





Abstract
Recent studies show that colonic vitamin D receptor (VDR) signaling protects the mucosal epithelial barrier and suppresses colonic inflammation, but the underlying molecular mechanism remains to be fully understood. To investigate the implication of colonic VDR downregulation seen in patients with inflammatory bowel disease, we assessed the effect of gut epithelial VDR deletion on colonic inflammatory responses in an experimental colitis model. In a 2,4,6-trinitrobenzenesulfonic acid–induced colitis model, mice carrying VDR deletion in gut epithelial cells [VDRflox/flox (VDRf/f);Villin-Cre or VDRΔIEC] or in colonic epithelial cells (VDRf/f;CDX2-Cre or VDRΔCEC) developed more severe clinical colitis than VDRf/f control mice, characterized by more robust T-helper (TH)1 and TH17 responses, with greater increases in mucosal interferon (IFN)-γ+, interleukin (IL)-17+, and IFN-γ+IL-17+ T cells. Accompanying the severe mucosal inflammation was more profound colonic epithelial cell apoptosis in the mutant mice. Treatment with caspase inhibitor Q-VD-OPh dramatically reduced colitis severity and attenuated TH1 and TH17 responses in VDRΔCEC mice. The blockade of cell apoptosis also prevented the increase in mucosal CD11b+CD103+ dendritic cells (DCs), known to be critical for TH17-cell activation. Moreover, depletion of gut commensal bacteria with antibiotics eliminated the robust TH1 and TH17 responses and CD11b+CD103+ DC induction. Taken together, these observations demonstrate that gut epithelial VDR deletion aggravates epithelial cell apoptosis, resulting in increases in mucosal barrier permeability. Consequently, invading luminal bacteria activate CD11b+CD103+ DCs, which promote mucosal TH1 and TH17 responses. Therefore, gut epithelial VDR signaling controls mucosal inflammation by suppressing epithelial cell apoptosis.
The colon is a complex system in which the mucosal epithelial barrier separates the commensal bacteria and luminal antigens from the mucosal immune system (1). The mucosal barrier consists of a monolayer of intestinal epithelial cells (IECs) and intercellular junctions that seal the paracellular space (2). Barrier dysfunction, which usually results from dysregulated tight junction and/or aberrant apoptosis of the IECs (3), can lead to increased barrier permeability and invasion of luminal antigens and bacteria into the lamina propria, triggering immune cell-mediated mucosal inflammation (4, 5). Impaired mucosal barrier function is thought to be a major pathogenic factor in the development of inflammatory bowel disease (IBD), one of the major disorders of the gastrointestinal system in humans (5).
The mucosal innate immune system constitutes an interface for monitoring the luminal environment and relaying signals to initiate adaptive immune responses. The antigen-presenting cells, including dendritic cells (DCs) and macrophages, can be activated by invading antigens and bacteria, and activated DCs play key roles in T-helper (TH)1 and TH17 responses in the mucosa (6). It has been reported that CD11c+CX3CR1+ DCs produce inflammatory mediators [e.g., interleukin (IL)-12, tumor necrosis factor (TNF)-α] and are a strong inducer of TH1 cells (7), and CD11b+CD103+ DCs drive mucosal TH17-cell differentiation and promote IL-17 responses (8, 9). Thus, excessive activation of the adaptive immunity is believed to be the more proximate driver of colitis (10). It is known that CD4+ TH1, TH2, and TH17 cells are important for the development of mucosal inflammation in the colon. TH1 cells produce interferon (IFN)-γ and IL-12, and are thought to be predominantly involved in Crohn disease. TH2 cells secret IL-4, IL-5, and IL-13, and are more involved in ulcerative colitis; and TH17 cells produce IL-17, IL-21, IL-22, IL-6, and TNF-α and are involved in both diseases (11). TH17 cells are highly plastic and can further differentiate to TH1/TH17 cells that coproduce IFN-γ and IL-17 (12).
Excessive IEC apoptosis is a major cause of increased mucosal permeability and results in focal disruption of the mucosal barrier that triggers mucosal immune response and inflammation. In addition, apoptotic cells can release inflammatory mediators such as high-mobility group protein 1 to drive DC maturation and activation, which activate adaptive immunity (13–15). It has been reported that p53-upregulated modulator of apoptosis (PUMA), a proapoptotic BCL-2 family member, is a key molecule mediating TNF-α–induced IEC apoptosis (16). In fact, increased IEC apoptosis is commonly seen in patients with IBD (17–19) and in murine models of colitis (20, 21).
Numerous reports have shown that vitamin D deficiency is associated with increased risk of IBD (22–24) and suggest vitamin D as an environmental risk factor for IBD. The biological activity of the vitamin D hormone is mediated by the vitamin D receptor (VDR), a nuclear hormone receptor (25) that is highly expressed in IECs. The abundance of VDR in the IECs suggests important roles of this nuclear receptor; until recently, however, the biological function of IEC VDR has been unclear. We reported that targeting human VDR overexpression to the IECs in transgenic mice rendered them highly resistant to colitis in several experimental colitis models (26, 27). Our studies showed that the gut epithelial VDR plays a critical role in protecting the integrity of the mucosal barrier by preventing tight junction dysfunction and by inhibiting IEC apoptosis (26, 28). The observation that reconstituting VDR-deficient IECs with the human VDR transgene in a VDR-null background was able to rescue VDR-null mice from severe colitis in the presence of a VDR-deficient immune system confirms a protective role of the epithelial VDR signaling against mucosal inflammation (26). The underlying protective mechanism of epithelial VDR signaling, however, remains to be fully elucidated.
Interestingly, we consistently observed ∼50% reduction in colonic VDR, mainly colonic epithelial VDR, in biopsy specimens from patients with IBD, including both Crohn disease and ulcerative colitis (26, 28), suggesting that gut VDR insufficiency may contribute to IBD pathogenesis. To explore the implication of this human observation, in the current study, we assessed the effect of epithelial VDR deletion on mucosal inflammation using the 2,4,6-trinitrobenzene sulfonic acid (TNBS)–induced colitis model, which is believed to mimic Crohn disease (29). Our data show that gut epithelial VDR controls microbiota-dependent TH1- and TH17-mediated mucosal inflammation by preventing excess epithelial cell apoptosis.
