiments were compared to identify commonly detected genes (384, 385 ). The objective of these two studies was to identify genes that are differentially expressed between prereceptive and receptive phases during the window of uterine receptivity for human implantation. Despite similarities in their approach, only a few genes were found to have common patterns of regulation during the putative window of human implantation. ApoD, Apolipoprotein D.
Identification of implantation-related genes in women
| Up-regulated genes | Down-regulated genes |
|---|---|
| Annexin II | AF1q |
| ApoD | Autotaxin |
| Dickkopf-1 (hdkk-1) | CAP2 adenylyl cyclase-associated protein |
| H2B/g | Centromere protein-A (CENP-A) |
| hCPE-R | Chromosome 1-specific transcript |
| ID4 DNA-binding protein | Cyclin B |
| IL-15 | Dipeptidyl aminopeptidase-like protein |
| IL-15 precursor | Endothelin 3 (EDN3) |
| Igλ gene locus | Fibroblast activation protein |
| Mammaglobin | Frizzled-related protein frpHE |
| Metallothionein-IG (MTIG) | GLI protein |
| NKG5 NK and T cell-specific gene | hFEN1 |
| Osteopontin | Matrilysin |
| Pyruvate carboxylase | Midline 1 fetal kidney isoform 3 (MID1) |
| TGFβ superfamily protein | MSX-2 |
| Neuronal olfactomedin-related ER localized protein | |
| RA-binding protein II (CRABP-II) | |
| Serine/threonine kinase (STK-1) | |
| TRAIL receptor 2 |
| Up-regulated genes | Down-regulated genes |
|---|---|
| Annexin II | AF1q |
| ApoD | Autotaxin |
| Dickkopf-1 (hdkk-1) | CAP2 adenylyl cyclase-associated protein |
| H2B/g | Centromere protein-A (CENP-A) |
| hCPE-R | Chromosome 1-specific transcript |
| ID4 DNA-binding protein | Cyclin B |
| IL-15 | Dipeptidyl aminopeptidase-like protein |
| IL-15 precursor | Endothelin 3 (EDN3) |
| Igλ gene locus | Fibroblast activation protein |
| Mammaglobin | Frizzled-related protein frpHE |
| Metallothionein-IG (MTIG) | GLI protein |
| NKG5 NK and T cell-specific gene | hFEN1 |
| Osteopontin | Matrilysin |
| Pyruvate carboxylase | Midline 1 fetal kidney isoform 3 (MID1) |
| TGFβ superfamily protein | MSX-2 |
| Neuronal olfactomedin-related ER localized protein | |
| RA-binding protein II (CRABP-II) | |
| Serine/threonine kinase (STK-1) | |
| TRAIL receptor 2 |
The results of two different human uterine microarray experiments were compared to identify commonly detected genes (384, 385 ). The objective of these two studies was to identify genes that are differentially expressed between prereceptive and receptive phases during the window of uterine receptivity for human implantation. Despite similarities in their approach, only a few genes were found to have common patterns of regulation during the putative window of human implantation. ApoD, Apolipoprotein D.
In addition to gene expression screening, numerous approaches have been taken to identify novel proteins that may be important for implantation. Overall uterine protein synthesis increases around the time of implantation and during artificially induced decidualization (386). The presence of unique proteins in implantation or decidual tissues was originally detected by immunological and radiolabeling experiments (387–390). Two-dimensional gel electrophoresis has improved the ability to detect implantation-related proteins (391–395). Currently, emerging techniques in proteomics have also led to applications in uterine biology (396–398) and will likely resolve the presence and function of novel implantation-specific molecules.
