




Abstract
It is unknown how the lack of insulin receptor (IR)/insulinlike growth factor I receptor (IGFIR) in a tissue-specific manner affects brown fat development and mitochondrial integrity and function, as well as its effect on the redistribution of the adipose organ and the metabolic status. To address this important issue, we developed IR/IGFIR double-knockout (DKO) in a brown adipose tissue–specific manner. Lack of those receptors caused severe brown fat atrophy, enhanced beige cell clusters in inguinal fat; loss of mitochondrial mass; mitochondrial damage related to cristae disruption; and the loss of proteins involved in autophagosome formation, mitophagy, mitochondrial quality control, and dynamics and thermogenesis. More important, DKO mice showed an impaired thermogenesis upon cold exposure, based on a failure in the mitochondrial fission mechanisms and a much lower uncoupling protein 1 transcription rate and content. As a result, DKO mice under normal conditions showed an obesity susceptibility, revealed by increased body fat mass and insulin resistance. Upon consumption of a high-fat diet, DKO mice displayed frank obesity, as shown by increased body weight, increased adiposity, insulin resistance, hyperinsulinemia, and hypertriglyceridemia, all consistent with a metabolic syndrome. Collectively, our data suggest a cause-and-effect relationship between failure in brown fat thermogenesis and increased adiposity and obesity.
The adipose organ includes two functionally and morphologically different types of adipose tissue in mammals: white adipose tissue (WAT), which is the primary site of dynamic triglyceride storage, and brown adipose tissue (BAT), which specializes in energy expenditure (1). In rodents, brown fat remains active for life. In humans, however, it largely disappears shortly after birth. However, the identification of active brown fat in human studies (2) has radically revised this concept, suggesting a critical role of brown fat in human adult energy metabolism and body weight control. It is now recognized that there are at least two distinct types of brown fat cells, located in the classic brown adipose depots or as resident cells mainly within the inguinal white fat, where the coregulatory protein PR domain containing 16 plays a specific role in fat browning (3, 4).
Mitochondrial biogenesis and bioenergetics are essential for the onset of nonshivering thermogenesis. Regarding mitochondrial biogenesis, the onset of the cytosolic quality control mechanisms assures mitochondrial integrity and functioning, maintaining a population of healthy mitochondria (5). Regarding mitochondrial bioenergetics, heat production in response to hypothermia or excess energy intake has been associated with the collapse of the proton gradient by the increased expression and anchorage of the tissue-specific protein uncoupling protein 1 (UCP-1) to the mitochondrial inner membrane. It is now recognized that the mitochondrial dynamics, an equilibrium between the mitochondrial fusion/fission process, is essential for the adaptation to cellular energy status. Thus, caloric restriction or starvation induces fusion and mitochondrial coupling (oxidative phosphorylation), and excess nutrient availability leads to fission and mitochondrial uncoupling. Thus, in brown adipocytes, mitochondrial fission has been proposed as an amplification pathway for energy expenditure (6, 7).
Insulinlike growth factor/insulin signaling plays a major role in brown fat development. Thus, brown adipose tissue–specific insulin receptor knockout (BATIRKO) mice showed brown fat lipoatrophy and moderate sporadic visceral adiposity upon aging. These animals showed an insulin secretion defect, but not insulin resistance (8). Unlike in BATIRKO mice, the lack of insulinlike growth factor I (IGFI) receptor (IGFIR) in BAT (BATIGFIRKO) mice showed no effect on brown fat mass development. However, these animals showed a moderate thermogenic impairment upon cold exposure. More important, BATIGFIRKO mice had mainly hepatic insulin resistance, without showing an insulin secretion defect (9). However, lack of insulin receptor (IR)/IGFIR in the whole adipose organ showed an almost complete absence of WAT and BAT, not previously described in brown fat–specific single knockouts regarding those receptors. Surprisingly, they showed a failure in brown fat thermogenesis associated with a lean phenotype upon consumption of a high-fat diet (HFD) (10, 11). Accordingly, it remained mostly unknown how the lack of IR/IGFIR in a tissue-specific manner affected brown fat mass development, its mitochondrial content and dynamics, thermogenic function, redistribution of the several compartments of the adipose organ, and metabolic status upon consumption of an HFD.
To address these important issues, we have generated the IR/IGFIR brown adipose tissue–specific double-knockout (BATIGFIRDKO) mice. Our results show that the lack of IR/IGFIR under normal conditions caused severe brown fat atrophy, thermogenic failure in response to a cold environment, enhancement of body fat mass, and global insulin resistance. Under HFD conditions, these mice showed insulin resistance, hyperinsulinemia, hypertriglyceridemia, and obesity, all consistent with metabolic syndrome.
Materials and Methods
Experimental model and genotyping
To obtain BATIGFIRDKO mice, we bred Ucp1-Cre−/−, double Igf1r (exon 3) and Ir (exon 4) floxed mice, previously described (12) with Ir/Igf1r double-floxed, Ucp1-Cre+/− transgenic mice (8). Experiments were performed on Igf1r/Ir double-floxed [hereafter referred to as control or wild-type (WT)] and BATIGFIRDKO male mice maintained on a mixed 129/SvJ C57BL/6 background. Mice were maintained in the Animal Care Facility under the standard conditions of temperature (23°C) and 12-hour light/dark cycle and were fed a standard diet (3% fat content, A04) and an HFD (63% fat content; Envigo, Cambridge, UK) for 8 weeks. Tail DNA (100 to 200 ng) was used for mice genotyping, as previously described (8, 9). All animal experimentation described in this report was conducted according to accepted standards of human animal care, as approved by the animal experimentation institutional committee.
Cell culture and brown preadipocyte cell line generation
Brown preadipocyte cells were isolated and immortalized from newborn mice with both floxed Ir and Igf1r alleles and were used as WT. Double-knockout (DKO) brown preadipocyte cell lines were generated from immortalized Ir and Igf1r floxed brown preadipocytes infected with an adenovirus encoding Cre recombinase, as previously described (13, 14). Cells were maintained in Dulbecco’s modified Eagle medium (4.5 g/L glucose) containing 10% (volume-to-volume ratio) fetal bovine serum, HEPES 10 mM, and penicillin/streptomycin (Lonza, Basel, Switzerland) at 37°C in a 5% CO2 environment. To study mitochondrial quality control mechanisms in WT and DKO cell lines, we stimulated those cells with carbonyl cyanide m-chlorophenyl hydrazine (CCCP) 20μM for 15 or 24 hours; in some experiments, 20 μM chloroquine (CQ) was used.
Protein extraction, Western blot, and plasma analysis
WT and DKO mice were euthanized by cervical dislocation, and the different tissues were lysed as described elsewhere (15). Western blot analyses were performed as previously described (16). All antibodies were prepared in 1× Tris-buffered saline, 0.1% Tween. The antibodies used were IGFIRβ (catalog no. sc-713) and IRβ (catalog no. sc-711) (Santa Cruz Biotechnology, Dallas, TX); hormone-sensitive lipase (HSL; catalog no. 4107), phospho–dynamin-related protein 1 [p-DRP1 (Ser616); catalog no. 3455], B-cell lymphoma-2 (BCL-2; catalog no. 2876), microtubule-associated protein 1 light chain 3 α/β (catalog no. 4108), peroxisome proliferator-activated receptor γ coactivator-1α (PGC1-α; catalog no. 2178), and immunoglobulin heavy chain-binding protein (BIP; catalog no. 3177) (Cell Signaling Technology, Danvers, MA); bone morphogenetic protein 7 (BMP7; catalog no. 5626-1; Epitomics); E3 ubiquitin-protein ligase parkin (Parkin; catalog no. ab15954), Mitofusin-2 (MFN-2; catalog no. ab56889), voltage-dependent anion-selective channel 1 (catalog no. ab15895), and UCP-1 (catalog no. ab10983) (Abcam, Cambridge, United Kingdom); dynaminlike protein 1 (catalog no. 611112) and optic atrophy 1 (OPA1; catalog no. 612606) (BD Biosciences); phosphatase and tensin homolog–induced putative kinase 1 (catalog no. BC100-494; Novusbio); and β-actin and α-tubulin (Sigma-Aldrich, Madrid, Spain) (Table 1). Immunoreactive bands were visualized using the ClarityTM enhanced chemiluminescence (Bio-Rad, Hercules, CA). Plasma insulin, leptin, adiponectin, tumor necrosis factor (TNF)-α, triiodothyronine (T3), IGFI, BMP7, triglycerides, and cholesterol were measured from blood collected from standard mice or mice fed an HFD for 8 weeks by using enzyme-linked immunosorbent assay or colorimetric assays as previously described (9).