Materials and Methods
Animals
VDRflox/flox (VDRf/f) mice that carry LoxP sites flanking exon 4 of the Vdr gene (30) were kindly provided by David Gardner (University of California, San Francisco). Villin-Cre transgenic mice (stock no. 021504) and CDX2-Cre transgenic mice (stock no. 009350) were obtained from Jackson Laboratory. The villin promoter targets transgene expression to intestinal epithelial cells, including small and large intestine (31), whereas the CDX2 promoter directs Cre recombinase expression only in colonic epithelial cells. The CDX2-Cre mice have been used to conditionally delete Apc gene from colonic epithelial cells (32). Mice that carry VDR deletion from small and large intestinal epithelial cells (VDRf/f;Villin-Cre; designated as VDRΔIEC) or from only the colonic epithelial cells (VDRf/f;CDX2-Cre; designated as VDRΔCEC) were generated by crossing VDRf/f mice with villin-Cre transgenic mice or CDX2-Cre transgenic mice, respectively. All mice were fed Teklad 2918 Global 18% protein rodent diet (Envigo), which contains 1% calcium, 0.7% phosphorus, and 1.5 IU/g vitamin D3.
Experimental colitis
Experimental colitis was induced in VDRΔIEC, VDRΔCEC, and control VDRf/f mice using TNBS according to a protocol previously described (26, 33). Briefly, mice were presensitized with 1% TNBS solution applied to the back skin for 8 days before they were treated by TNBS installation through the colon. Mouse body weight and vitality were monitored and disease score assessed daily. Mice were euthanized at appropriate times as indicated. Colons were collected immediately thereafter and fixed in 10% formalin for histology. Mucosa was scraped to isolate total RNAs or proteins. To address the effect of IEC apoptosis, mice were treated with Q-VD-OPh, a broad-spectrum caspase inhibitor (34). Q-VD-OPh was delivered by intraperitoneal injection at 8 mg/kg at 60 minutes and 24 hours (35) after TNBS instillation. To assess the contribution of commensals to mucosal immune responses, an antibiotic-induced microbiota-depleted (AIMD) model was established as described previously (36, 37). Mice were treated with a cocktail of antibiotics in drinking water (ampicillin, 1 g/L; vancomycin, 500 mg/L; neomycin sulfate, 1 g/L; and metronidazole, 1 g/L) for 4 weeks. Control mice were treated with regular water. TNBS treatment was started at the end of the third week in these mice. All animal study protocols were approved by the Institutional Animal Care and Use Committee at the University of Chicago.
Histology
Freshly harvested colons were fixed in 10% formalin overnight and processed as described previously (26). Colonic histology was examined by hematoxylin and eosin staining. Colonic macroscopic scores and histologic scores were recorded on the basis of a previously described scoring system (38–40). To assess intestinal cell apoptosis, colon sections were subjected to terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling staining using an In Situ Cell Death Detection Kit (Roche Life Science) according to the manufacturer’s instruction.
Flow cytometry
Lamina propria cells were isolated from the colon as described previously (41). In brief, mice were euthanized, their colons were dissected, cut open longitudinally, and washed in cold phosphate-buffered saline (PBS). The colons were cut into 1.5-cm pieces and washed in PBS containing 1 mM dithiothreitol for 10 minutes at room temperature on a shaker, followed by two washes with shaking in PBS containing 30 mM EDTA and 10 mM HEPES at 37°C for 10 minutes. The tissues were then digested in RPMI 1640 medium containing DNase I (150 μg/mL; Sigma-Aldrich) and collagenase VIII (150 U/mL; Sigma-Aldrich) with 10% fetal bovine serum at 37°C in a 5% CO2 incubator for 1.5 hours. Digested cell suspensions were passed through a 70-μm cell strainer and separated by centrifugation on a discontinuous 40%/80% Percoll gradient at 2500 rpm for 20 minutes at room temperature. Cells were harvested for flow cytometry analyses.
Before cell staining, anti-CD16/32 antibody (eBioscience) was used to block nonspecific binding to Fc receptors. For intracellular staining, cells were fixed and permeabilized using a Mouse Regulatory T-Cell Staining Kit (eBioscience) according to the manufacturer’s protocol. For cytokine production, cells were stimulated with phorbol 12-myristate 13-acetate (PMA; 50 ng/mL) and ionomycin (500 ng/mL) for 4 hours. Brefeldin A (2 µg/mL) was added 2 hours before cells were harvested for analysis. Dead cells were excluded from the analysis using a Live and Dead Violet Viability Kit (Invitrogen).
Anti-mouse CD3e FITC, anti-mouse CD4 Percp-Cy5.5, anti-mouse/rat IL-17A PE, anti-mouse/rat Foxp3 FITC, anti-mouse/human RORγt PE, anti-CD103-APC, and anti-FoxP3-FITC were purchased from eBioscience. Anti-mouse IFN-γ Percp-Cy5.5, anti-mouse IL-10 PE, anti-mouse CD25 Pecy7, anti-mouse CD4 Pecy7, and anti-mouse TCR-β FITC were purchased from BD Pharmingen. Anti-CD11c-Pecy7, anti-CD11b-PerCP/Cy5.5, and anti-MHCII-FITC were obtained from BioLegend. Fluorescence-activated cell sorting (FACS) was performed in BD LSRFortessa (BD Biosciences) and data analyzed by FlowJo software, version 10 (Tree Star).
Real-time polymerase chain reaction
Mucosal total RNAs were extracted using TRIzol reagents (Life Technologies). First-strand complementary DNAs were synthesized using a ThermoScript RT kit (Life Technologies). Mucosal cytokine transcripts were quantified by real-time polymerase chain reaction (PCR) in a Roche 480 Real-Time PCR System, using SensiFAST SYBR No-Rox kits (Bioline). The amount of transcripts was calculated using the 2ΔΔCt formula, with the endogenous β-actin transcript as the internal control. PCR primer sequences were as reported previously (26, 42).
Western blot
Mucosal lysates were dissolved in Laemmli sample buffer Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electroblotted onto Immobilon-P (Thermo Fisher Scientific) membranes. The membranes were incubated with primary antibodies, and then with horseradish peroxidase-conjugated secondary antibodies. Antigens were visualized using a chemiluminescence kit (Thermo Fisher Scientific) as described (26). The primary antibodies against the following proteins were used: VDR (catalog no. sc-13133; Santa Cruz Biotechnology), PUMA (catalog no. 7467), caspase 3 (catalog no. 9662), and p53 (catalog no. 9282; Cell Signaling), and β-actin (catalog no. A5316; Sigma-Aldrich).
