





Abstract
As the inflammatory enzyme myeloperoxidase (MPO) is abundant in ruptured human atherosclerotic plaques, we aimed to investigate the role of MPO as a potential diagnostic and therapeutic target for high-risk plaque.
We employed the tandem stenosis model of atherosclerotic plaque instability in apolipoprotein E gene knockout (Apoe−/−) mice. To test the role of MPO, we used Mpo−/−Apoe−/− mice and the 2-thioxanthine MPO inhibitor AZM198. In vivo MPO activity was assessed by liquid chromatography-tandem mass spectrometry detection of 2-chloroethidium generation from hydroethidine and by bis-5HT-DTPA-Gd (MPO-Gd) molecular magnetic resonance imaging (MRI), while plaque phenotype was verified histologically. Myeloperoxidase activity was two-fold greater in plaque with unstable compared with stable phenotype. Genetic deletion of MPO significantly increased fibrous cap thickness, and decreased plaque fibrin and haemosiderin content in plaque with unstable phenotype. AZM198 inhibited MPO activity and it also increased fibrous cap thickness and decreased fibrin and haemosiderin in plaque with unstable phenotype, without affecting lesion monocytes and red blood cell markers or circulating leukocytes and lipids. MPO-Gd MRI demonstrated sustained enhancement of plaque with unstable phenotype on T1-weighted imaging that was two-fold greater than stable plaque and was significantly attenuated by both AZM198 treatment and deletion of the Mpo gene.
Our data implicate MPO in atherosclerotic plaque instability and suggest that non-invasive imaging and pharmacological inhibition of plaque MPO activity hold promise for clinical translation in the management of high-risk coronary artery disease.
Inflammation is a therapeutic target for the prevention of major adverse cardiac events following acute myocardial infarction, and myeloperoxidase (MPO) is an inflammatory enzyme released by neutrophils and macrophages that are abundant at sites of plaque rupture. We demonstrate elevated MPO activity in unstable compared with stable plaque and show that pharmacological inhibition of MPO increases fibrous cap thickness and reduces indices of intraplaque haemorrhage indicative of a more stable plaque phenotype. We also show that MPO activity can be detected non-invasively using molecular imaging, suggesting that MPO represents a potential diagnostic and therapeutic target for the management of high-risk patients.
Introduction
The rupture of vulnerable atherosclerotic plaque is the initiating event for the majority of myocardial infarctions. Culprit lesions are characterized by thin fibrous caps, high inflammatory cell content, and lipid-rich necrotic cores.1 Inflammation is a key driver of plaque rupture as it enhances collagen degradation and impairs fibrous cap formation.2 Myeloperoxidase (MPO) is an inflammatory enzyme abundantly expressed in culprit coronary plaque in fatal cases of acute myocardial infarction.3 Furthermore, circulating concentrations of MPO predict major adverse cardiac events in patients presenting with chest pain and in the post-infarct setting.4–6 Possible mechanistic links between MPO and plaque instability include induction of endothelial cell apoptosis, activation of latent proteinases, and the promotion of pro-coagulant and pro-thrombotic effects.7 However, despite its involvement in numerous pathological pathways and its strong association with major adverse cardiac events, a causal relationship between MPO and plaque rupture has not been demonstrated. This is partially due to the absence of unstable plaque in standard models of murine atherosclerosis including low density lipoprotein receptor (Ldlr− / −) and apolipoprotein E gene-deficient (Apoe− / −) mice.8
The recent development of the ‘tandem stenosis’ (TS) mouse model of plaque instability9 has provided a tool to assess the specific mechanisms contributing to the development of high-risk atherosclerotic plaque and the efficacy of potential therapies targeting such disease. In this model, the right carotid artery of Apoe− / − mice is subjected to low shear and high tensile stress, resulting in plaques with unstable phenotype (hereafter referred to as unstable plaque) that share many features of culprit human lesions.9 Moreover, 2-thioxanthines have been characterized recently as a novel class of effective suicide inhibitors of MPO10 that irreversibly inactivate the enzyme through covalent attachment to the enzyme’s haem prosthetic group as MPO uses hydrogen peroxide (H2O2).10 In the present study, we used this model of plaque instability to evaluate the impact of genetic deletion of Mpo or pharmacological inhibition of MPO using 2-thioxanthine, to assess the relationship between MPO activity and unstable plaque. Myeloperoxidase activity in plaque was evaluated by molecular imaging using the MPO-activatable probe MPO sensor bis-5HT-DTPA-Gd (MPO-Gd)11 and by mass spectrometry.12 The results obtained support a role for MPO as a potential diagnostic and therapeutic target for the identification and stabilization of high-risk plaque.
Methods
All experiments were approved by the relevant Animal Ethics Committees. For expanded methods, please see the Supplementary material online. Numeric data was analysed for normality using the Shapiro–Wilk normality test, the significance determined using the appropriate parametric or non-parametric test, and individual data then shown with mean ± standard deviation (SD).