Antibody Table
| RRID | Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|
| AB_671792 | IGF1R β subunit | C-terminus of IGFIRβ of human origin | IGF-IRβ antibody (C-20) | Santa Cruz Biotechnology, sc-713 | Rabbit; polyclonal | 1:500 |
| AB_631835 | IR β subunit | C-terminus of IRβ of human origin | IR β antibody (C-19) | Santa Cruz Biotechnology, sc-711 | Rabbit; polyclonal | 1:1000 |
| AB_2296900 | HSL | Total HSL protein | HSL antibody | Cell Signaling Technology, 4107 | Rabbit; polyclonal | 1:1000 |
| AB_329825 | phospho AKT (Ser473) | Endogenous levels of Akt1 only when phosphorylated at Ser473 | Phospho-Akt (Ser473) antibody | Cell Signaling Technology, 9271 | Rabbit; polyclonal | 1:1000 |
| AB_329827 | AKT | Endogenous levels of total Akt1, Akt2, and Akt3 proteins | Akt antibody | Cell Signaling Technology, 9272 | Rabbit; polyclonal | 1:1000 |
| AB_11000305 | BMP7 | Synthetic peptide corresponding to residues in human BMP7 | Anti-BMP7 antibody [EPR5897] | Epitomics, 5626-1 | Rabbit; monoclonal | 1:500 |
| AB_2241462 | UCP1 | Synthetic peptide conjugated to KLH, corresponding to amino acids 145-159 of human UCP1, with N-terminal cysteine added | Anti-UCP1 antibody | Abcam, ab10983 | Rabbit; polyclonal | WB, 1:1000; IHC, 2 mg/mL |
| AB_2085352 | phospho-DRP1 (Ser616) | Endogenous levels of DRP1 only when phosphorylated at Ser 616 | Phospho-DRP1 (Ser616) antibody | Cell Signaling, 3455 | Rabbit; polyclonal | 1:1000 |
| AB_2064177 | BCL-2 | Endogenous levels of total Bcl-2α protein | Bcl-2 antibody | Cell Signaling, 2876 | Rabbit; polyclonal | 1:1000 |
| AB_2137703 | LC3A/B | Endogenous levels of total LC3A and LC3B proteins | LC3A/B antibody | Cell Signaling, 4108 | Rabbit; polyclonal | 1:1000 |
| AB_2166218 | PGC1-α | Epitope corresponding to amino acids 1–300 mapping at the N-terminus of PGC-1 of human origin | PGC-1α antibody (H-300) | Santa Cruz Biotechnology, sc-13067 | Rabbit; polyclonal | 1:500 |
| AB_2119845 | BIP | Endogenous levels of total BIP protein | BIP (C50B12) rabbit mAb | Cell Signaling, 3177 | Rabbit; monoclonal | 1:1000 |
| AB_443270 | Parkin | Synthetic peptide: RILGEEQYTRYQQYGAEEC conjugated to KLH, corresponding to amino acids 304-322 of mouse Parkin | Anti-Parkin antibody | Abcam, ab15954 | Rabbit; polyclonal | 1:1000 |
| AB_2142629 | MFN-2 | Recombinant fragment corresponding to human MFN-2 amino acids 661–758 | Anti-MFN=2 antibody | Abcam, ab56889 | Mouse; monoclonal | 1:1000 |
| AB_2214787 | VDAC1/Porin | Synthetic peptide derived from residues 150–250 of human VDAC1/Porin | Anti-VDAC1/Porin antibody-mitochondrial loading control | Abcam, ab15895 | Rabbit; polyclonal | 1:1000 |
| AB_945896 | TOMM20 | Recombinant full-length protein (GST-tag) corresponding to human TOMM20 amino acids 1–145 | Anti-TOMM20 antibody | Abcam, ab56783 | Mouse; monoclonal | IF, 1:75 |
| AB_2263836 | HADHA | Synthetic peptide surrounding amino acid 750 (human) | Anti-HADHA antibody | Abcam, ab54477 | Rabbit; polyclonal | IF, 1:75 |
| AB_398423 | DLP1 | Rat DLP1 amino acids 601–722 | Purified mouse anti-DLP1 | BD Biosciences, 611112 | Mouse; monoclonal | 1:1000 |
| AB_399888 | OPA1 | Human OPA1 amino acids 708–830 | Purified mouse anti-OPA1 | BD Biosciences, 612606 | Mouse; monoclonal | 1:1000 |
| AB_10127658 | PINK1 | Synthetic peptide made to the human PINK1 protein sequence (between residues 175 and 250) | PINK1 antibody | Novusbio, BC100-494 | Rabbit; polyclonal | 1:1000 |
| AB_476697 | β-actin | Recognizes an epitope located on the N-terminal end of the β-isoform of actin | Monoclonal Anti–β-actin, clone AC-74 | Sigma-Aldrich, A2228 | Mouse; monoclonal | 1:5000 |
| AB_477583 | α-tubulin | Recognizes an epitope located at the C-terminal end of the α-tubulin isoform (amino acids 426–430) in a variety of organisms (e.g., human, bovine, mouse, and chicken). | α-Tubulin | Sigma-Aldrich, T6199 | Mouse; monoclonal | 1:5000 |
| AB_1542690 | p62 | C-terminal domain (20 amino acids: C-NYD IGA ALD TIQ YSK HPP PL) of human p62 protein, coupled to KLH; this peptide sequence is identical in human, monkey, bovine, mouse and rat | Pab to p62 protein, C-terminal specific | Progen Biotechnik, GP62-C | Guinea pig; polyclonal | IF, 1:75 |
| RRID | Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|
| AB_671792 | IGF1R β subunit | C-terminus of IGFIRβ of human origin | IGF-IRβ antibody (C-20) | Santa Cruz Biotechnology, sc-713 | Rabbit; polyclonal | 1:500 |
| AB_631835 | IR β subunit | C-terminus of IRβ of human origin | IR β antibody (C-19) | Santa Cruz Biotechnology, sc-711 | Rabbit; polyclonal | 1:1000 |
| AB_2296900 | HSL | Total HSL protein | HSL antibody | Cell Signaling Technology, 4107 | Rabbit; polyclonal | 1:1000 |
| AB_329825 | phospho AKT (Ser473) | Endogenous levels of Akt1 only when phosphorylated at Ser473 | Phospho-Akt (Ser473) antibody | Cell Signaling Technology, 9271 | Rabbit; polyclonal | 1:1000 |
| AB_329827 | AKT | Endogenous levels of total Akt1, Akt2, and Akt3 proteins | Akt antibody | Cell Signaling Technology, 9272 | Rabbit; polyclonal | 1:1000 |
| AB_11000305 | BMP7 | Synthetic peptide corresponding to residues in human BMP7 | Anti-BMP7 antibody [EPR5897] | Epitomics, 5626-1 | Rabbit; monoclonal | 1:500 |
| AB_2241462 | UCP1 | Synthetic peptide conjugated to KLH, corresponding to amino acids 145-159 of human UCP1, with N-terminal cysteine added | Anti-UCP1 antibody | Abcam, ab10983 | Rabbit; polyclonal | WB, 1:1000; IHC, 2 mg/mL |
| AB_2085352 | phospho-DRP1 (Ser616) | Endogenous levels of DRP1 only when phosphorylated at Ser 616 | Phospho-DRP1 (Ser616) antibody | Cell Signaling, 3455 | Rabbit; polyclonal | 1:1000 |
| AB_2064177 | BCL-2 | Endogenous levels of total Bcl-2α protein | Bcl-2 antibody | Cell Signaling, 2876 | Rabbit; polyclonal | 1:1000 |
| AB_2137703 | LC3A/B | Endogenous levels of total LC3A and LC3B proteins | LC3A/B antibody | Cell Signaling, 4108 | Rabbit; polyclonal | 1:1000 |
| AB_2166218 | PGC1-α | Epitope corresponding to amino acids 1–300 mapping at the N-terminus of PGC-1 of human origin | PGC-1α antibody (H-300) | Santa Cruz Biotechnology, sc-13067 | Rabbit; polyclonal | 1:500 |
| AB_2119845 | BIP | Endogenous levels of total BIP protein | BIP (C50B12) rabbit mAb | Cell Signaling, 3177 | Rabbit; monoclonal | 1:1000 |
| AB_443270 | Parkin | Synthetic peptide: RILGEEQYTRYQQYGAEEC conjugated to KLH, corresponding to amino acids 304-322 of mouse Parkin | Anti-Parkin antibody | Abcam, ab15954 | Rabbit; polyclonal | 1:1000 |
| AB_2142629 | MFN-2 | Recombinant fragment corresponding to human MFN-2 amino acids 661–758 | Anti-MFN=2 antibody | Abcam, ab56889 | Mouse; monoclonal | 1:1000 |
| AB_2214787 | VDAC1/Porin | Synthetic peptide derived from residues 150–250 of human VDAC1/Porin | Anti-VDAC1/Porin antibody-mitochondrial loading control | Abcam, ab15895 | Rabbit; polyclonal | 1:1000 |
| AB_945896 | TOMM20 | Recombinant full-length protein (GST-tag) corresponding to human TOMM20 amino acids 1–145 | Anti-TOMM20 antibody | Abcam, ab56783 | Mouse; monoclonal | IF, 1:75 |
| AB_2263836 | HADHA | Synthetic peptide surrounding amino acid 750 (human) | Anti-HADHA antibody | Abcam, ab54477 | Rabbit; polyclonal | IF, 1:75 |
| AB_398423 | DLP1 | Rat DLP1 amino acids 601–722 | Purified mouse anti-DLP1 | BD Biosciences, 611112 | Mouse; monoclonal | 1:1000 |
| AB_399888 | OPA1 | Human OPA1 amino acids 708–830 | Purified mouse anti-OPA1 | BD Biosciences, 612606 | Mouse; monoclonal | 1:1000 |
| AB_10127658 | PINK1 | Synthetic peptide made to the human PINK1 protein sequence (between residues 175 and 250) | PINK1 antibody | Novusbio, BC100-494 | Rabbit; polyclonal | 1:1000 |
| AB_476697 | β-actin | Recognizes an epitope located on the N-terminal end of the β-isoform of actin | Monoclonal Anti–β-actin, clone AC-74 | Sigma-Aldrich, A2228 | Mouse; monoclonal | 1:5000 |
| AB_477583 | α-tubulin | Recognizes an epitope located at the C-terminal end of the α-tubulin isoform (amino acids 426–430) in a variety of organisms (e.g., human, bovine, mouse, and chicken). | α-Tubulin | Sigma-Aldrich, T6199 | Mouse; monoclonal | 1:5000 |
| AB_1542690 | p62 | C-terminal domain (20 amino acids: C-NYD IGA ALD TIQ YSK HPP PL) of human p62 protein, coupled to KLH; this peptide sequence is identical in human, monkey, bovine, mouse and rat | Pab to p62 protein, C-terminal specific | Progen Biotechnik, GP62-C | Guinea pig; polyclonal | IF, 1:75 |
Abbreviations: IF, immunofluorescence; IHC, immunohistochemistry; KLH, keyhole limpet hemocyanin; LC3A/B, microtubule-associated protein 1 light chain 3 α/β; mAB, monoclonal antibody; PINK1, phosphatase and tensin homolog–induced putative kinase 1; RRID, Research Resource Identifier; WB, Western blot.