Serum parameters
Serum total calcium was measured using the Stanbio Calcium LiquiColor Test (Stanbio Laboratory). Serum phosphorus was measured using the Stanbio Laboratory Phosphorus Liqui-ultraviolet Test (Stanbio Laboratory). Serum parathyroid hormone (PTH) levels were measured using Immutopics Mouse PTH 1-84 enzyme-linked immunosorbent assay kit (Immutopics). Serum 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] concentrations were measured using a 1,25(OH)2 vitamin D enzyme-linked immunosorbent assay kit (ALPCO).
Statistical analysis
Data values are presented as mean ± standard deviation. Statistical comparisons were carried out using unpaired two-tailed Student t test for two-group comparisons; for three or more group comparisons, two-way analysis of variance was used with a Student-Newman-Keuls post hoc test. Animal body weight changes and survival rate were analyzed by log-rank test. P < 0.05 was considered statistically significant.
Results
VDR deletion from gut epithelial cells exacerbated colitis
Adult VDRΔIEC mice and VDRΔCEC mice were used for the study. Western blot analyses confirmed that gut mucosal VDR protein was reduced by >90% in mucosal lysates of VDRΔIEC mice compared with VDRf/f control mice (Fig. 1A), whereas VDR deletion was detected only in colonic mucosal lysates but not seen in the small intestine in VDRΔCEC mice (Fig. 2A). Immunostaining data confirmed that most mucosal epithelial cells in VDRΔIEC mice were VDR negative in the intestines relative to the VDRf/f counterparts (Supplemental Fig. 1A).
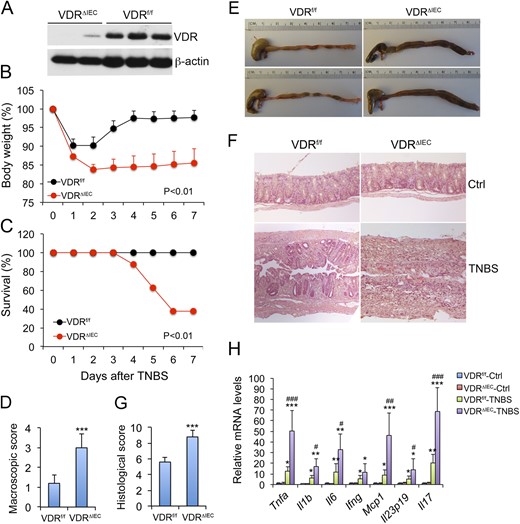
Effects of gut epithelial VDR deletion on the development of TNBS-induced colitis. (A) Western blot analysis of intestinal VDR in VDRf/f and VDRΔIEC mice; (B) Body weight changes of VDRf/f and VDRΔIEC mice over time following TNBS treatment; n = 5 to 7 mice; P < 0.01 by log rank test. (C) Survival curve of TNBS-treated VDRf/f and VDRΔIEC mice; n = 5 to 7 mice; P < 0.01 by log rank test. (D) Macroscopic disease scores on day 7 following TNBS treatment; ***P < 0.001 vs. VDRf/f; n = 5 mice. (E) Gross morphology of large intestines from TNBS-treated VDRf/f and VDRΔIEC mice on day 7. (F) Representative hematoxylin and eosin–stained distal colon sections cut longitudinally from VDRf/f and VDRΔIEC mice on day 4 after TNBS treatment. (G) Histological damage scores of colon sections on day 7 after TBNS treatment; ***P < 0.001 vs. VDRf/f; n = 5 mice. (H) Real-time PCR quantitation of mucosal proinflammatory cytokine transcripts on day 4 after TNBS treatment. *P < 0.05; **P < 0.001; ***P < 0.001 vs. corresponding control; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. VDRf/f-TNBS; n = 5 mice. Ctrl, control; Ifng, interferon γ, Il1b, interleukin 1B; Il6, interleukin 6; Il17, interleukin 17; Mcp1, monocyte chemotactic protein-1; Tnfa, tumor necrosis factor α.
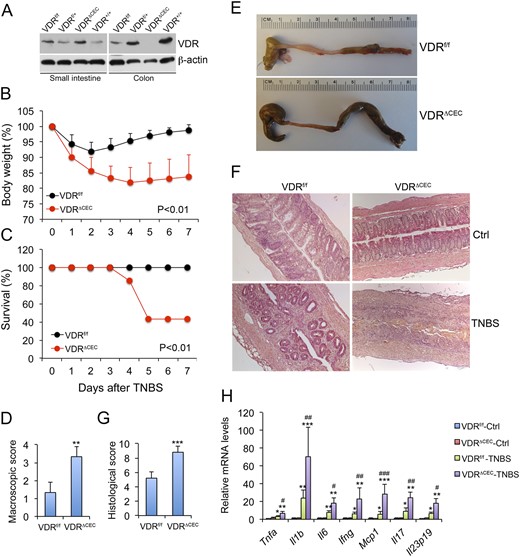
Colonic epithelial VDR deletion aggravates colonic inflammation in TNBS-induced colitis model. (A) Western blots showing VDR expression in the small intestine but not in the colon of VDRΔCEC mice. (B) Body weight changes following TNBS treatment; n = 6 to 8 mice; P < 0.01 by log rank test. (C) Survival curve; n = 6 to 8 mice; P < 0.01 by log rank test. (D) Scores of colonic macroscopic damage. (E) Gross images of the large intestine on day 7. (F) Hematoxylin and eosin staining of distal colon sections cut longitudinally on day 4. (G) Histological scores of the colons; **P < 0.01; ***P < 0.001 vs. VDRf/f; n = 4 to 5 mice. (H) Real-time RT-PCR quantitation of mucosal proinflammatory cytokines and chemokines in VDRf/f and VDRΔCEC mice with or without TNBS treatment on day 4. *P < 0.05; **P < 0.001; ***P < 0.001 vs. corresponding control; #P < 0.05; ##P < 0.01; ###P < 0.001 vs. VDRf/f-TNBS; n = 3 to 5 mice. Ctrl, control; Ifng, interferon γ; Il1b, interleukin 1B; Il6, interleukin 6; Il17, interleukin 17; Mcp1, monocyte chemotactic protein-1; Tnfa, tumor necrosis factor α.