VIII. Perspectives and Future Directions
Infertility and rapid population growth are two pressing global reproductive health issues. Events of preimplantation embryo development and uterine preparation for implantation are two major determinants of these concerns. Basic and clinical research to better understand these events will help alleviate problems of female infertility, improve fertility regulation in women, and lead to the development of new and improved contraceptive methods. Interactions between the heterogeneous cell types of the uterus and embryo in relation to endocrine, paracrine, juxtacrine, and autocrine factors during implantation are extremely complex (4). Thus, exploring and defining the molecular road map during the critical time of implantation necessitates well-thought out experimental designs in the context of both embryonic and uterine contributions to formulate a more meaningful blueprint. The objectives are not easily achievable in the human due to experimental difficulties, ethical considerations, and current restrictions on research with human embryos. Therefore, various animal models will continue to be used for studying embryo-uterine interactions during implantation. This may have relevance to human implantation. In addition, efforts should be directed to establish reliable in vitro systems, which are not currently available, to study implantation. However, experiments using endometrial biopsy samples to identify molecules associated with human uterine receptivity (window of implantation) during the menstrual cycle with changing estrogen and progesterone levels should continue to be pursued to obtain a better insight into human implantation.
Although a wealth of knowledge on the roles of growth factors, cytokines, homeotic genes, transcription factors, and lipid mediators in embryo-uterine interactions during implantation has been generated, their hierarchical blueprint in directing uterine and embryonic function during implantation remains to be deciphered. An arduous task before us is to unravel the intricate nature of the signaling pathways in implantation. We need to understand whether these pathways function independently, in parallel, or converge to a common signaling pathway to establish the network of cross-talk between the embryo and uterus that is necessary for implantation. Gene-targeting experiments in mice have identified a large number of genes that are important for female fertility; reproductive phenotypes of a select number of gene mutations are shown in Table 3. This list is not exhaustive and is likely to expand enormously with emergence of the rapidly evolving new technologies and results. Although some molecules involved in defining the pathways regulating implantation have been identified, our understanding of the implantation process is still far from complete. For example, many of the genes that are expressed in an implantation-specific manner and appear to be important for implantation cannot be studied in depth because deletion of these genes results in embryonic lethality. Uterine- or embryo-specific conditional knockout of genes of interest is urgently needed to better understand the definitive roles of these genes in uterine biology and implantation. Our failure to identify suitable uterine cell-specific promoters has been a hindrance to the achievement of this objective. One difficulty in identifying the critical roles of signaling molecules within a gene family is the redundant or compensatory functions of the gene products within the family.
Female reproductive performances in gene-targeted mouse models
| Gene | Phenotype | Ref. |
|---|---|---|
| Phenotypes related to embryo development | ||
| α-Catenin | Retarded blastocyst development with disrupted trophectoderm leading to embryonic lethality | 401 |
| ATP-binding cassette transporter 1 | Defective steroid hormone synthesis; placental malformations and impaired embryo growth in mutant females leading to subfertility | 402 |
| Cannabinoid receptor (CB1) | Asynchronous preimplantation development in female mutants leading to subfertility | 60 |
| E-cadherin | Failure of blastocyst formation leading to early embryonic death | 150 |
| FGF-4 | Defective proliferation of inner cell mass causing embryonic demise | 403 |
| Heat shock transcription factor 1 | Normal ovulation and fertilization in female mutants, but failure of preimplantation embryo development (maternal effect) leading to female infertility | 404, 405 |
| Maternal effect gene (Mater) | Defective preimplantation embryo development in female mutants leading to infertility | 406 |
| Nucleoplasmin 2 | Defective chromatin remodeling and preimplantation embryo development (subfertility) | 407 |
| Synaptonemal complex protein 3 | Defective meiotic chromosomal segmentation resulting in aneuploidy and embryonic death (subfertility) | 408 |