Antibody Table
| RRID | Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|
| AB_671792 | IGF1R β subunit | C-terminus of IGFIRβ of human origin | IGF-IRβ antibody (C-20) | Santa Cruz Biotechnology, sc-713 | Rabbit; polyclonal | 1:500 |
| AB_631835 | IR β subunit | C-terminus of IRβ of human origin | IR β antibody (C-19) | Santa Cruz Biotechnology, sc-711 | Rabbit; polyclonal | 1:1000 |
| AB_2296900 | HSL | Total HSL protein | HSL antibody | Cell Signaling Technology, 4107 | Rabbit; polyclonal | 1:1000 |
| AB_329825 | phospho AKT (Ser473) | Endogenous levels of Akt1 only when phosphorylated at Ser473 | Phospho-Akt (Ser473) antibody | Cell Signaling Technology, 9271 | Rabbit; polyclonal | 1:1000 |
| AB_329827 | AKT | Endogenous levels of total Akt1, Akt2, and Akt3 proteins | Akt antibody | Cell Signaling Technology, 9272 | Rabbit; polyclonal | 1:1000 |
| AB_11000305 | BMP7 | Synthetic peptide corresponding to residues in human BMP7 | Anti-BMP7 antibody [EPR5897] | Epitomics, 5626-1 | Rabbit; monoclonal | 1:500 |
| AB_2241462 | UCP1 | Synthetic peptide conjugated to KLH, corresponding to amino acids 145-159 of human UCP1, with N-terminal cysteine added | Anti-UCP1 antibody | Abcam, ab10983 | Rabbit; polyclonal | WB, 1:1000; IHC, 2 mg/mL |
| AB_2085352 | phospho-DRP1 (Ser616) | Endogenous levels of DRP1 only when phosphorylated at Ser 616 | Phospho-DRP1 (Ser616) antibody | Cell Signaling, 3455 | Rabbit; polyclonal | 1:1000 |
| AB_2064177 | BCL-2 | Endogenous levels of total Bcl-2α protein | Bcl-2 antibody | Cell Signaling, 2876 | Rabbit; polyclonal | 1:1000 |
| AB_2137703 | LC3A/B | Endogenous levels of total LC3A and LC3B proteins | LC3A/B antibody | Cell Signaling, 4108 | Rabbit; polyclonal | 1:1000 |
| AB_2166218 | PGC1-α | Epitope corresponding to amino acids 1–300 mapping at the N-terminus of PGC-1 of human origin | PGC-1α antibody (H-300) | Santa Cruz Biotechnology, sc-13067 | Rabbit; polyclonal | 1:500 |
| AB_2119845 | BIP | Endogenous levels of total BIP protein | BIP (C50B12) rabbit mAb | Cell Signaling, 3177 | Rabbit; monoclonal | 1:1000 |
| AB_443270 | Parkin | Synthetic peptide: RILGEEQYTRYQQYGAEEC conjugated to KLH, corresponding to amino acids 304-322 of mouse Parkin | Anti-Parkin antibody | Abcam, ab15954 | Rabbit; polyclonal | 1:1000 |
| AB_2142629 | MFN-2 | Recombinant fragment corresponding to human MFN-2 amino acids 661–758 | Anti-MFN=2 antibody | Abcam, ab56889 | Mouse; monoclonal | 1:1000 |
| AB_2214787 | VDAC1/Porin | Synthetic peptide derived from residues 150–250 of human VDAC1/Porin | Anti-VDAC1/Porin antibody-mitochondrial loading control | Abcam, ab15895 | Rabbit; polyclonal | 1:1000 |
| AB_945896 | TOMM20 | Recombinant full-length protein (GST-tag) corresponding to human TOMM20 amino acids 1–145 | Anti-TOMM20 antibody | Abcam, ab56783 | Mouse; monoclonal | IF, 1:75 |
| AB_2263836 | HADHA | Synthetic peptide surrounding amino acid 750 (human) | Anti-HADHA antibody | Abcam, ab54477 | Rabbit; polyclonal | IF, 1:75 |
| AB_398423 | DLP1 | Rat DLP1 amino acids 601–722 | Purified mouse anti-DLP1 | BD Biosciences, 611112 | Mouse; monoclonal | 1:1000 |
| AB_399888 | OPA1 | Human OPA1 amino acids 708–830 | Purified mouse anti-OPA1 | BD Biosciences, 612606 | Mouse; monoclonal | 1:1000 |
| AB_10127658 | PINK1 | Synthetic peptide made to the human PINK1 protein sequence (between residues 175 and 250) | PINK1 antibody | Novusbio, BC100-494 | Rabbit; polyclonal | 1:1000 |
| AB_476697 | β-actin | Recognizes an epitope located on the N-terminal end of the β-isoform of actin | Monoclonal Anti–β-actin, clone AC-74 | Sigma-Aldrich, A2228 | Mouse; monoclonal | 1:5000 |
| AB_477583 | α-tubulin | Recognizes an epitope located at the C-terminal end of the α-tubulin isoform (amino acids 426–430) in a variety of organisms (e.g., human, bovine, mouse, and chicken). | α-Tubulin | Sigma-Aldrich, T6199 | Mouse; monoclonal | 1:5000 |
| AB_1542690 | p62 | C-terminal domain (20 amino acids: C-NYD IGA ALD TIQ YSK HPP PL) of human p62 protein, coupled to KLH; this peptide sequence is identical in human, monkey, bovine, mouse and rat | Pab to p62 protein, C-terminal specific | Progen Biotechnik, GP62-C | Guinea pig; polyclonal | IF, 1:75 |
| RRID | Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog No. | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|
| AB_671792 | IGF1R β subunit | C-terminus of IGFIRβ of human origin | IGF-IRβ antibody (C-20) | Santa Cruz Biotechnology, sc-713 | Rabbit; polyclonal | 1:500 |
| AB_631835 | IR β subunit | C-terminus of IRβ of human origin | IR β antibody (C-19) | Santa Cruz Biotechnology, sc-711 | Rabbit; polyclonal | 1:1000 |
| AB_2296900 | HSL | Total HSL protein | HSL antibody | Cell Signaling Technology, 4107 | Rabbit; polyclonal | 1:1000 |
| AB_329825 | phospho AKT (Ser473) | Endogenous levels of Akt1 only when phosphorylated at Ser473 | Phospho-Akt (Ser473) antibody | Cell Signaling Technology, 9271 | Rabbit; polyclonal | 1:1000 |
| AB_329827 | AKT | Endogenous levels of total Akt1, Akt2, and Akt3 proteins | Akt antibody | Cell Signaling Technology, 9272 | Rabbit; polyclonal | 1:1000 |
| AB_11000305 | BMP7 | Synthetic peptide corresponding to residues in human BMP7 | Anti-BMP7 antibody [EPR5897] | Epitomics, 5626-1 | Rabbit; monoclonal | 1:500 |
| AB_2241462 | UCP1 | Synthetic peptide conjugated to KLH, corresponding to amino acids 145-159 of human UCP1, with N-terminal cysteine added | Anti-UCP1 antibody | Abcam, ab10983 | Rabbit; polyclonal | WB, 1:1000; IHC, 2 mg/mL |
| AB_2085352 | phospho-DRP1 (Ser616) | Endogenous levels of DRP1 only when phosphorylated at Ser 616 | Phospho-DRP1 (Ser616) antibody | Cell Signaling, 3455 | Rabbit; polyclonal | 1:1000 |
| AB_2064177 | BCL-2 | Endogenous levels of total Bcl-2α protein | Bcl-2 antibody | Cell Signaling, 2876 | Rabbit; polyclonal | 1:1000 |
| AB_2137703 | LC3A/B | Endogenous levels of total LC3A and LC3B proteins | LC3A/B antibody | Cell Signaling, 4108 | Rabbit; polyclonal | 1:1000 |
| AB_2166218 | PGC1-α | Epitope corresponding to amino acids 1–300 mapping at the N-terminus of PGC-1 of human origin | PGC-1α antibody (H-300) | Santa Cruz Biotechnology, sc-13067 | Rabbit; polyclonal | 1:500 |
| AB_2119845 | BIP | Endogenous levels of total BIP protein | BIP (C50B12) rabbit mAb | Cell Signaling, 3177 | Rabbit; monoclonal | 1:1000 |
| AB_443270 | Parkin | Synthetic peptide: RILGEEQYTRYQQYGAEEC conjugated to KLH, corresponding to amino acids 304-322 of mouse Parkin | Anti-Parkin antibody | Abcam, ab15954 | Rabbit; polyclonal | 1:1000 |
| AB_2142629 | MFN-2 | Recombinant fragment corresponding to human MFN-2 amino acids 661–758 | Anti-MFN=2 antibody | Abcam, ab56889 | Mouse; monoclonal | 1:1000 |
| AB_2214787 | VDAC1/Porin | Synthetic peptide derived from residues 150–250 of human VDAC1/Porin | Anti-VDAC1/Porin antibody-mitochondrial loading control | Abcam, ab15895 | Rabbit; polyclonal | 1:1000 |
| AB_945896 | TOMM20 | Recombinant full-length protein (GST-tag) corresponding to human TOMM20 amino acids 1–145 | Anti-TOMM20 antibody | Abcam, ab56783 | Mouse; monoclonal | IF, 1:75 |
| AB_2263836 | HADHA | Synthetic peptide surrounding amino acid 750 (human) | Anti-HADHA antibody | Abcam, ab54477 | Rabbit; polyclonal | IF, 1:75 |
| AB_398423 | DLP1 | Rat DLP1 amino acids 601–722 | Purified mouse anti-DLP1 | BD Biosciences, 611112 | Mouse; monoclonal | 1:1000 |
| AB_399888 | OPA1 | Human OPA1 amino acids 708–830 | Purified mouse anti-OPA1 | BD Biosciences, 612606 | Mouse; monoclonal | 1:1000 |
| AB_10127658 | PINK1 | Synthetic peptide made to the human PINK1 protein sequence (between residues 175 and 250) | PINK1 antibody | Novusbio, BC100-494 | Rabbit; polyclonal | 1:1000 |
| AB_476697 | β-actin | Recognizes an epitope located on the N-terminal end of the β-isoform of actin | Monoclonal Anti–β-actin, clone AC-74 | Sigma-Aldrich, A2228 | Mouse; monoclonal | 1:5000 |
| AB_477583 | α-tubulin | Recognizes an epitope located at the C-terminal end of the α-tubulin isoform (amino acids 426–430) in a variety of organisms (e.g., human, bovine, mouse, and chicken). | α-Tubulin | Sigma-Aldrich, T6199 | Mouse; monoclonal | 1:5000 |
| AB_1542690 | p62 | C-terminal domain (20 amino acids: C-NYD IGA ALD TIQ YSK HPP PL) of human p62 protein, coupled to KLH; this peptide sequence is identical in human, monkey, bovine, mouse and rat | Pab to p62 protein, C-terminal specific | Progen Biotechnik, GP62-C | Guinea pig; polyclonal | IF, 1:75 |
Abbreviations: IF, immunofluorescence; IHC, immunohistochemistry; KLH, keyhole limpet hemocyanin; LC3A/B, microtubule-associated protein 1 light chain 3 α/β; mAB, monoclonal antibody; PINK1, phosphatase and tensin homolog–induced putative kinase 1; RRID, Research Resource Identifier; WB, Western blot.
Ex vivo lipolysis assay
BAT from WT and DKO 3-month-old mice was removed, weighed, and placed in prewarmed Krebs buffer for 5 minutes. The samples were used for ex vivo lipolysis assays as previously described (17).
Quantitative reverse transcriptase polymerase chain reaction analysis
BAT RNA (3 μg) was reverse transcribed with a high-capacity complementary DNA reverse transcription kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. The gene expression of Ppargc1a, Tfam, Adrb3, Dio2, Ucp1, and Fgf21 were analyzed by real-time quantitative polymerase chain reaction using Taqman probes and glyceraldehyde 3-phosphate dehydrogenase as endogenous control using a StepOnePlus™ Real-Time PCR System (Applied Biosystems). The results were calculated by using the 2−ΔΔCt method (18).
Immunofluorescence and colocalization analysis by confocal microscopy
Cells were grown on glass coverslips in normal culture dishes. Then, cells were fixed by using paraformaldehyde 4% for 15 minutes at room temperature. After fixing, cells were permeabilized in phosphate-buffered saline (PBS) with 0.5% Triton X-100 for 10 minutes and then blocked (3% bovine serum albumin, 0.1% Tween 20 in PBS) for 1 hour. Cells were incubated overnight at 4°C with primary antibodies (1:75 dilution in blocking solution): anti–translocase of outer mitochondrial membrane 2 (catalog no. ab56783) and anti–trifunctional enzyme subunit α (HADHA; catalog no. ab54477) were from Abcam, and anti-p62 (GP62-C) was purchased from Progen Biotechnik GmbH (Heidelberg, Germany). After incubation, coverslips were incubated with the corresponding secondary antibodies (dilution 1:100) for 1 hour at room temperature and DNA was stained with 4′,6-diamidino-2-phenylindole. For analyzing HADHA clearance by immunofluorescence, we used ImageJ software, version 1.48 (National Institutes of Health, Bethesda, MD); an outline was drawn around each cell. Circularity, area, and mean fluorescence were measured, along with several adjacent background readings. Then, we measured total corrected cellular fluorescence as integrated density − (area of selected cell × mean fluorescence of background readings) as previously described (19). For colocalization analysis, an SP-2 AOBS inverted microscope (Leica) was used. Images were collected by using a 63× Zeiss objective (numerical aperture, 1.4). All images were obtained in a 1.024 × 1.024-pixel format. Images were processed with Coloc2 (plugin for colocalization analysis; Fiji). The threshold was obtained automatically by using Costes automatic threshold, and Manders coefficient was determined (20, 21).
Thermogenic response to cold exposure
For the acute cold exposure experiment, 3- and 12-month-old WT and DKO male littermates that had been acclimatized to thermoneutrality (28°C) for 3 days were transferred to 4°C for 12 hours with full access to water and food. Body temperature was measured periodically by using a digital thermometer with a colonic probe (BIO-9882; Bioseb, Vitrolles, France) (9). Because hypothermic animals could not survive, those animals were withdrawn from the cold exposure at 4 hours; their controls remained cold exposed up to 12 hours. Alternatively, 12-month-old WT or DKO mice were euthanized after 4 hours of cold exposure to obtain plasma and BAT, which were processed for subsequent analysis.