The small intestine is critically involved in calcium and phosphorus absorption; however, consistent with a previous report (43), serum calcium and phosphorus levels in adult VDRΔIEC mice were normal (calcium: 8.98 ± 0.89 mg/dL vs 9.08 ± 0.19 mg/dL in VDRf/f mice; phosphorus: 8.36 ± 2.09 mg/dL vs 9.22 ± 1.48 mg/dL in VDRf/f mice; Supplemental Fig. 1B and 1C), which is in contrast to global VDR-null mice that are severely hypocalcemic (44). However, serum 1,25(OH)2D3 and PTH levels were significantly elevated in VDRΔIEC mice compared with the VDRf/f counterparts [1,25(OH)2D3: 279.89 ± 38.96 pmol/L vs 124.57 ± 24.78 pmol/L; PTH: 199.78 ± 47.98 pg/mL vs 96.97 ± 19.86 pg/mL; Supplemental Fig. 1D and 1E], consistent with previously reported findings (43). These data suggest these mice maintain their serum calcium levels by increasing bone resorption. VDRΔCEC mice also had normal serum calcium levels (9.23 ± 1.84 mg/dL vs 9.49 ± 1.23 mg/dL in VDRf/f mice; Supplemental Fig. 2A), but their serum 1,25(OH)2D3 levels were moderately increased, whereas serum PTH levels were slightly suppressed [1,25(OH)2D3: 154.78 ± 21.47 pmol/L vs 104.67 ± 15.76 pmol/L in VDRf/f mice; PTH: 56.89 ± 10.34 pg/mL vs 87.45 ± 7.98 pg/mL in VDRf/f mice; Supplemental Fig. 2B and 2C], consistent with previously reported results (45). Thus, data interpretation would not be confounded by the calcium and phosphorus status in VDRΔIEC or VDRΔCEC mice, but we cannot exclude the potential confounding effects resulting from changes in the hormonal environment of the genetically modified mice.
Because it was reported that VDR messenger RNA was absent in the kidney when VDR deletion was driven by CDX2-Cre (45), we measured VDR protein levels in the kidney of VDRΔIEC and VDRΔCEC mice, using VDRf/f and VDR−/− mice as positive and negative controls, respectively. Indeed, VDR was deleted from the kidney of VDRΔCEC mice, but not VDRΔIEC mice (Supplemental Fig. 2D). These results are consistent with the previous conclusion that VDRΔIEC and VDRΔCEC mice maintain normal blood calcium levels by different mechanisms (43, 45).
To assess the role of intestinal epithelial VDR in the development of colitis, we studied VDRΔIEC and VDRΔCEC mice in parallel with VDRf/f control mice, using the TNBS-induced colitis model. After intrarectal TNBS instillation, VDRf/f mice started to recover their body weight on day 3, and full recovery was usually seen by days 6 or 7. In contrast, neither VDRΔIEC nor VDRΔCEC mice were able to fully recover their weight (Fig. 1B and Fig. 2B), and 50% to 60% of these mice died within this 7-day period (Fig. 1C and Fig. 2C). Indeed, VDRΔIEC and VDRΔCEC mice developed more severe diarrhea and rectal bleeding than did VDRf/f mice. Compared with VDRf/f mice, VDRΔIEC and VDRΔCEC mice had higher macroscopic disease activity scores (Fig. 1D and Fig. 2D), and their colons remained shortened and markedly swollen with no visible feces pellet formation inside, even on day 6 (Fig. 1E and Fig. 2E). On day 4, massive crypt regeneration was seen in the colon of TNBS-treated VDRf/f mice, but new crypt formation was absent in the distal colon of VDRΔIEC and VDRΔCEC mice, and only massive ulceration was seen (Fig. 1F and Fig. 2F). Overall VDRΔIEC and VDRΔCEC mice had a much higher histologic damage score in the colon (Fig. 1G and Fig. 2G) and showed much more severe mucosal inflammation, manifested by much greater robust proinflammatory cytokine production (TNF-α, IL-1β, IL-6, IL-17, IL-23, IFN-γ, and MCP-1) after TNBS treatment (Fig. 1H and Fig. 2H). Overall, VDRΔIEC and VDRΔCEC mice developed much more severe colitis compared with their normal controls, and they basically phenocopied each other in terms of colitis development.
Gut epithelial VDR deficiency enhanced mucosal inflammatory responses
To understand why gut epithelial VDR loss causatively promoted colitis in this experimental model, we examined lamina propria leukocytes by FACS analysis. As shown in Fig. 3, TNBS treatment induced IL-17-producing CD4+ TH17 cells (Fig. 3A and 3B), IFN-γ–producing CD4+ TH1 cells (Fig. 3C and 3D), and TH1/TH17 cells that produced both IFN-γ and IL-17 (Fig. 3E and 3F) in VDRf/f mice on day 2, but the TH1, TH17, and TH1/ TH17 responses were much more robust in VDRΔIEC mice. Overall, there was a more dramatic increase in CD4+ T-helper cells that produce IFN-γ, IL-17, and both cytokines in VDRΔIEC mice (Fig. 3A–3F); however, there were no significant differences in mucosal FoxP3+ T regulatory (Treg) cells or IL-10–producing FoxP3+ Treg cells between VDRf/f and VDRΔIEC mice (Supplemental Fig. 3). With no differences in Treg cells but more dramatic increases in the TH1, TH17, and TH1/TH17 populations, the balance shifted to favor inflammation in the mucosal microenvironment of VDRΔIEC mice. These observations indicate that defects in VDR signaling in gut epithelial cells promote mucosal inflammation, which is believed to drive colitis development.