| Phenotypes related to uterine biology and implantation | ||
| ADAMTS-1 | Impaired fertilization with ovarian histological changes (uterine cysts) leading to subfertility | 409 |
| α-Tocopherol | Fetal resorption due to vitamin D deficiency | 410 |
| Basigin | Defective fertilization and implantation | 411, 412 |
| Centromere protein B | Disrupted luminal and glandular uterine epithelium leading to subfertility (genetic background dependent) | 413 |
| Colony-stimulating factor-1 | Defective implantation and reduced fertility | 227, 414 |
| COX-2 | Impaired ovulation and fertilization; defective attachment reaction and decidualization associated with reduced angiogenic response | 186, 188 |
| Cytosolic phospholipase A2 | On-time implantation is deferred and gives rise to postimplantation defects leading to small litter size (maternal effect) | 54 |
| EGF-R | Implantation or postimplantation failures of (−/−) embryos (genetic background dependent) | 222 |
| ERα | Ovarian cysts with uterine hypoplasia leading to infertility | 99, 415–417 |
| Hmx3 | Failure of implantation (maternal effect) | 255 |
| HoxA 10 | Primarily defective decidualization leading to reduced fertility (maternal effect) | 248, 251, 418 |
| HoxA 11 | Lack of uterine glands with defective implantation and decidualization and infertility | 249, 250 |
| 25-Hydroxyvitamin D 1α-hydroxylase enzyme | Uterine hypoplasia and absence of corpus luteum leading to infertility | 419 |
| IGF-I | Defective ovarian and uterine functions with infertility | 420 |
| IL 11 receptor IL-11Rα | Impaired decidualization leading to infertility | 229 |
| LIF | Failure in implantation and decidualization (maternal effect) | 228 |
| PPARδ | Placental defects in the majority of (−/−) embryos leading to lethality; uterine defects causing subfertility | 190, 259 |
| PPARγ | Placental defects leading to embryonic lethality | 263 |
| PR | Unopposed estrogen action and uterine hyperplasia with failure in implantation and decidualization | 103 |
| Ubiquitin protein ligase E6AP | Ovarian and uterine hypoplasia leading to subfertility | 421 |
| Wnt7a | Abnormal oviduct and uterine development leading to infertility | 422 |
| Gene | Phenotype | Ref. |
|---|---|---|
| Phenotypes related to embryo development | ||
| α-Catenin | Retarded blastocyst development with disrupted trophectoderm leading to embryonic lethality | 401 |
| ATP-binding cassette transporter 1 | Defective steroid hormone synthesis; placental malformations and impaired embryo growth in mutant females leading to subfertility | 402 |
| Cannabinoid receptor (CB1) | Asynchronous preimplantation development in female mutants leading to subfertility | 60 |
| E-cadherin | Failure of blastocyst formation leading to early embryonic death | 150 |
| FGF-4 | Defective proliferation of inner cell mass causing embryonic demise | 403 |
| Heat shock transcription factor 1 | Normal ovulation and fertilization in female mutants, but failure of preimplantation embryo development (maternal effect) leading to female infertility | 404, 405 |
| Maternal effect gene (Mater) | Defective preimplantation embryo development in female mutants leading to infertility | 406 |
| Nucleoplasmin 2 | Defective chromatin remodeling and preimplantation embryo development (subfertility) | 407 |
| Synaptonemal complex protein 3 | Defective meiotic chromosomal segmentation resulting in aneuploidy and embryonic death (subfertility) | 408 |
| Phenotypes related to uterine biology and implantation | ||
| ADAMTS-1 | Impaired fertilization with ovarian histological changes (uterine cysts) leading to subfertility | 409 |
| α-Tocopherol | Fetal resorption due to vitamin D deficiency | 410 |
| Basigin | Defective fertilization and implantation | 411, 412 |
| Centromere protein B | Disrupted luminal and glandular uterine epithelium leading to subfertility (genetic background dependent) | 413 |
| Colony-stimulating factor-1 | Defective implantation and reduced fertility | 227, 414 |
| COX-2 | Impaired ovulation and fertilization; defective attachment reaction and decidualization associated with reduced angiogenic response | 186, 188 |
| Cytosolic phospholipase A2 | On-time implantation is deferred and gives rise to postimplantation defects leading to small litter size (maternal effect) | 54 |
| EGF-R | Implantation or postimplantation failures of (−/−) embryos (genetic background dependent) | 222 |
| ERα | Ovarian cysts with uterine hypoplasia leading to infertility | 99, 415–417 |
| Hmx3 | Failure of implantation (maternal effect) | 255 |
| HoxA 10 | Primarily defective decidualization leading to reduced fertility (maternal effect) | 248, 251, 418 |
| HoxA 11 | Lack of uterine glands with defective implantation and decidualization and infertility | 249, 250 |