Immunohistochemistry and adipocyte quantification
For UCP-1 immunohistochemistry, inguinal WAT and epididymal WAT sections from 3-month-old mice were incubated with rabbit polyclonal UCP-1 antibody (catalog no. ab10983) at 2 mg/mL in PBS-Tween/1% bovine serum antigen overnight at 4°C. Secondary antibody incubation and development using a diaminobenzidine substrate kit was performed as previously described (4). Freshly isolated brown and white fat depots collected from WT or DKO mice were fixed in 10% formalin for 24 hours and embedded in paraffin for histological analysis. Different sections (5 to 7 µm thick) were deparaffinized and rehydrated to be stained with hematoxylin and eosin. All images were taken at ×20 magnification, and individual adipocyte area and cell number were determined by using image analysis software (ImageJ Launcher, version 1.46). Relative adipocyte size from four animals (WATs) or three [interscapular brown adipose tissue (iBAT)] per group was calculated in square micrometers . Individual adipocyte area was selected with the ImageJ freehand selection tool, and areas of 40 adipocytes of each tissue were quantified in five different sections per mouse. Adipocyte number was calculated by using the ImageJ cell counter plugin, and all adipocytes from at least six different sections of each tissue were quantified per mouse (9).
Metabolic efficiency: energy storage by nuclear magnetic resonance
WT and DKO 3-month-old mice fed with standard diet or mice fed with an HFD for 8 weeks were anesthetized with isoflurane; respiration was continuously monitored, and body fat was measured by using a BioSpin (Bruker), as previously shown (22). The results were represented as fat body volume vs total body volume by using ImageJ software.
Glucose tolerance test, insulin tolerance test, and insulin secretion test
Glucose and insulin tolerance tests were performed on 3- or 12-month-old WT and DKO mice as described elsewhere (23). Mice were fasted the previous day for the glucose tolerance test, and all tests were done in the morning at the same time.
Electron microscopy
BAT samples from three WT or DKO mice were fixed in 4% paraformaldehyde (Electron Microscopy Tech), 2.5% glutaraldehyde grade I (Sigma-Aldrich) in 0.1 M sodium phosphate buffer (pH 7.3) for 4 hours at 4°C. Samples were postfixed in 1% OsO4 (Electron Microscopy Sciences), 1.5% K4 [Fe (CN)6] for 1 hour, dehydrated with acetone, and embedded in epon‐812 (Taab). Thin sections (60 to 70 nm) were obtained with an Ultracut E (Leica) ultramicrotome, stained with lead citrate, and examined under a JEM‐1010 transmission electron microscope (JEOL) in the Electron Microscopy Center at Complutense University of Madrid (Madrid, Spain). For quantification, we measured the number and mitochondria area from at least 18 cells per animal.
Positron emission tomography-computed tomography
Positron emission tomography (PET)/computed tomography (CT) studies were performed in the Brain Cartography Center at Complutense University of Madrid (Madrid, Spain). To assess the iBAT glucose uptake in WT and DKO mice, animals were anesthetized with isoflurane and then were injected intraperitoneally with 11.1 MBq of [18F]-2-fluor-d-2-deoxy-d-glucose (18F-FDG) used as a radioactive tracer. We measured the glucose uptake at room temperature (26°C) or at 4°C. Different images of PET and CT were taken with the hybrid PET/CT scan from Oncovision (Valencia, Spain), and results were analyzed with PMOD software, version 3.0 (Pmod Technologies, Zurich, Sweden). Results are shown as standard uptake values.
Data analysis
All values are expressed as means ± standard error of the mean. Statistical significance was tested with unpaired Student t tests or with one-way or two-way analyses of variance, followed by Tukey tests if differences were noted (Prism 5.0; GraphPad Software, La Jolla, CA). The null hypothesis was rejected when the P value was <0.05.
Results
Brown fat atrophy and mitochondrial damage in DKO vs control mice
Mice carrying floxed Igf1r and Ir alleles were bred with mice carrying a Cre transgene driven by the Ucp1 promoter to generate BATIGFIRDKO mice. Although recombination of the floxed alleles was observed in the DKO vs control mice, a faint expression of IGFIR or IR was detected in the iBAT taken from DKO mice as compared with their littermate controls, probably due to a white fat infiltration into the remnant iBAT tissue. By contrast, IGFIR or IR levels were unchanged in inguinal white fat, gastrocnemius skeletal muscle, heart, and liver (Supplemental Fig. 1). Although collectively body weight was not affected [Fig. 1(a)] and serum IGFI levels were significantly increased (Supplemental Table 1), iBAT mass was severely atrophied in DKO mice at 3 months (remnant brown fat) [Fig. 1(b) and 1(c)]. In the remnant iBAT, we observed a small number of hypertrophic brown adipocytes [Fig. 1(d) and 1(e)] that showed an enhanced lipid content [Fig. 1(b), lower panels] associated with a decrease in ex vivo lipolysis rate [Fig. 1(f)].
![The lack of IGFIR/IR caused severe brown fat atrophy, loss of mitochondrial mass, and mitochondrial cristae disruption. (a) Body weight of male WT (n = 58/52/24/14/23) and BATIGFIRDKO (n = 61/36/15/10/29) mice fed a standard diet. (b) Representative image of interscapular brown adipose tissue depot in 3-month-old WT and DKO mice (upper panels). Hematoxylin and eosin–stained sections of iBAT from 3-month-old WT (n = 3) and DKO (n = 3) mice (lower panels) (magnification, ×20). (c) Graph indicating the iBAT weight/body weight ratio in 3-month-old WT (n = 15) and DKO (n = 19) mice. (d) Brown adipocyte size (square micrometers) from iBAT compartment is shown in 3-month-old WT (n = 3) and DKO (n = 3) mice (200 adipocytes per group at magnification of ×20). (e) Adipocyte number quantification from iBAT compartment comparing 3-month-old WT (n = 3) and DKO (n = 3) mice (six images per group). (f) Ex vivo iBAT lipolysis experiment comparing WT (n = 5) and DKO (n = 3) mice. (g) Representative electron micrographs from iBAT samples of 3-month-old WT (n = 3) and DKO (n = 3) mice [scale bars: 5 μm (upper panels), 0.5 μm (middle panels), and 0.2 μm (bottom panels)]. (h) Number of mitochondria per cell quantification from 3-month-old WT (n = 3) and DKO (n = 3) mice, with 15 cells per animal. (i) Quantification of mean mitochondrial area (square micrometers) from 3-month-old WT (n = 3) and DKO (n = 3) mice, with 15 cells per animal. All results are presented as mean ± standard error of the mean. Statistical significance was assessed by two-tailed Student t test. For WT vs DKO groups: *P < 0.05; **P < 0.01; ***P < 0.001.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/159/1/10.1210_en.2017-00738/1/m_en.2017-00738f1.jpeg?Expires=1716362913&Signature=euSaNYGXXj-em0WWBD25pUZ9XBdnxcb0VALdeF8XV1pnPQpgy9wOkL-mUfgHnpQc2vxpzgPbUel~SVuXgipmxNuh8l77gif~XH6Chm1~FLa1mT6JoLEpb5qaq~BWmIV8KAC2NEU~TQBltJ0DnXq3XM0ByGIFFB~BB5Zx~p7wEtlBmAsxCVslYLU0upd~MJwzYoVt9WG7~qfq52cyHpomcobOdmbee4qBnIf~F~z9KTg6lBUjBX61I0bOyQK6a18b60b9yqen5yH2YF2K2KXR9SHYcXh28~glaFYhct0FYY8DTgXD59lojBh9Aezjv6OQgTQsGldkz31JFNAQEaoFnA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The lack of IGFIR/IR caused severe brown fat atrophy, loss of mitochondrial mass, and mitochondrial cristae disruption. (a) Body weight of male WT (n = 58/52/24/14/23) and BATIGFIRDKO (n = 61/36/15/10/29) mice fed a standard diet. (b) Representative image of interscapular brown adipose tissue depot in 3-month-old WT and DKO mice (upper panels). Hematoxylin and eosin–stained sections of iBAT from 3-month-old WT (n = 3) and DKO (n = 3) mice (lower panels) (magnification, ×20). (c) Graph indicating the iBAT weight/body weight ratio in 3-month-old WT (n = 15) and DKO (n = 19) mice. (d) Brown adipocyte size (square micrometers) from iBAT compartment is shown in 3-month-old WT (n = 3) and DKO (n = 3) mice (200 adipocytes per group at magnification of ×20). (e) Adipocyte number quantification from iBAT compartment comparing 3-month-old WT (n = 3) and DKO (n = 3) mice (six images per group). (f) Ex vivo iBAT lipolysis experiment comparing WT (n = 5) and DKO (n = 3) mice. (g) Representative electron micrographs from iBAT samples of 3-month-old WT (n = 3) and DKO (n = 3) mice [scale bars: 5 μm (upper panels), 0.5 μm (middle panels), and 0.2 μm (bottom panels)]. (h) Number of mitochondria per cell quantification from 3-month-old WT (n = 3) and DKO (n = 3) mice, with 15 cells per animal. (i) Quantification of mean mitochondrial area (square micrometers) from 3-month-old WT (n = 3) and DKO (n = 3) mice, with 15 cells per animal. All results are presented as mean ± standard error of the mean. Statistical significance was assessed by two-tailed Student t test. For WT vs DKO groups: *P < 0.05; **P < 0.01; ***P < 0.001.
Next, we studied mitochondrial biogenesis markers. Thus, messenger RNA (mRNA) expression of Tfam (transcription factor A, mitochondrial) and protein expression of PGC1-α were decreased in remnant iBAT [Fig. 2(b) and 2(c)]. Alteration in mitochondrial biogenesis markers was associated with a decrease in number and size of mitochondria [Fig. 1(h)] and with a severe mitochondrial cristae disruption observed by electron microscopy in DKO mice [Fig. 1(g)]. However, no autophagosome formation was observed in the remnant iBAT tissue bearing damaged mitochondria. Then, we hypothesized a failure in the autophagic/mitophagic process in the atrophic brown fat from DKO mice. Thus, we explored the genetic expression of protein markers potentially involved in those processes in the remnant iBAT. Although expression of LC3B-II was unchanged in DKO mice, we observed a drastic decrease in mitophagy/autophagy ensemble machinery proteins, such as PINK-1, Parkin, and p62. In addition, we observed a decrease in MFN-2, optic atrophy protein 1 L/S ratio, p-DRP1 (Ser616)/dynamin-related protein 1 (DRP1) ratio, and UCP-1. Collectively, our data strongly suggest that the damaged mitochondria observed in remnant iBAT from DKO mice present alterations in mitochondrial dynamics and in thermogenic machinery. In addition, expression of BIP, a reticulum stress pathway marker, and BCL-2, an antiapoptotic marker, were significantly decreased in DKO vs control mice [Fig. 2(a) and 2(b)].