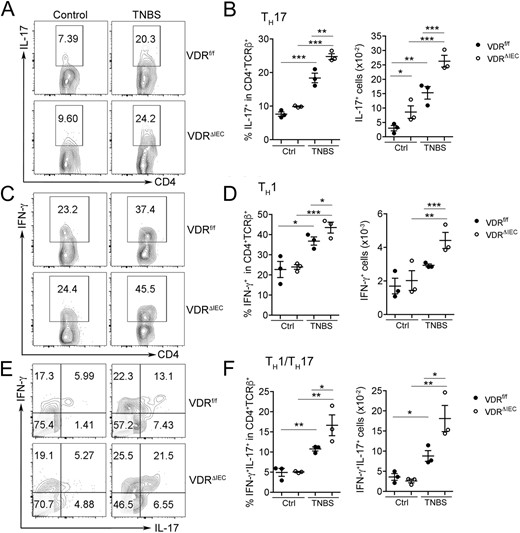
Gut epithelial VDR deletion exacerbated mucosal inflammatory responses in colitis. Lamina propria cells isolated from VDRf/f or VDRΔIEC mice on day 2 after TNBS treatment were analyzed by FACS. Control mice received ethanol only. (A,C,E) Representative FACS plots gated on CD4+TCRβ+ cells for (A) IL-17+ T cells, (C) IFN-γ+ T cells, and (E) IFN-γ+IL-17+ T cells. (B,D,F) FACS quantitation of cell percentage and cell number for (B) TH17 cells, (D) TH1 cells, and (F) TH1/TH17 cells in the lamina propria. *P < 0.05, **P < 0.01, ***P < 0.001. Ctrl, control.
Epithelial VDR deletion increased epithelial cell apoptosis
Results of our previous studies indicated VDR overexpression in gut epithelial cells inhibits epithelial cell apoptosis, because VDR downregulates PUMA, a key apoptotic mediator (26). Thus, we tested the hypothesis that epithelial VDR deficiency promotes epithelial cell apoptosis, hence increasing mucosal barrier permeability in colitis models. As shown in Fig. 4, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling staining revealed that deletion of VDR from the epithelial cells drastically increased epithelial cell apoptosis in VDRΔIEC mice relative to VDRf/f control mice after TNBS instillation (Fig. 4A). At the molecular level, TNBS-treated VDRΔIEC mice had more robust induction of mucosal PUMA as well as caspase 3 activation compared with VDRf/f control mice (Fig. 4B and 4C). Consistently, claudin-2, a tight junction protein that is highly induced during colonic inflammation (46), was also induced more in TNBS-treated VDRΔIEC mice than in their VDRf/f counterparts (Fig. 4B and 4C). Therefore, excessive IEC apoptosis due to overactivation of the PUMA-mediated proapoptotic pathway appears to account for the elevated mucosal inflammatory responses and severe colitis development seen in mice without epithelial VDR.
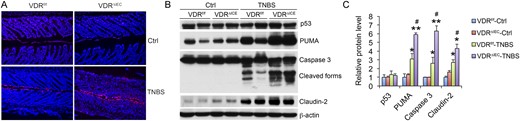
Gut epithelial VDR deletion enhances apoptosis of epithelial cells in colitis development. (A) Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling staining of distal colon sections from VDRf/f and VDRΔIEC mice on day 4 after TNBS treatment. (B) Western blot analyses and (C) densitometric quantitation of mucosal p53, PUMA, caspase 3 activation, and claudin-2 in VDRf/f and VDRΔIEC mice on day 4. *P < 0.05; **P < 0.01 vs corresponding control; #P < 0.05 vs VDRf/f-TNBS; n = 3 to 4 mice. Ctrl, control.
Blockade of apoptosis attenuated mucosal inflammation
To further test the hypothesis that excessive IEC apoptosis is the main cause of increased mucosal inflammation in mice with epithelial VDR deficiency, we treated the mice with Q-VD-OPh, a caspase inhibitor that has been used to block IEC apoptosis in mice (35). As shown in Fig. 5, treatment with Q-VD-OPh after TNBS instillation markedly attenuated colonic damage and ameliorated colitis in both VDRΔIEC and VDRΔCEC mice (Fig. 5A and 5B), as the shortened and swollen morphology of the VDRΔIEC or VDRΔCEC colons induced by TNBS diminished after Q-VD-OPh treatment. At the histologic level, Q-VD-OPh almost completely blocked the development of ulceration in the colon of VDRΔIEC mice (Fig. 5C). FACS analyses of the lamina propria cells demonstrated that Q-VD-OPh treatment substantially attenuated the TH1 and TH17 responses induced by TNBS in VDRf/f and VDRΔCEC mice. The TH1, TH17, and TH1/TH17 populations were significantly decreased in mice treated with TNBS/Q-VD-OPh compared with these cell populations in TNBS-treated groups (Fig. 5D–5I). Moreover, lamina propria CD11b+CD103+ DCs that are known to promote TH17 polarization, but not CD11b-CD103+ DCs, were induced to a much greater extent in VDRΔCEC mice compared with VDRf/f mice after TNBS instillation, and the induction of this CD11b+CD103+ DC population was also substantially blocked by Q-VD-OPh (Fig. 6A–6C). These observations suggest epithelial VDR deletion leads to CD11b+CD103+ DC activation, which promotes TH17 and TH1/TH17 differentiation.
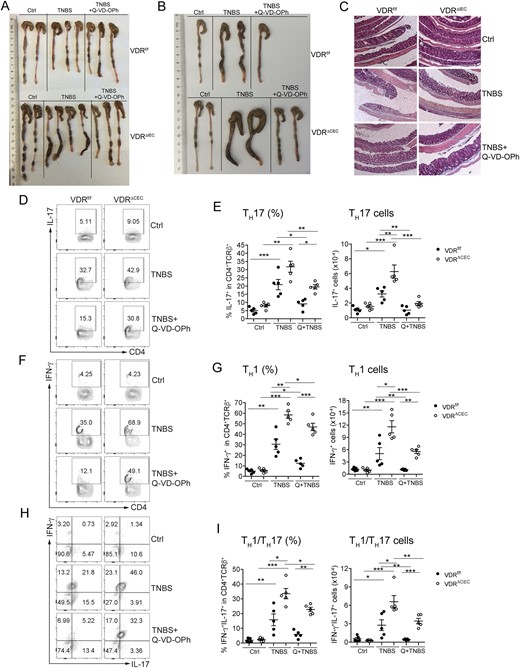
Blockade of cell apoptosis attenuates colitis and mucosal inflammatory responses. Mice were treated with vehicle, TNBS, or TNBS+Q-VD-OPh, as indicated. (A) Gross images of colons from VDRf/f and VDRΔIEC mice on day 3. (B) Gross images of colons from VDRf/f and VDRΔCEC mice on day 3. (C) Representative hematoxylin- and eosin-stained sections of distal colons prepared by the Swiss roll method from VDRf/f and VDRΔIEC mice on day 4. (D,F,H) Representative FACS plots gated on CD4+TCRβ+ cells for (D) IL-17+ T cells, (F) IFN-γ+ T cells, and (H) IFN-γ+IL-17+ T cells; and (E,G,I) FACS quantitation of percentage and cell number for (E) TH17 cells, (G) TH1 cells, and (I) TH1/TH17 cells in the lamina propria on day 3. *P < 0.05, **P < 0.01, ***P < 0.001. Ctrl, vehicle; Q, Q-VD-OPh.