| 25-Hydroxyvitamin D 1α-hydroxylase enzyme | Uterine hypoplasia and absence of corpus luteum leading to infertility | 419 |
| IGF-I | Defective ovarian and uterine functions with infertility | 420 |
| IL 11 receptor IL-11Rα | Impaired decidualization leading to infertility | 229 |
| LIF | Failure in implantation and decidualization (maternal effect) | 228 |
| PPARδ | Placental defects in the majority of (−/−) embryos leading to lethality; uterine defects causing subfertility | 190, 259 |
| PPARγ | Placental defects leading to embryonic lethality | 263 |
| PR | Unopposed estrogen action and uterine hyperplasia with failure in implantation and decidualization | 103 |
| Ubiquitin protein ligase E6AP | Ovarian and uterine hypoplasia leading to subfertility | 421 |
| Wnt7a | Abnormal oviduct and uterine development leading to infertility | 422 |
Female reproductive performances in gene-targeted mouse models
| Gene | Phenotype | Ref. |
|---|---|---|
| Phenotypes related to embryo development | ||
| α-Catenin | Retarded blastocyst development with disrupted trophectoderm leading to embryonic lethality | 401 |
| ATP-binding cassette transporter 1 | Defective steroid hormone synthesis; placental malformations and impaired embryo growth in mutant females leading to subfertility | 402 |
| Cannabinoid receptor (CB1) | Asynchronous preimplantation development in female mutants leading to subfertility | 60 |
| E-cadherin | Failure of blastocyst formation leading to early embryonic death | 150 |
| FGF-4 | Defective proliferation of inner cell mass causing embryonic demise | 403 |
| Heat shock transcription factor 1 | Normal ovulation and fertilization in female mutants, but failure of preimplantation embryo development (maternal effect) leading to female infertility | 404, 405 |
| Maternal effect gene (Mater) | Defective preimplantation embryo development in female mutants leading to infertility | 406 |
| Nucleoplasmin 2 | Defective chromatin remodeling and preimplantation embryo development (subfertility) | 407 |
| Synaptonemal complex protein 3 | Defective meiotic chromosomal segmentation resulting in aneuploidy and embryonic death (subfertility) | 408 |
| Phenotypes related to uterine biology and implantation | ||
| ADAMTS-1 | Impaired fertilization with ovarian histological changes (uterine cysts) leading to subfertility | 409 |
| α-Tocopherol | Fetal resorption due to vitamin D deficiency | 410 |
| Basigin | Defective fertilization and implantation | 411, 412 |
| Centromere protein B | Disrupted luminal and glandular uterine epithelium leading to subfertility (genetic background dependent) | 413 |
| Colony-stimulating factor-1 | Defective implantation and reduced fertility | 227, 414 |
| COX-2 | Impaired ovulation and fertilization; defective attachment reaction and decidualization associated with reduced angiogenic response | 186, 188 |
| Cytosolic phospholipase A2 | On-time implantation is deferred and gives rise to postimplantation defects leading to small litter size (maternal effect) | 54 |
| EGF-R | Implantation or postimplantation failures of (−/−) embryos (genetic background dependent) | 222 |
| ERα | Ovarian cysts with uterine hypoplasia leading to infertility | 99, 415–417 |
| Hmx3 | Failure of implantation (maternal effect) | 255 |
| HoxA 10 | Primarily defective decidualization leading to reduced fertility (maternal effect) | 248, 251, 418 |
| HoxA 11 | Lack of uterine glands with defective implantation and decidualization and infertility | 249, 250 |
| 25-Hydroxyvitamin D 1α-hydroxylase enzyme | Uterine hypoplasia and absence of corpus luteum leading to infertility | 419 |
| IGF-I | Defective ovarian and uterine functions with infertility | 420 |
| IL 11 receptor IL-11Rα | Impaired decidualization leading to infertility | 229 |
| LIF | Failure in implantation and decidualization (maternal effect) | 228 |
| PPARδ | Placental defects in the majority of (−/−) embryos leading to lethality; uterine defects causing subfertility | 190, 259 |
| PPARγ | Placental defects leading to embryonic lethality | 263 |
| PR | Unopposed estrogen action and uterine hyperplasia with failure in implantation and decidualization | 103 |
| Ubiquitin protein ligase E6AP | Ovarian and uterine hypoplasia leading to subfertility | 421 |
| Wnt7a | Abnormal oviduct and uterine development leading to infertility | 422 |
| Gene | Phenotype | Ref. |
|---|---|---|
| Phenotypes related to embryo development | ||
| α-Catenin | Retarded blastocyst development with disrupted trophectoderm leading to embryonic lethality | 401 |
| ATP-binding cassette transporter 1 | Defective steroid hormone synthesis; placental malformations and impaired embryo growth in mutant females leading to subfertility | 402 |