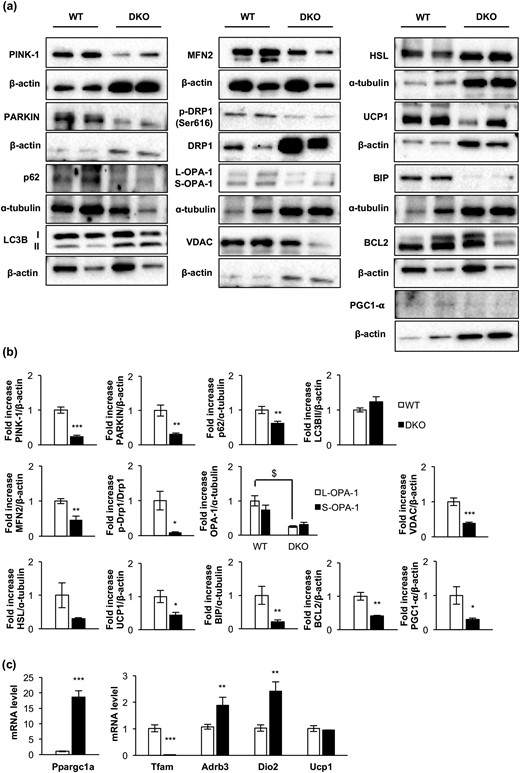
Loss of essential protein components of the cytosolic quality control mechanisms, mitochondrial dynamics, mitochondrial biogenesis, uncoupling mechanisms, endoplasmic reticulum, and apoptosis in DKO mice. (a) Representative Western blots from iBAT showing different regulators of cytosolic quality control mechanism, mitochondrial dynamics and biogenesis, and brown fat functionality between 3-month-old WT and DKO mice. (b) Western blot quantification of BCL2, HSL, and PGC1-α (WT, n = 3; DKO, n = 3); p-Drp1/Drp1, OPA-1 (WT, n = 4; DKO, n = 4); LC3B (WT, n = 6; DKO, n = 5); UCP1, MFN2, and PARKIN (WT, n = 6; DKO, n = 6); PINK1 (WT, n = 6; DKO, n = 7); P62 (WT, n = 7; DKO, n = 6); and BIP (WT, n = 8; DKO, n = 7) and VDAC (WT, n = 8; DKO, n = 8). (c) Plot indicating fold-increased mRNA levels of Dio2 (WT, n = 6; DKO, n = 3); Ppargc1a, Tfam, and Ucp1 (WT, n = 7; DKO, n = 3) and Adrb3 (WT, n = 9; DKO, n = 3) genes in iBAT from 3-month-old mice. All results are presented as mean ± standard error of the mean. Statistical significance assessed by two-tailed Student t test. WT vs DKO groups: *P < 0.05; **P < 0.01; ***P < 0.001. Statistical significance of OPA-1 was assessed by one-way analysis of variance followed by the Tukey test. $P < 0.05 L-OPA-1 between WT and DKO groups. PINK1, phosphatase and tensin homolog–induced putative kinase 1.
Failure of mitophagy in DKO vs control brown preadipocytes
As stated previously, we hypothesized that the autophagic/mitophagic mechanisms failed in the remnant iBAT tissue from DKO mice. To address this important issue, we investigated two kinds of cytosolic quality control mechanisms in control and DKO neonatal brown preadipocytes: First, we investigated the autophagic response, as revealed by the expression of the membrane-bound isoform of L3CB-II in response to 15 hours of treatment with CCCP, a mitochondrial uncoupler, in the absence or presence of CQ, an inhibitor of autophagic flux at the autolysosome formation. Our results showed that the expression of the membrane-bound isoform of L3CB-II in response to 15 hours of CCCP was further increased in the presence of CQ in DKO and in control cells, as quantified in Fig. 3(a). Second, we analyzed HADHA mitochondrial labeling by immunofluorescence in response to CCCP in the presence or absence of CQ. In cells treated with CCCP for 15 to 24 hours, we observed a significant decrease of HADHA (30% of the initial value) in WT vs 60% of the initial value in DKO cells, as quantified in Fig. 3(b). However, we detected a 60% significant increase in HADHA fluorescence signal in WT cells, but not in DKO, in cells treated with CCCP and CQ [Fig. 3(b)]. These data suggested that DKO brown preadipocytes display a specific block at the mitophagic flux. Hence, we explored the mitochondrial labeling for mitophagy initiation in both cell lines. In WT cells, confocal microscopy studies revealed a colocalization of TOM20, a mitochondrial marker, and p62, the mitophagy/autophagy ensemble protein, in response to 4 hours of CCCP treatment. However, in DKO cells, colocalization of TOM20/p62 was significantly inhibited [Fig. 4(b)]. These results suggested a failure in the mitochondrial labeling for mitophagy initiation in the brown preadipocytes lacking IGFIR/IR receptors. Altogether, our results suggested a lower mitophagic flux, without changes in the autophagic flux, in DKO vs control brown preadipocytes.
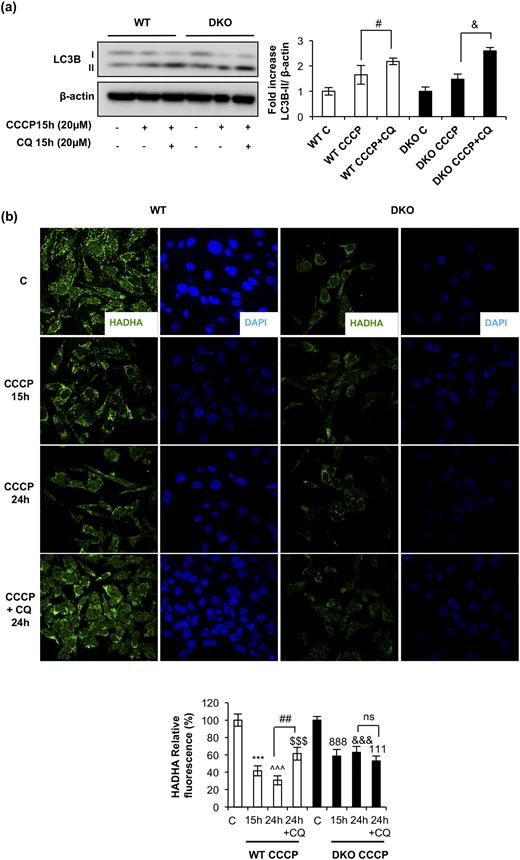
Impaired mitophagic flux in DKO brown preadipocytes. (a) Representative Western blots analyzing LC3B protein levels in response to CCCP for 15 hours in the presence or absence of CQ, comparing brown preadipocytes WT and DKO, with the respective quantitation. Results are presented as mean ± standard error of the mean (SEM). Statistical significance was assessed by two-tailed Student t test; #P < 0.05 between CCCP and CCCP+CQ groups in WT preadipocytes (n = 3) and &P < 0.05 between CCCP and CCCP+CQ groups in DKO preadipocytes (n = 3). (b) Immunofluorescence for HADHA protein from brown preadipocytes WT and DKO in response to the indicated CCCP treatment, followed by CQ stimulation where indicated. All images were taken at ×63 magnification. The relative fluorescence intensity, as detailed in the Materials and Methods section, was quantified and represented in the contiguous graph. Results were represented as mean ± SEM. Statistical significance was assessed by two-tailed Student t test; ***P < 0.001 between WT control and WT with CCCP for 15 hours (n = 4); ^^^P < 0.001 between WT control and WT with CCCP for 24 hours (n = 4); $$$P < 0.001 between WT control and WT with CCCP and CQ for 24 hours (n = 4); ##P < 0.01 between WT with CCCP for 24 hours and WT with CCCP and CQ for 24 hours (n = 4); 888P < 0.001 between DKO control and DKO with CCCP for 15 hours (n = 4); &&&P < 0.001 between DKO control and DKO with CCCP for 24 hours (n = 4); 111P < 0.001 between DKO control and DKO with CCCP and CQ for 24 hours (n = 4); not significant (ns) between DKO with CCCP for 24 hours and DKO with CCCP and CQ for 24 hours (n = 4).
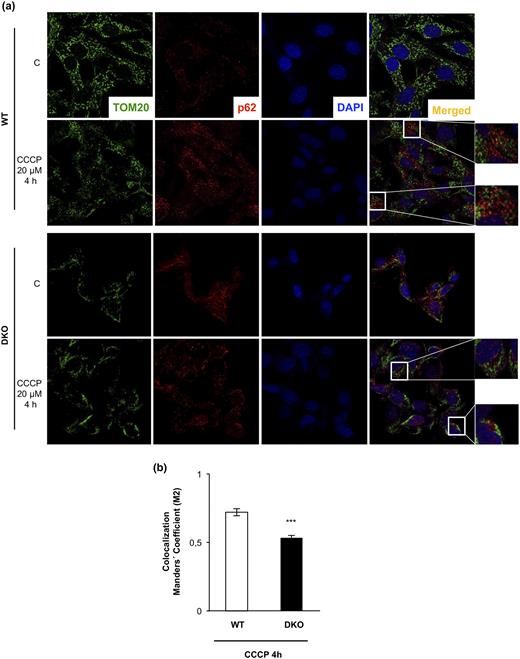
DKO brown preadipocytes showed an impaired p62-TOM20 colocalization upon 4 hour of CCCP treatment. (a) Representative immunofluorescence of TOM20, p62, and 4′,6-diamidino-2-phenylindole (DAPI) from brown preadipocytes obtained from WT or DKO mice under control conditions or CCCP treatment. In the CCCP treatment of both cell lines, two different regions are amplified in the insets. (b) After use of the Coloc2 plug-in (as explained in the Materials and Methods section), colocalization Manders coefficient after CCCP treatment is represented in the graph. Results are presented as mean ± standard error of the mean. Statistical significance was assessed by two-tailed Student t test; ***P < 0.001 between WT (n = 3) and DKO (n = 3) cells treated with CCCP for 4 hours.
Cold-induced thermogenic failure in DKO vs control mice
To assess the metabolic activity of BAT, DKO and control mice were submitted to PET analysis using 18F-FDG as a tracer for measuring the glucose uptake. As depicted in Fig. 5(a), the metabolic activity in brown fat depots increased in response to a cold environment in control mice, as quantified in Fig. 5(b). However, DKO mice failed to enhance their BAT glucose uptake under the same experimental conditions, which is consistent with the deletion of the IR, essential for insulin-dependent glucose uptake in brown fat. Next, we studied the thermogenic functionality. Thus, mice maintained 72 hours at thermoneutrality (28°C) were exposed to cold acclimation at 4°C. At 3 or 12 months, DKO mice showed a severe failure in maintaining their body temperature as compared with their controls. Thus, DKO mice decrease their body temperature around 32°C vs 35°C to 36°C observed in control mice upon 4 hours of cold exposure [Fig. 5(c)]. In this regard, control mice maintained their body temperature at 36°C during the 12 hours they were studied. To assess whether the thermogenic failure observed in DKO mice was related to alterations in hormones that are implicated in BAT activation, we measured T3 and BMP7 plasma levels.