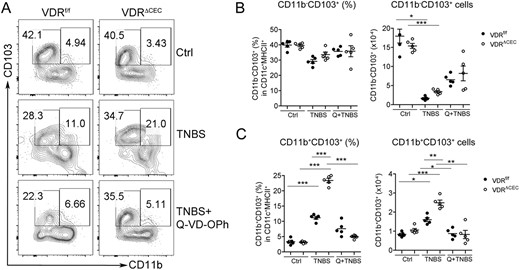
Blockade of cell apoptosis attenuates the increase of CD11b+CD103+ DCs in lamina propria in TNBS-induced colitis. (A) Representative FACS plots gated on CD11c+MHCII+ for CD11b+CD103+ cells in VDRf/f and VDRΔCEC mice treated with vehicle, TNBS, or TNBS+Q-VD-OPh on day 3. (B,C) FACS quantitation of cell percentage and cell number for (B) CD11b−CD103+ DCs and (C) CD11b+CD103+ DCs in the lamina propria. *P < 0.05, **P < 0.01, ***P < 0.001. Ctrl, vehicle; Q, Q-VD-OPh.
Depletion of commensal bacteria markedly ameliorated mucosal inflammation regardless of epithelial VDR status
Previous studies demonstrated that the microbiota in the colon is required for TH17 differentiation (47). To address whether commensal bacteria are required for the robust mucosal inflammatory response in the absence of epithelial VDR signaling, we treated the mice with an antibiotic cocktail to deplete the microbiota, as reported previously (36, 37), before TNBS instillation. As shown in Fig. 7, the magnitude of body weight loss caused by TNBS treatment was the same in AIMD VDRf/f and VDRΔCEC mice (Fig. 7A), suggesting there is no difference in colonic inflammatory response, if any, between in the AIMD VDRf/f and VDRΔCEC mice. Consistently, TNBS had little effect on CD11b+CD103+ or CD11b-CD103+ DCs under this AIMD condition. Although the percentage of CD11b+CD103+ DCs appeared to be increased after TNBS treatment, the absolute cell number did not change compared with the mice without TNBS treatment. Importantly, there was no significant difference in this cell population between AIMD VDRf/f and VDRΔCEC mice (Fig. 7B and 7C). Also TNBS failed to induce TH1 or TH17 inflammatory responses in these AIMD mice, regardless of their epithelial VDR status (Fig. 7D–7F). These data indicate that even though epithelial VDR deletion can cause mucosal barrier dysfunction, the luminal commensal bacteria are still required for the initiation of mucosal inflammatory responses under TNBS induction.
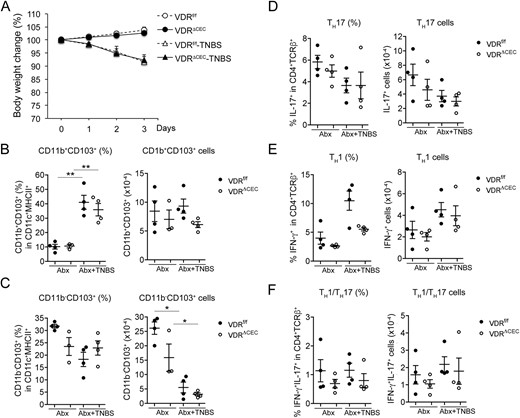
Depletion of microbiota ameliorated mucosal inflammatory responses in VDRf/f and VDRΔCEC mice. VDRf/f and VDRΔCEC mice received an antibiotic cocktail dissolved in drinking water for 3 weeks before being treated with TNBS instillation. Antibiotic treatment was continued after TNBS treatment. Lamina propria leukocytes were analyzed by FACS on day 3 after TNBS treatment, and cell percentage and cell numbers were quantified. (A) Body weight changes after TNBS treatment. (B) CD11b+CD103+ cells. (C) CD11b−CD103+ cells. (D) TH17 cells. (E) TH1 cells. (F) TH1/TH17 cells. *P < 0.05; **P < 0.001. Abx, antibiotic.
Discussion
In this study, we investigated the role of gut epithelial VDR signaling in the regulation of mucosal inflammation in an experimental colitis model resembling Crohn disease, using two lines of transgenic mice that are depleted of VDR from gut epithelial cells. We showed that epithelial VDR deficiency in the gut or only in the colon led to severe colitis and high mortality in the TNBS model as a result of exaggerated TH1 and TH17 responses in the mucosa. The primary defect caused by epithelial VDR depletion appears to be excessive apoptosis in the colonic epithelia, due to overactivation of the PUMA-mediated proapoptotic pathway, which results in epithelial barrier dysfunction. Consequently, the impaired mucosal barrier leads to increased invasion of luminal bacteria and antigens, which trigger robust mucosal inflammatory responses. The evidence to support this notion is that blockade of apoptosis with a caspase inhibitor markedly ameliorated colitis and attenuated mucosal TH1 and TH17 responses in the mice lacking epithelial VDR, and depletion of luminal commensal bacteria with antibiotics eliminated mucosal inflammation not only in normal mice but also in these epithelial VDR-deficient mice. In other words, epithelial apoptosis and commensal bacteria are required for the development of severe colitis in the mice with epithelial VDR deletion. Collectively, these data demonstrate that the epithelial VDR signaling pathway suppresses microbiota-dependent mucosal inflammation by inhibiting gut epithelial cell apoptosis. Thus, it is concluded that the epithelial VDR plays an essential role in protecting the integrity of the mucosal barrier against inflammation-inflicted injury.