| Cannabinoid receptor (CB1) | Asynchronous preimplantation development in female mutants leading to subfertility | 60 |
| E-cadherin | Failure of blastocyst formation leading to early embryonic death | 150 |
| FGF-4 | Defective proliferation of inner cell mass causing embryonic demise | 403 |
| Heat shock transcription factor 1 | Normal ovulation and fertilization in female mutants, but failure of preimplantation embryo development (maternal effect) leading to female infertility | 404, 405 |
| Maternal effect gene (Mater) | Defective preimplantation embryo development in female mutants leading to infertility | 406 |
| Nucleoplasmin 2 | Defective chromatin remodeling and preimplantation embryo development (subfertility) | 407 |
| Synaptonemal complex protein 3 | Defective meiotic chromosomal segmentation resulting in aneuploidy and embryonic death (subfertility) | 408 |
| Phenotypes related to uterine biology and implantation | ||
| ADAMTS-1 | Impaired fertilization with ovarian histological changes (uterine cysts) leading to subfertility | 409 |
| α-Tocopherol | Fetal resorption due to vitamin D deficiency | 410 |
| Basigin | Defective fertilization and implantation | 411, 412 |
| Centromere protein B | Disrupted luminal and glandular uterine epithelium leading to subfertility (genetic background dependent) | 413 |
| Colony-stimulating factor-1 | Defective implantation and reduced fertility | 227, 414 |
| COX-2 | Impaired ovulation and fertilization; defective attachment reaction and decidualization associated with reduced angiogenic response | 186, 188 |
| Cytosolic phospholipase A2 | On-time implantation is deferred and gives rise to postimplantation defects leading to small litter size (maternal effect) | 54 |
| EGF-R | Implantation or postimplantation failures of (−/−) embryos (genetic background dependent) | 222 |
| ERα | Ovarian cysts with uterine hypoplasia leading to infertility | 99, 415–417 |
| Hmx3 | Failure of implantation (maternal effect) | 255 |
| HoxA 10 | Primarily defective decidualization leading to reduced fertility (maternal effect) | 248, 251, 418 |
| HoxA 11 | Lack of uterine glands with defective implantation and decidualization and infertility | 249, 250 |
| 25-Hydroxyvitamin D 1α-hydroxylase enzyme | Uterine hypoplasia and absence of corpus luteum leading to infertility | 419 |
| IGF-I | Defective ovarian and uterine functions with infertility | 420 |
| IL 11 receptor IL-11Rα | Impaired decidualization leading to infertility | 229 |
| LIF | Failure in implantation and decidualization (maternal effect) | 228 |
| PPARδ | Placental defects in the majority of (−/−) embryos leading to lethality; uterine defects causing subfertility | 190, 259 |
| PPARγ | Placental defects leading to embryonic lethality | 263 |
| PR | Unopposed estrogen action and uterine hyperplasia with failure in implantation and decidualization | 103 |
| Ubiquitin protein ligase E6AP | Ovarian and uterine hypoplasia leading to subfertility | 421 |
| Wnt7a | Abnormal oviduct and uterine development leading to infertility | 422 |
Strategies comparing global gene expression profiles between the implantation and interimplantation sites have identified novel genes in the implantation process. Thus, a genome-wide screening approach coupled with functional assays will help elucidate these complex signaling pathways. In addition, further experiments should be pursued to compare gene expression patterns between the uterine-receptive and nonreceptive phases, and between the active and dormant blastocysts under defined experimental conditions. The results obtained from these experiments may help uncover new signaling molecules and pathways not previously identified. The application of proteomics is also likely to provide information regarding interactions among various molecular pathways in specifying the molecular road map to implantation. Another area of research that deserves particular attention is the identification of embryonic signaling molecules that influence uterine functions for implantation. Although chorionic gonadotropins in primates are well known for their role in pregnancy establishment, it is not yet clear whether they also function as implantation initiators. The situation is different in large animals in which embryo-derived interferons and estrogens are known to function as important signaling molecules for pregnancy recognition (12, 399, 400). These studies have been possible due to the availability of relatively large amounts of blastocyst tissue in these species. In other species, including rodents and humans, the most limiting factor has been the availability of an adequate amount of tissue for biochemical and molecular biology experiments. With the advent of microscale proteomics and genomic approaches, it is hoped that more information on embryonic signals is likely to be forthcoming.