![Impaired brown fat thermogenesis and mitochondrial fission mechanisms upon cold exposure in DKO mice. (a) Representative PET images of iBAT 18F-FDG uptake from 3-month-old WT (n = 2) and DKO (n = 2) mice at room temperature (RT; left panels) or at 4°C (right panels). (b) PET quantification shown as standard uptake value (SUV). Results are presented as mean ± standard error of the mean (SEM). Statistical significance was assessed by two-way analysis of variance (ANOVA), followed by the Tukey test; &&&P < 0.001 between WT RT and DKO RT groups; $$$P < 0.001 between WT 4°C and DKO 4°C groups; and ***P < 0.001 between WT RT and WT 4°C groups. (c) Representative plots of cold-exposure analysis showing the temperature from 0 to 12 hours comparing 3-month-old (WT, n = 8; n = 7 DKO) and 12-month-old (WT, n = 4; DKO, n = 4) mice. Results are presented as mean ± SEM. Statistical significance was assessed by two-way ANOVA, followed by the Tukey test; *P < 0.05 and **P < 0.01 between 3-month-old WT and DKO groups; ##P < 0.01 and ###P < 0.001 between 12-month-old WT and DKO groups. (d) BMP7 plasma levels from 12-month-old WT [control (Ct), n = 8 ; 4 hours (4h), n = 8 ] and DKO (Ct, n = 4; 4h, n = 4) mice. (e) T3 plasma levels from 12-month-old WT (Ct, n = 9; 4h, n = 4) and DKO (Ct, n = 6; 4h, n = 4) mice. (f) Representative Western blots showing different regulators of mitochondrial dynamics and brown fat functionality of iBAT from 12-month-old WT and DKO mice at thermoneutrality (WT, n = 4; DKO, n = 3) or after 4 hours of 4°C exposure (WT, n = 4; DKO, n = 3) and its corresponding quantification. Results are presented as mean ± SEM. Statistical significance was assessed by one-way ANOVA (BMP7, T3 plasma levels, p-Drp1/Drp1, PGC1-α, UCP1, and BMP7) or two-way ANOVA (OPA-1), followed by the Tukey test; *P < 0.05, **P < 0.01, and ***P < 0.001 between WT Ct and WT 4h groups; ##P < 0.01 between DKO Ct and DKO 4h; $P < 0.05 and $$P < 0.01 between WT 4h and DKO 4h; &P < 0.05 between WT Ct and DKO Ct; and ^P < 0.05 between L-OPA-1 WT 4h and S-OPA-1 WT 4h.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/159/1/10.1210_en.2017-00738/1/m_en.2017-00738f5.jpeg?Expires=1716362913&Signature=HCKPrb~6PWaOhjcNfzjIQ4ov6RH1pTz8A00TbS7zZ9AdcnLk7sVxnN7VBQYJ49LMl0eBUC3TTJYXBysp3C9w8GbUrAhSB2oguJPq-Zo9DyAM5oL2tzbuksHfuKaaTxcllsSCoBFLHY~cShDyu8M4G3wCowP6w-2DbL6flSec43d~1bIzcHCj7pbKexL~ucKRYiXS4orJoufB0nV4im4SOlVJz-Wzs3UCFqMuglVFRzhX6p-66e-~tOahtWj1t6LCtreFlFJIFhH42mXgl8aXAFHXqeuVtIZyw5CN77oKkesqZ1Ipl7F-mi8QA1upnnGCUYJPkZ95RgSwb~EiGO5kUA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Impaired brown fat thermogenesis and mitochondrial fission mechanisms upon cold exposure in DKO mice. (a) Representative PET images of iBAT 18F-FDG uptake from 3-month-old WT (n = 2) and DKO (n = 2) mice at room temperature (RT; left panels) or at 4°C (right panels). (b) PET quantification shown as standard uptake value (SUV). Results are presented as mean ± standard error of the mean (SEM). Statistical significance was assessed by two-way analysis of variance (ANOVA), followed by the Tukey test; &&&P < 0.001 between WT RT and DKO RT groups; $$$P < 0.001 between WT 4°C and DKO 4°C groups; and ***P < 0.001 between WT RT and WT 4°C groups. (c) Representative plots of cold-exposure analysis showing the temperature from 0 to 12 hours comparing 3-month-old (WT, n = 8; n = 7 DKO) and 12-month-old (WT, n = 4; DKO, n = 4) mice. Results are presented as mean ± SEM. Statistical significance was assessed by two-way ANOVA, followed by the Tukey test; *P < 0.05 and **P < 0.01 between 3-month-old WT and DKO groups; ##P < 0.01 and ###P < 0.001 between 12-month-old WT and DKO groups. (d) BMP7 plasma levels from 12-month-old WT [control (Ct), n = 8 ; 4 hours (4h), n = 8 ] and DKO (Ct, n = 4; 4h, n = 4) mice. (e) T3 plasma levels from 12-month-old WT (Ct, n = 9; 4h, n = 4) and DKO (Ct, n = 6; 4h, n = 4) mice. (f) Representative Western blots showing different regulators of mitochondrial dynamics and brown fat functionality of iBAT from 12-month-old WT and DKO mice at thermoneutrality (WT, n = 4; DKO, n = 3) or after 4 hours of 4°C exposure (WT, n = 4; DKO, n = 3) and its corresponding quantification. Results are presented as mean ± SEM. Statistical significance was assessed by one-way ANOVA (BMP7, T3 plasma levels, p-Drp1/Drp1, PGC1-α, UCP1, and BMP7) or two-way ANOVA (OPA-1), followed by the Tukey test; *P < 0.05, **P < 0.01, and ***P < 0.001 between WT Ct and WT 4h groups; ##P < 0.01 between DKO Ct and DKO 4h; $P < 0.05 and $$P < 0.01 between WT 4h and DKO 4h; &P < 0.05 between WT Ct and DKO Ct; and ^P < 0.05 between L-OPA-1 WT 4h and S-OPA-1 WT 4h.
Upon 4 hours of cold exposure, a significant increase in T3 plasma levels (a thermogenic hormone) in control, but not in DKO mice, was observed [Fig. 5(e)]. However, we found no significant changes in the mRNA expression of Dio2 deiodinase in DKO vs control mice under the same experimental conditions [Fig. 6(a)]. In DKO mice, plasma levels of BMP7 (a browning signal) increased [Fig. 5(d)] but its protein levels in iBAT did not [Fig. 5(f)], upon 4 hours of cold exposure, suggesting that the rise in circulating BMP7 was likely due to increased secretion from peripheral tissues.
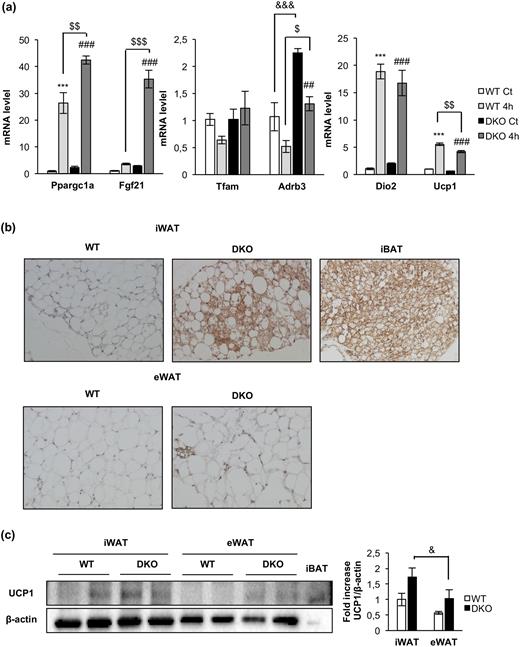
Acute cold-induced gene expression and increased browning in inguinal fat depots in DKO mice. (a) Plot indicating fold-increased mRNA levels of iBAT genes from 12-month-old WT and DKO mice at thermoneutrality (WT, n = 3; DKO, n = 3) or after 4 hours (4h) of 4°C exposure (WT, n = 3; DKO n = 3). Results are presented as mean ± standard error of the mean. Statistical significance was assessed by one-way analysis of variance, followed by the Tukey test; ***P < 0.001 between WT control (Ct) and WT 4h groups; $P < 0.05, $$P < 0.01, and $$$P < 0.001 between WT 4h and DKO 4h; ##P < 0.01 and ###P < 0.001 between DKO Ct and DKO 4h; and &&&P < 0.001 between WT Ct and DKO Ct. (b) Immunohistochemistry for Ucp-1 protein (brown stain) in sections of inguinal WAT (iWAT) and epididymal WAT (eWAT) from 3-month-old WT (n = 3) and DKO (n = 3) mice. iBAT was used as a positive control. All images were taken at magnification of ×20. (c) Representative blots from UCP-1 protein expression in iWAT and eWAT (45 μg of protein was loaded) from 3-month-old WT (n = 4 animals per tissue) and DKO (n = 4 animals per tissue) and its corresponding quantitation. In this blot, 5 μg of iBAT was used as a positive control of UCP-1 protein expression. Results are presented as mean ± standard error of the mean. Statistical significance was assessed by one-way analysis of variance, followed by the Tukey test; &P < 0.05 between iWAT DKO and eWAT DKO groups.
Regarding the molecular mechanisms that may contribute to energy expenditure in brown fat, we explored the triggering of the mitochondrial fission mechanism and the uncoupling status of the mitochondria as revealed by UCP-1 expression. Upon cold exposure, the mitochondrial pro-fission p-DRP1 (Ser616)/DRP1 protein ratio was much higher in control vs DKO mice. In addition, OPA1 protein expression and the mitochondrial pro-fission S-OPA-1/L-OPA-1 protein ratio were higher in control vs DKO mice [Fig. 5(f)]. Both results were significant. Interestingly, PGC1-α (a mitochondrial biogenesis marker) and UCP-1 (an uncoupling marker) were expressed at much higher protein levels in control vs DKO mice [Fig. 5(f)]. In response to 4 hours of cold exposure, expression of these markers was not increased at the protein level [Fig. 5(f)], but both were upregulated at the mRNA transcriptional level [Fig. 6(a)], suggesting a failure in protein synthesis of these genes at posttranscriptional level in DKO vs control mice. No changes in Tfam mRNA expression were found upon cold exposure in control or DKO mice.
Finally, two essentials genes in brown fat functionality, Fgf21 or Adrb3, were studied. Fgf21 showed a very low basal transcription in control mice. Upon 4 hours of cold exposure, a significant increase in the mRNA expression was observed in DKO vs control mice. Expression of Adrb3, the β3 adrenoceptors, markedly increased in DKO vs control mice. However, upon cold exposure, their rates of transcription were downregulated in control and DKO mice [Fig. 6(a)].
Inguinal WAT browning and adipose organ redistribution in DKO vs control mice
We extended our studies on the primary effect of the lack of IR/IGFIR receptors on brown adipocytes to the brown adipocyte resident cells, located within visceral or inguinal fat depots (beige cells) in 3-month-old mice. In this regard, we found more clusters of brown adipocytes within the inguinal fat depots within DKO than in control mice [Fig. 6(b), upper panel]. More important, UCP-1 protein expression was higher in DKO than in control mice [Fig. 6(c)]. However, no brown adipocyte clusters were found (although a faint UCP-1 protein expression was found) in epididymal fat depots from control or DKO mice [Fig. 6(b), lower panel and 6(c)]. Then, we investigated how the thermogenic failure observed in DKO mice affects the adipose organ at different levels. In this regard, we observed a significant increase in the body fat mass in 3-month-old DKO mice, as revealed by nuclear magnetic resonance (NMR) quantification [Fig. 7(a) and 7(b)]. To assess whether this fat increase was related to redistribution among several fat depots, we studied tissue weight, cell size, and number of adipocytes in inguinal, epididymal, retroperitoneal, and mesenteric fat depots. Thus, epididymal, retroperitoneal, and mesenteric fat depots mass were enhanced in DKO vs control mice. In addition, retroperitoneal and mesenteric fat depots showed no changes in cell size and cell number in WT or DKO mice. However, epididymal white depots showed larger cell size and lower cell number in DKO vs control mice. In contrast, inguinal fat mass was diminished, showing larger cell size and lower cell adipocyte number, in DKO vs control mice [Fig. 7(c) and (d)]. However, these alterations did not correlate with changes in plasma levels of leptin, or adiponectin, or TNF-α, in 3-month-old DKO mice (Supplemental Table 1).