The intestine, especially the colon, contains many commensal bacteria that are separated from the body by the mucosal epithelial barrier. Gut epithelial cells abundantly express VDR (they probably are among the highest expressers of VDR in the body); however, except for the regulation of transepithelial calcium transport in the small intestine, the role of the epithelial VDR remains elusive. Using a gain-of-function transgenic mouse model, we previously demonstrated that gut epithelial VDR signaling inhibits colitis development (26); here, we confirmed this conclusion by using a loss-of-function model with epithelial VDR deletion. Mechanistically, epithelial VDR signaling attenuates PUMA induction in mucosal inflammation by blocking NF-κB activation, thereby suppressing epithelial cell apoptosis. When VDR was ablated from the epithelial cells, PUMA induction was exaggerated, leading to excess apoptosis. To directly link the excessive apoptosis to the severe mucosal inflammation and colonic injury observed in the mice without epithelial VDR, we pharmacologically blocked apoptosis in these mice, which led to marked attenuation of colonic injury and TH1 and TH17 inflammatory responses. The observation that the Q-VD-OPh compound treatment did not block the induction of TH1 and TH17 responses completely (Fig. 5) may be explained by a low efficacy of inhibition on epithelial cell apoptosis at the particular dose of this caspase inhibitor. Optimal doses or dose-response experiments likely are needed to achieve better results, but the current experimental evidence is sufficient to support the conclusion that epithelial barrier dysfunction was the primary cause for the severe colitis and high mortality seen in the mice with epithelial VDR deletion.
How the VDR status in the epithelium influences mucosal immune responses in the colon was not investigated in our previous studies. In the current study, we demonstrated that VDR ablation in the gut epithelial cells exacerbated mucosal inflammatory responses with a robust induction of IFN-γ– and IL-17–producing TH1, TH17, and TH1/TH17 cell populations in the lamina propria. These robust responses were due, at least in part, to an increase in the CD11b+CD103+ DC population, which has been identified as a potent inducer of TH17 differentiation and activation. In fact, IL-6 produced by these CD11b+CD103+ DCs is required for intestinal TH17 differentiation (8). On the other hand, the FoxP3+ Treg cell population, which produces IL-10, was not changed in these epithelial VDR-deficient mice. Treg cells are essential to maintain intestinal homeostasis, because IL-10 is a key anti-inflammatory cytokine that targets TH17 cells to suppress mucosal inflammation (48, 49). Therefore, these aberrant immune responses caused by epithelial VDR deletion lead to an imbalance in the mucosal microenvironment that favors TH1- and TH17-mediated inflammation. It is well established that the vitamin D hormone has potent immune-regulatory activities on innate and adaptive immunity (50, 51). Indeed, 1,25(OH)2D3 can suppress DC differentiation and activation, promote FoxP3+ Treg cell differentiation, and inhibit CD4+ TH1 and TH17 cell differentiation and functions (51). However, given that the vitamin D–VDR signaling is intact in all immune cells in these epithelial VDR-deficient mice, it is certain that their aberrant immune responses are secondary, as a result of mucosal barrier dysfunction caused by VDR deletion.
Previous studies reported that the commensal bacteria in the gut are necessary for the development of colitis (52). Our data from the bacterial depletion experiment confirmed the requirement of the commensal bacteria for the development of mucosal inflammation, even in the presence of an impaired epithelial barrier caused by epithelial VDR ablation. This result provides additional proof that the defects in the epithelial barrier are the primary cause of exaggerated mucosal inflammation in the absence of epithelial VDR. Although a treatment with the antibiotic cocktail may not have been able to completely deplete all gut bacteria in the mice, the AIMD mouse model is widely used and well characterized model in microbiota research (36, 37). Indeed, that TNBS still induced moderate body weight loss in the AIMD mice (10%, compared with ∼20% in regular VDRΔCEC mice after TNBS treatment) suggests the remaining microbiota might be able to initiate limited colonic inflammation after TNBS instillation, but this inflammatory response, if any, is independent of epithelial VDR status and TH1 and TH17 cells. Recently we showed that accompanying the loss of epithelial VDR expression in patients with IBD or in experimental colitis models is a dramatic induction Cyp27b1, the 1α-hydroxylase enzyme catalyzing 1,25(OH)2D3 biosynthesis, suggesting an important role of locally produced 1,25(OH)2D3 in the protection against mucosal barrier injury during colonic inflammation (53). Interestingly, the induction of Cyp27b1 in the colon depends on commensal bacterial in the colon. On the other hand, previous studies have shown that VDR gene variations identified in a genome-wide association analysis are associated with changes in gut microbiota, and VDR deletion can cause dysbiosis (54, 55). Moreover, vitamin D can regulate antimicrobial peptide production in the gut (56, 57). Additional studies are needed to assess the effect of epithelial VDR depletion on gut microbiota in the TNBS model, which may or may not confound the results seen in the AIMD mice.
A few recent studies have reported conflicting results in the study of mice with epithelial VDR deletion, using the dextran sodium sulfate (DSS)–induced experimental colitis model. Kim et al. (58) showed that epithelial VDR deletion exacerbated colitis symptoms and increased colonic inflammation after 3% DSS treatment, and Wu et al. (59) reported that epithelial VDR deletion led to increased susceptibility to DSS-induced colitis and caused defective autophagy (59), whereas Wang et al. (60) and Leyssens et al. (61) observed few effects of epithelial VDR deletion on colonic injury and colitis development after DSS treatment. It should be noted that severe colitis and high rate of mortality were reported in global VDR knockout mice after DSS treatment (62). The reasons underlying the discrepancy among these reports are unclear, but differences in mouse genetic background and/or in mouse diets could contribute to the discrepancy. It is noted that Kim et al. (58) reported that the mice with villin-Cre–driven VDR deletion developed hypocalcemia, whereas Leyssens et al. (61) reported normal serum calcium levels in this mouse model when the mice were fed the high-calcium (2%), high-lactose (20%) rescue diet, and Lieben et al. (43) observed normal serum calcium levels in this model, as we did in the current study, even when the mice were fed regular diet containing 1% calcium. Regardless of these reports, the data in our current study, based on the TNBS colitis model, support a critical role for epithelial VDR signaling in suppressing colonic inflammation.
There are some fundamental differences between DSS and TNBS colitis models. DSS is toxic to the gut epithelial cells and induces colitis by disruption of the mucosal epithelial barrier. Mice treated with DSS develop colitis that resembles ulcerative colitis (63). TNBS is thought to haptenize colonic autologous or microbiota proteins, rendering them immunogenic to the host immune system, inducing TH1 and TH17 cell–mediated colonic inflammation (29). Thus, the TNBS model is believed to resemble Crohn disease. It is unclear whether DSS and TNBS have different effects on epithelial VDR signaling in the colon. Studies are warranted to address this issue. It also would be interesting to study the epithelial VDR-ablated mice using other colitis models such as the adoptive T-cell transfer model, which is thought to be more relevant to human IBD (64).