Finally, although the mechanics and cellular architecture of the implantation process vary, certain basic similarities do exist among species. For example, implantation occurs at the blastocyst stage, there is a defined window of uterine receptivity for implantation, a reciprocal interaction between the blastocyst and the uterus is essential for implantation, and a localized increase in uterine vascular permeability occurs at the site of the blastocyst during the attachment reaction. Thus, recognition and characterization of signaling pathways in these steps may give rise to a unifying scheme relevant to understanding the mechanism of human implantation.
Acknowledgement
We thank Ben Danzo for his critical reading and editing of the manuscript and Stan Fernald of the Cell Imaging Facility at the Center for Reproductive Sciences at the University of Kansas Medical Center for the artwork. We apologize for unintended omission of any relevant references.
Studies by the authors incorporated in this review were supported by National Institutes of Health Grants HD29968, HD12304, HD33994, DA 06668 (to S.K.De.), HD 40810 (to H.L.), HD37830 (to S.K.Da.), HD40221 (to J.R.), and HD37394 (to B.C.P.). S.K.De. is recipient of MERIT Awards from the National Institute of Child Health and Human Development and the National Institute on Drug Abuse. H.W. is Lalor Foundation postdoctoral fellow.
Abbreviations
- 2-AG,
2-Arachidonoylglycerol;
- Ang1,
angiopoietin-1;
- ARNT,
aryl hydrocarbon nuclear translocator;
- BMP,
bone morphogenetic protein;
- CBP,
cAMP response element-binding protein (CREB)-binding protein;
- cdk,
cyclin-dependent kinase;
- CKI,
cyclin-dependent kinase inhibitor;
- COX-2,
cyclooxygenase-2;
- DDRT-PCR,
differential display RT-PCR;
- ECM,
extracellular matrix;
- EGF,
epidermal growth factor;
- EGF-R,
EGF receptor;
- ER,
estrogen receptor;
- ERE,
estrogen response element;
- FGF,
fibroblast growth factor;
- FLK1,
fetal liver kinase 1;
- FLT1,
fms-like tyrosine kinase 1;
- H2,
histamine type 2 receptor;
- HB-EGF,
heparin-binding EFG-like growth factor;
- HDC,
histidine decarboxylase;
- HIF,
hypoxia-inducible factor;
- ICM,
inner cell mass;
- IHH,
Indian hedgehog;
- LIF,
leukemia inhibitory factor;
- MMP,
matix metalloproteinase;
- NRP1,
Neuropilin-1;
- 4-OH-E2,
4-hydroxy-17β-estradiol;
- PBP,
PPAR-binding protein;
- PDZ,
primary decidual zone;
- PG,
prostaglandin;
- PGI2,
prostacyclin;
- PlGF,
placental growth factor;
- PPAR,
peroxisome proliferator-activated receptor;
- PR,
progesterone receptor;
- PRIP,
PPAR-interacting protein;
- PTC,
Patched;
- RGD,
Arg-Gly-Asp sequence;
- RIP,
receptor interacting protein;
- RXR,
retinoid X receptor;
- SAGE,
serial analysis of gene expression;
- SDZ,
secondary decidual zone;
- SRC,
steroid receptor coactivator;
- TIF,
transcriptional intermediary factor;
- TIMP,
tissue inhibitor of MMPs;
- TR,
thyroid hormone receptor;
- VEGF,
vascular endothelial growth factor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}