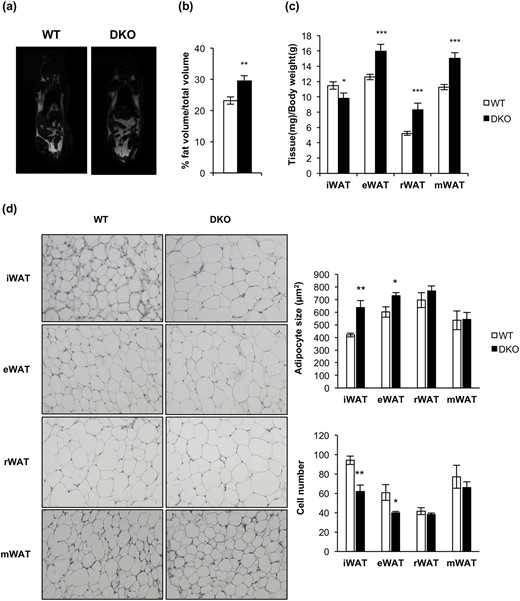
Loss of IGFIR/IR increased obesity susceptibility and induced a redistribution of the adipose organ. (a) Representative images from NMR comparing 3-month-old WT (n = 15) and DKO (n = 9) mice. (b) Quantification of NMR images. The results are presented as the percentage of fat volume per total volume. (c) Graph representing the epididymal WAT (eWAT), inguinal WAT (iWAT), retroperitoneal WAT (rWAT), and mesenteric WAT (mWAT) weights per total body weight comparing 3-month-old WT (n = 18) and DKO (n = 15) mice. (d) Hematoxylin and eosin–stained sections of different WAT depots from 3-month-old WT (n = 3) and DKO (n = 3) mice (magnification, ×20). White adipocyte size (square micrometers) from WAT depots is shown comparing 3-month-old WT (n = 3) and DKO (n = 3) mice (200 adipocytes per group) at magnification of ×20. Adipocyte number quantification from iBAT compartment comparing 3-month-old WT (n = 3) and DKO (n = 3) mice (six images per group). All results were presented as mean ± standard error of the mean. Statistical significance was assessed by two-tailed Student t test; *P < 0.05, **P < 0.01, and ***P < 0.001 between WT and DKO groups.
Metabolic and endocrine status in DKO vs control mice
To assess the glycemic homeostasis and whole-body insulin sensitivity of DKO vs control mice, we studied glucose and insulin tolerance tests. Glucose tolerance remained unchanged upon development in 3- and 12-month-old DKO or control mice [Fig. 8(a) and 8(b)]. These results are consistent with the unchanged postprandial glycemia found in DKO vs control mice (Supplemental Table 1). However, insulin tolerance was impaired at 3 months or 12 months in DKO vs control mice. In fact, insulin secretion tests revealed that the plasma insulin levels were higher in DKO vs control mice [Fig. 8(a) and (b)]. Thus, the loss of insulin sensitivity did not correlate with the unchanged leptin/adiponectin ratio or TNF-α plasma levels observed in DKO vs control mice. In fact, we did not find any correlation between enhanced fat mass and elevation of those cytokines (Supplemental Table 1).
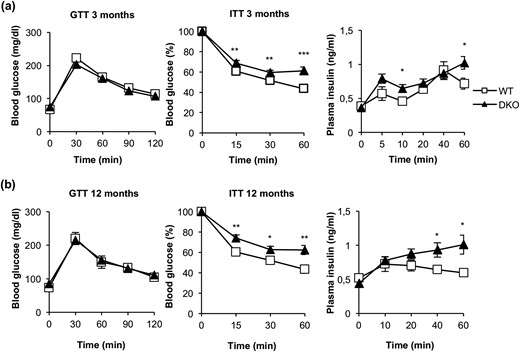
DKO mice showed insulin resistance and moderate hyperinsulinemia. (a) Glucose tolerance test (GTT), insulin tolerance test (ITT), and insulin plasma levels from 3-month-old WT (GTT, n = 22; ITT, n = 23; insulin plasma levels, n = 7) and DKO (GTT, n = 13; ITT, n = 19; insulin plasma levels, n = 8) mice. (b) GTT, ITT, and insulin plasma levels from 12-month-old WT (GTT, n = 9; ITT, n = 10; insulin plasma levels, n = 4 to 7) and DKO (GTT, n = 15; ITT, n = 18; insulin plasma levels, n = 5 to 9) mice. Results are presented as mean ± standard error of the mean. Statistical significance was assessed by two-tailed Student t test; *P < 0.05, **P < 0.01, and ***P < 0.001 between WT and DKO groups.
Effect of HFD on mitochondrial fission, adipose organ function, and metabolic status in DKO mice
On the basis of the severe thermogenic impairment observed in DKO mice upon cold exposure, we hypothesized that high-fat feeding would provoke an energy imbalance in these mice. To address this important issue, we submitted DKO or control mice to an HFD for 8 weeks after weaning. DKO mice weighed on average 20% more than control animals [Fig. 9(b)], without changes in food intake [Fig. 9(d)]. NMR revealed a 70% increase in the body fat mass in DKO vs controls [Fig. 9(c)]. In fact, epididymal, retroperitoneal, and mesenteric fat depots moderately increased in DKO vs control mice, as quantified in Fig. 9(e). However, moderate inguinal fat and a severe decrease of iBAT were observed under the same experimental conditions [Fig. 9(e) and 9(f)]. These results are entirely consistent with the pronounced increase in the plasma leptin/adiponectin ratio under an HFD, without significant changes in circulating TNF-α (Supplemental Table 1). Subsequently, we explored the contribution of the mitochondrial fission mechanisms to brown fat thermogenesis in response to an HFD. The pro-fission p-DRP1/DRP1 protein ratio was slightly lower in DKO than in controls. The S-OPA-1/L-OPA-1 protein ratio increased and the protein level of the pro-fission isoform S-OPA-1 was much lower in DKO vs control mice with HFD, as quantified in Fig. 9(a). Mitochondrial content, as revealed by the expression of the membrane integral protein marker VDAC (porin), was lower in DKO than in control mice, as quantified in Fig. 9(a). Regarding metabolic status, DKO and control mice on an HFD displayed moderate glucose intolerance. However, insulin resistance was more severe in DKO mice than in corresponding controls. An insulin secretion test revealed a significant compensatory insulin secretion upon glucose injection in DKO vs controls [Fig. 9(g–i)]. In fact, DKO mice displayed severe hyperinsulinemia, without changes in their postprandial glycemia (Supplemental Table 1). In addition, we observed significant hypertriglyceridemia in DKO mice, consistent with the very high leptin/adiponectin ratio observed in DKO vs control mice (Supplemental Table 1) and suggestive of a mobilization of lipids from the adipose organ to peripheral tissues.
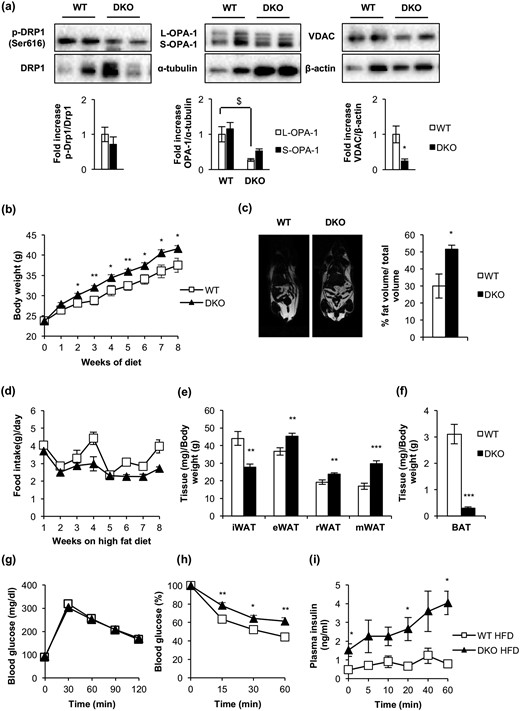
BATIGFIRDKO progressed into a manifest obesity upon consumption of an HFD. (a) Representative Western blots from iBAT showing different regulators of mitochondrial dynamics comparing 3-month-old WT (OPA-1, n = 3; p-Drp1/Drp1 and VDAC, n = 4) and DKO (OPA-1, n = 3; p-Drp1/Drp1 and VDAC, n = 4) mice fed with HFD for 8 weeks and its corresponding quantitation. (b) Body weight of male WT (n = 8) and DKO (n = 13) mice fed with HFD for 8 weeks. (c) Representative images from NMR comparing WT (n = 2) and DKO (n = 3) mice fed with HFD for 8 weeks. Quantification of NMR images is represented as the percentage of fat volume per total volume. (d) Food intake of WT (n = 3) and DKO (n = 3) mice fed with HFD for 8 weeks. (e) Graph representing the epididymal WAT (eWAT), inguinal WAT (iWAT), retroperitoneal WAT (rWAT), and mesenteric WAT (mWAT) weights per total body weight comparing WT (n = 6) and DKO (n = 8) mice fed with HFD for 8 weeks. (f) Graph indicating the iBAT weight/body weight ratio from WT (n = 6) and DKO (n = 8) mice fed with HFD for 8 weeks. (g) Glucose tolerance test from WT (n = 8) and DKO (n = 11) mice fed with HFD for 8 weeks. (h) Insulin tolerance test from WT (n = 9) and DKO (n = 12) mice fed with HFD for 8 weeks. (i) Insulin plasma levels from WT (n = 6) and DKO (n = 5) mice fed with HFD for 8 weeks. Results are presented as mean ± standard error of the mean. Statistical significance was assessed by two-tailed Student t test; *P < 0.05, **P < 0.01, and ***P < 0.001 between WT and DKO groups. Statistical significance of OPA-1 was assessed by one-way analysis of variance, followed by the Tukey test; $P < 0.05 L-OPA-1 between WT and DKO groups.
Discussion
BATIRKO was previously described as a mouse model of brown fat lipoatrophy related to its lipid content depletion. In addition, BATIGFIRKO animals showed a loss of the large fat droplets owing to enhanced HSL expression, and an increased basal rate of lipolysis in the iBAT. However, BATIGFIRDKO mice showed severe brown fat atrophy, which is consistent with the 50% atrophy of skeletal muscles observed in skeletal muscle-specific IGFIR/IR in DKO mice (24) and, in contrast, with normal pancreatic islet development in pancreatic β cells in DKO mice (12). Regarding lipid content, DKO mice showed enhanced lipid content and diminished rate of ex vivo lipolysis in BAT. In this regard, protein kinase A–mediated lipolysis is mediated by the interaction between perilipin-1 and OPA-1 (25). Thus, defects of lipolysis in brown fat of MFN-2 knockout mice have recently been attributed to mitochondria-lipid interaction between MFN-2 and perilipins (26). Both mitochondrial proteins MFN-2 and OPA-1 are poorly expressed in DKO mice, which may contribute to the inhibition of the rate of lipolysis observed in those mice. In addition, the inhibition of the lipolytic rate concurred with a lower HSL expression and hypertrophic brown adipocytes in the remnant brown fat tissue. In this regard, targeted disruption of HSL resulted in brown adipocyte hypertrophy (27).