In this study, we examined two mouse models of VDR deletion in gut epithelial cells. Although VDRΔIEC mice and VDRΔCEC mice have normal serum calcium levels, they are distinct in that they have different skeletal phenotypes and maintain the blood calcium level by different mechanisms. VDRΔIEC mice maintain a normal blood calcium level at the expense of bone loss, reflected by their markedly elevated levels of serum 1,25(OH)2D3 and PTH (Supplemental Fig. 1), whereas VDRΔCEC mice do so through duodenal compensation of calcium absorption (43, 45). It is unclear whether these different compensatory mechanisms affect the colonic inflammatory phenotype differently. In fact, except for the AIMD experiment that was conducted only in the VDRΔCEC mice, all other experiments were performed in the VDRΔIEC and the VDRΔCEC models with similar results. Therefore, these two distinct models actually validated the experimental outcomes reciprocally. It is noted that claudin-2 protein was highly induced in the TNBS model, shown here and in other studies (28, 65), but claudin-2 transcript was not seen induced in a previous study (26). This messenger RNA and protein discrepancy is intriguing, but the reason is unknown.
In conclusion, the evidence from this study demonstrates that epithelial VDR signaling regulates mucosal inflammation by suppressing epithelial cell apoptosis. Given the importance of epithelial VDR in the mucosal barrier function and our previous observation that gut epithelial VDR was downregulated by >50% in patients with IBD (26), it is speculated that maintaining or upregulating the epithelial VDR could be an effective therapeutic strategy for colitis. It is well known that ligand stimulation can increase VDR expression (66). Our prior work showed that TNF-α, a major proinflammatory cytokine that drives the development of IBD, downregulates VDR in intestinal epithelial cells by a microRNA-mediated mechanism (42). Therefore, vitamin D therapy and anti-TNF therapy are predicted to increase epithelial VDR levels. In this regard, developing novel active vitamin D analogs for the treatment of IBD might be an attractive strategy.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer; Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| β-Actin | Sigma-Aldrich; A5316 | Mouse; monoclonal (clone AC-74) | WB (1:1000) | AB_476743 | ||
| Caspase 3 | Cell Signaling Technology; 9662 | Rabbit; polyclonal | WB (1:1000) | AB_10839261 | ||
| VDR | Santa Cruz Biotechnology; sc-13133 | Mouse; monoclonal | WB (1:1000) | AB_628040 | ||
| PUMA | Cell Signaling Technology; 7467 | Rabbit; monoclonal | WB (1:1000) | AB_10829605 | ||
| p53 | Cell Signaling Technology; 9282 | Rabbit; monoclonal | WB (1:1000) | AB_331476 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer; Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| β-Actin | Sigma-Aldrich; A5316 | Mouse; monoclonal (clone AC-74) | WB (1:1000) | AB_476743 | ||
| Caspase 3 | Cell Signaling Technology; 9662 | Rabbit; polyclonal | WB (1:1000) | AB_10839261 | ||
| VDR | Santa Cruz Biotechnology; sc-13133 | Mouse; monoclonal | WB (1:1000) | AB_628040 | ||
| PUMA | Cell Signaling Technology; 7467 | Rabbit; monoclonal | WB (1:1000) | AB_10829605 | ||
| p53 | Cell Signaling Technology; 9282 | Rabbit; monoclonal | WB (1:1000) | AB_331476 |
Abbreviations: RRID, Research Resource Identifier; WB, Western blot.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer; Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| β-Actin | Sigma-Aldrich; A5316 | Mouse; monoclonal (clone AC-74) | WB (1:1000) | AB_476743 | ||
| Caspase 3 | Cell Signaling Technology; 9662 | Rabbit; polyclonal | WB (1:1000) | AB_10839261 | ||
| VDR | Santa Cruz Biotechnology; sc-13133 | Mouse; monoclonal | WB (1:1000) | AB_628040 | ||
| PUMA | Cell Signaling Technology; 7467 | Rabbit; monoclonal | WB (1:1000) | AB_10829605 | ||
| p53 | Cell Signaling Technology; 9282 | Rabbit; monoclonal | WB (1:1000) | AB_331476 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer; Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used | RRID |
|---|---|---|---|---|---|---|
| β-Actin | Sigma-Aldrich; A5316 | Mouse; monoclonal (clone AC-74) | WB (1:1000) | AB_476743 | ||
| Caspase 3 | Cell Signaling Technology; 9662 | Rabbit; polyclonal | WB (1:1000) | AB_10839261 | ||
| VDR | Santa Cruz Biotechnology; sc-13133 | Mouse; monoclonal | WB (1:1000) | AB_628040 | ||
| PUMA | Cell Signaling Technology; 7467 | Rabbit; monoclonal | WB (1:1000) | AB_10829605 | ||
| p53 | Cell Signaling Technology; 9282 | Rabbit; monoclonal | WB (1:1000) | AB_331476 |
Abbreviations: RRID, Research Resource Identifier; WB, Western blot.
Abbreviations:
- 1,25(OH)2D3
1,25-dihydroxyvitamin D3
- AIMD
antibiotic-induced microbiota-depleted
- DC
dendritic cell
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- FACS
fluorescence-activated cell sorting
- IEC
intestinal epithelial cell
- IFN
interferon
- IL
interleukin
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PTH
parathyroid hormone
- PUMA
p53-upregulated modulator of apoptosis
- TH
T helper
- TNBS
2,4,6-trinitrobenzenesulfonic acid
- TNF
tumor necrosis factor
- VDR
vitamin D receptor
- VDRf/f
VDRflox/flox.
Acknowledgments
Financial Support: This work was supported in part by National Institutes of Health Grant CA180087 (to Y.C.L.), National Institute of Diabetes and Digestive and Kidney Diseases Grant P30DK42086, and a research grant from the Foundation for Clinical Research in Inflammatory Bowel Disease.
Author Contributions: Y.C.L. designed the research, acquired funding, supervised the research, and wrote the manuscript; L.H., T.L., Y.S., F.T., H.H., D.K.D., and Y.C. performed the research; L.H. and Y.C.L. analyzed the data; M.B. provided reagents and critically read the manuscript.
Disclosure Summary: The authors have nothing to disclose.
References
Author notes
These authors contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}