Ligand activation of the IGFIR or IR induces mTORC1/p70S6 kinase pathway, which negatively regulates the autophagy-signaling pathway. Thus, an upregulation of the formation of autophagosomes and the autophagic flux upon IGFIR blockade might be expected. However, the inhibition of the IGFIR signaling attenuated the autophagosome formation, owing to, at least in part, the reduced formation of the autophagosome precursors at the plasma membrane (28). In the remnant iBAT from DKO mice, we found severe mitochondria cristae disruption concurrently with the loss of expression of MFN-2, as recently described in adipose-specific MFN-2 knockout mice (26), or with the loss of OPA-1, a protein required for mitochondrial cristae assembly (29). However, the complete lack of autophagosome formation in DKO suggests a failure in the mitochondrial quality control mechanisms (mitophagy). To address this important issue, we explored mitophagy in DKO brown preadipocytes. This analysis of DKO revealed a mitophagic flux failure, but not an autophagic failure, in response to an oxidative uncoupler. More important, the mitophagic failure was associated with an impairment of the mitochondrial labeling with p62 for mitophagy initiation, a major mechanism for regulation of mitophagy in mammals (5). Along these lines, p62 has been described as a link between β-adrenergic input and mitochondrial function and thermogenesis (30). In addition, PINK-1 expression was profoundly reduced in DKO cells (as described previously in DKO mice), which is an essential component of the mitochondrial labeling for mitophagy initiation (5), and required for optimal IGFI and insulin-dependent signaling (31).
Cold exposure induces hypothermia and triggers nonshivering thermogenesis. This mechanism, which implies an enhanced metabolic activity by brown fat, is severely impaired in DKO mice, as revealed by PET analysis or cold acclimation. In the case of BATIRKO, we observed enhanced UCP-1 expression. In BATIGFIRKO, however, a loss of UCP-1 expression related to moderate impairment of cold thermogenesis was observed. Our data on DKO mice suggested that brown fat–mediated, nonshivering thermogenesis upon cold exposure or HFD depends on at least two mechanistic components. One mechanism depends on the contribution of UCP-1 to the uncoupling mechanisms, which was impaired in DKO mice at two levels: lower UCP-1 protein content and lower rate of Ucp1 transcription following cold exposure. In this regard, the ablation of Ucp1 has been shown to induce obesity and impair thermogenesis in mice (32). In addition, there was a second mechanism regarding the contribution of the mitochondrial fission machinery to the energy expenditure. That mechanism was visualized by the impaired DRP1 phosphorylation in DKO upon cold exposure. In addition, the balance between L-OPA-1 and S-OPA-1 protein expression mediated by the mitochondrial proteases YME1L and OMA1 leads to mitochondrial fusion or fission (33). Thus, higher protein expression of S-OPA-1 likely triggers mitochondrial fragmentation, mitochondrial uncoupling, and energy expenditure in control mice. This mechanism was impaired in DKO mice.
Regarding transcriptional activity of key thermogenic genes, levels of Adrb3, the target for the sympathetic response, were significantly higher at basal temperature or upon cold exposure in DKO mice, suggesting the existence of a compensatory mechanism in DKO mice. Similarly, genes such as Fgf21 enhanced their rate of transcription in DKO mice upon acute cold exposure. Conversely, genes such as Dio2 or Ucp1 slightly decreased under the same conditions. The rate of transcription of Ppargc1a significantly increased after 4 hours of cold exposure in both DKO and control mice. However, at the protein level, expression of PGC1-α was reduced upon cold exposure in DKO mice. In this regard, the lack of IR in the remnant iBAT may mimic the attenuated insulin signaling observed under caloric restriction status, which may result in the upregulation of glycogen synthase kinase β, targeting PGC1-α for intranuclear proteasomal degradation (34).
It is currently recognized that there are two distinct types of brown fat cells localized either in the canonical brown adipose depot or as resident cells mainly within the inguinal fat. Given the severe brown fat atrophy observed in DKO mice, we explored the presence of beige cells in the epididymal or inguinal fat pads from those mice. Surprisingly, we found more beige cell clusters in DKO mice. Those beige cell clusters were almost missing in our comparative immunohistochemical studies carried out from epididymal fat pads. These results were corroborated by the enhanced UCP-1 protein expression in the inguinal fat in DKO vs control mice. Thus, an enhanced number of beige cell clusters within the inguinal fat pads may be interpreted as a compensatory mechanism to the severe brown fat atrophy observed in DKO mice. Consistent with this, BMP7 plasma levels increased upon cold exposure in the DKO vs control mice. Thus, we cannot exclude the possibility that BMP7 functions as a circulating signal to stimulate beige cells formation in DKO mice.
Whether other browning signals, such as natriuretic peptide (35) or meteorinlike factor (36), may be involved in the compensatory mechanism described previously remains to be established. However, the enhanced presence of beige cells within the inguinal fat did not ameliorate the thermogenic failure observed in DKO mice. These results contrasted with those previously described in BATIGFIRKO mice, where we observed an absence of beige cells formation in inguinal white fat depots (9). These results suggest that only the joint lack of IGFIR/IR signaling in the emerging beige cells within the inguinal fact gives rise to a compensatory signaling. We hypothesized that the impaired brown thermogenesis would enhance susceptibility to increase body fat mass. Indeed, DKO mice had more body fat and displayed a redistribution of the adipose organ as compared with controls. These mice also presented insulin resistance, which was compensated by an increased insulin secretion; thus, these mice had normal glucose tolerance. However, at 3 months, the leptin/adiponectin ratio was slightly increased without changes in circulating TNF-α.
An HFD was used to explore the contribution of the mitochondrial dynamics to brown fat energy expenditure. DKO mice, with their lower mitochondrial content in the brown fat, also displayed a reduction in mitochondrial pro-fission p-DRP1 and S-OPA-1 proteins. Thus, the susceptibility to adiposity observed under the standard diet in the DKO model was manifest as obesity on an HFD, as revealed by the increased body weight, increased body fat mass, insulin resistance, severe hyperinsulinemia, hyperleptinemia, and enhanced hypertriglyceridemia in DKO mice. Collectively, our data suggest a cause and effect relationship between a failure in the brown fat thermogenesis and an increased adiposity and obesity. Consistent with our observations, the ablation of the BAT by means of toxins induces obesity in rodents (37). However, FIGIRKO mice, paradoxically, although bearing severe brown fat atrophy and having impaired thermogenesis, have a lean phenotype and obesity resistance when subjected to HFD (10, 11).
In conclusion, the lack of both IR and IGFIR receptors caused severe brown fat atrophy and mitochondrial damage related to cristae disruption. However, DKO mice increased the number of beige cell clusters observed within the inguinal fat. DKO mice displayed impaired thermogenesis upon cold exposure based on a failure in the mitochondrial fission mechanisms and a much lower UCP-1 transcription rate and content. DKO mice under normal conditions are susceptible as demonstrated by increased body fat mass and insulin resistance. In response to HFD, DKO mice display frank obesity as revealed by increased body weight, increased adiposity, increased circulating leptin, insulin resistance, hyperinsulinemia, and hypertriglyceridemia, all consistent with metabolic syndrome (Fig. 10).
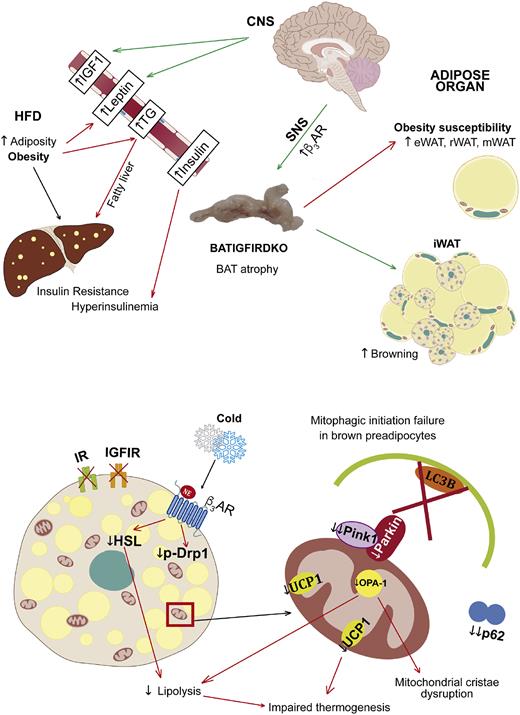
Primary phenotype and the underlying mechanisms involved and secondary phenotype in BATIGFIRDKO mice. Lack of IGFIR/IR caused severe brown fat atrophy and mitochondrial damage related to cristae disruption and also the loss of essential components of the protein machinery involved in the mitochondrial quality control, such as PINK-1, and mitochondrial dynamics, such as MFN-2 and OPA-1. More important, DKO showed impaired brown fat thermogenesis upon cold exposure based on a failure in the mitochondrial fission mechanisms and a much lower UCP1 content. However, DKO mice increased the number of beige cell clusters observed within the inguinal fat. As a result, DKO mice showed obesity susceptibility, as revealed by increased body fat mass and insulin resistance. Upon consumption of an HFD, DKO vs control mice showed manifest obesity, revealed by increased body weight, increased adiposity, insulin resistance, hyperinsulinemia, and hypertriglyceridemia, mobilizing lipids to peripheral tissues as the liver. CNS, central nervous system; SNS, sympathetic nervous system; TG, triglycerides; β3AR, β-3 adrenergic receptor.
Abbreviations:
- 18F-FDG
[18F]-2-fluor-d-2-deoxy-d-glucose
- BAT
brown adipose tissue
- BATIGFIRDKO
insulin receptor/insulinlike growth factor I receptor brown adipose tissue–specific double-knockout
- BATIGFIRKO
insulinlike growth factor receptor in brown adipose tissue knockout
- BATIRKO
brown adipose tissue–specific insulin receptor knockout
- BCL-2
B-cell lymphoma-2
- BIP
immunoglobulin heavy chain-binding protein
- BMP7
bone morphogenetic protein 7
- CCCP
carbonyl cyanide m-chlorophenyl hydrazine
- CQ
chloroquine
- CT
computed tomography
- DKO
double-knockout
- DRP1
dynamin-related protein 1
- HADHA
trifunctional enzyme subunit α
- HFD
high-fat diet
- HSL
hormone-sensitive lipase
- iBAT
interscapular brown adipose tissue
- IGFI
insulinlike growth factor I
- IGFIR
insulinlike growth factor receptor
- IR
insulin receptor
- MFN-2
Mitofusin-2
- mRNA
messenger RNA
- NMR
nuclear magnetic resonance
- OPA1
optic atrophy 1
- Parkin
E3 ubiquitin-protein ligase parkin
- PBS
phosphate-buffered saline
- p-DRP1
phospho–dynamin-related protein 1
- PET
positron emission tomography
- PGC1-α
peroxisome proliferator-activated receptor γ coactivator-1α
- T3
triiodothyronine
- TNF
tumor necrosis factor
- UCP-1
uncoupling protein 1
- WAT
white adipose tissue
- WT
wild-type.
Acknowledgments
The authors thank Deborah Burks for the extensive revision of the manuscript.
Financial Support: Grants SAF2011/22555 and SAF2014-51795-R (to M.B.) from the Ministerio de Economia e Innovación, and CIBER de Diabetes y Enfermedades Metabólicas Asociadas, Institution de Salud Carlos III, Spain supported this work.
Author Contributions: V.V.-H. participated in the design of the study, acquisition of data, analysis and interpretation of data, and statistical analysis and helped to draft the manuscript. C.G., G.G., S.F., and A.G.-A. participated in the acquisition of data, statistical analysis, and interpretation of data. C.R.K. participated in the discussion of the study. M.B. participated in the design of the study and its coordination, analysis and interpretation of data, and writing the manuscript. All authors have read and approved the final manuscript.
Disclosure Summary: The authors have nothing to disclose.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}