Results
The recently described TS mouse model of plaque instability9 reflects human atherosclerotic plaque instability including consistent thinning of the fibrous cap with occasional cap disruption and intraplaque haemorrhage. This novel model also represents a discovery tool for the development and testing of therapeutic strategies aimed at preventing plaque rupture.9
Myeloperoxidase activity is higher in plaque with unstable than stable phenotype
MPO-Gd has been applied recently to detect MPO activity in the aortic root of Apoe− / − mice.13 We, therefore, first employed MPO-Gd and T1-weighted turbo spin echo imaging (T1-TSE) (Supplementary material online, Figure S1) to assess MPO activity in unstable and stable atherosclerotic plaques in TS Apoe−/− mice. Time-of-flight angiography showed decreased signal in the right common carotid artery at the site of unstable plaque due to luminal stenosis and decreased arterial flow, whereas the angiographic appearance of the brachiocephalic artery that contained stable plaque was unchanged compared with WT mice (Figure 1A). The latter was ascribed to preservation of blood flow via the subclavian artery, as predicted by computational fluid dynamics, effectively shielding the brachiocephalic artery from alterations in shear stress following TS surgery.9 For contrast-enhanced imaging, 0.3 mmol/kg MPO-Gd or DTPA-Gd was administered intravenously following acquisition of baseline magnetic resonance imaging (MRI) sequences (Supplementary material online, Figure S1B). There was no significant enhancement of the non-diseased vessel wall of the left carotid artery following administration of MPO-Gd (Figure 1B). In contrast, administration of MPO-Gd resulted in enhancement of unstable plaque in the right carotid artery at 30 min after contrast injection (CNR = 23.7 ± 6.8, mean ± SD), which was sustained at 60 min (CNR = 25 ± 6.4) (Figure 1B, right), consistent with activation and retention of this tracer in unstable plaque. Administration of DTPA-Gd, a contrast agent with no molecular specificity, resulted in enhancement of unstable plaque on T1-weighted imaging at 30 min following injection (CNR = 19.2 ± 7.2), which decreased at 60 min (CNR = 13.4 ± 3.6) (Figure 1B), consistent with movement into and out of the extracellular space of atherosclerotic plaque. In unstable plaque, administration of MPO-Gd did not result in significant sustained enhancement of the brachiocephalic artery that contains stable plaque (Figure 1B). Determination of plaque Gd content by nuclear microscopy confirmed significant accumulation of Gd in unstable plaque following administration of MPO-Gd compared with DTPA-Gd (Figure 1C).
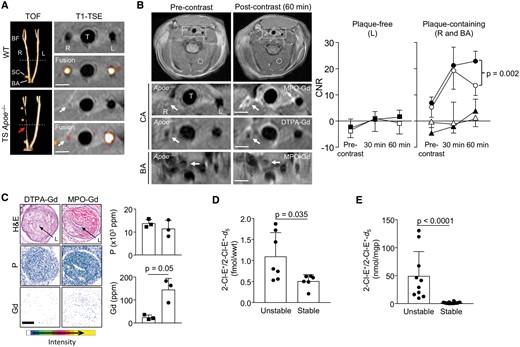
MPO sensor bis-5HT-DTPA-Gd (MPO-Gd) selectively enhances unstable atherosclerotic plaque that contains higher myeloperoxidase (MPO) activity than stable plaque. (A) Time-of-flight angiography (TOF, 3D rendered) of the right (R) and left (L) carotid arteries, the carotid bifurcation (BF), subclavian (SC), and brachiocephalic arteries (BA) in a wild-type (WT) and TS Apoe− / − mouse after 13 weeks of Western Diet (WD), with corresponding (broken line) cross-sectional T1-weighted turbo spin echo (T1-TSE) images. Asterisks indicate the tandem stenosis, while red arrow depicts the site of unstable plaque with reduced signal due to luminal stenosis and reduced blood flow (visualized with TOF/T1-TSE fusion). (B) Pre- and post-MPO-Gd and DTPA-Gd T1-TSE imaging in TS Apoe− / − mice. Representative as-acquired (2-cm field-of-view) MR images and higher-magnification images of unstable plaque (R), plaque-free artery (L) and stable plaque (BA) before and 60 min after probe administration, with corresponding time-course of CNR (MPO-Gd, filled symbols; DTPA-Gd, open symbols) in L (squares), R (circles), and BA (triangles). Broken rectangles and circles indicate regions of interest for vessel wall and skeletal muscle, respectively; Scale bar = 1 mm. Data show mean ± standard deviation of n = 6 (Mpo-Gd) and n = 5 (DTPA-Gd) separate experiments with difference at 60 min post-contrast assessed by unpaired t-test. (C) Quantification of tissue gadolinium (Gd) and phosphorus (P) by nuclear microscopy in unstable plaque following administration of MPO-Gd (circles) or DTPA-Gd (squares). Corresponding H&E sections indicate the arterial lumen (L). (D) In vivo and (E) ex vivo MPO activity in unstable and stable plaque of TS Apoe− / − mice after 13 weeks of WD. MPO activity was assessed by the conversion of hydroethidine to 2-chloroethidium (2-Cl-E+) determined by LC–MS/MS as described in Methods section. Results show individual values of separate experiments with mean + standard deviation.
We next assessed MPO activity by quantifying the MPO-specific conversion of hydroethidine (administered in vivo or ex vivo) to 2-chloroethidium, determined by liquid chromatography with tandem mass spectrometry (LC-MS/MS).12 Myeloperoxidase activity, both in vivo (Figure 1D) and ex vivo (Figure 1E), was significantly higher in unstable than stable plaque. We also attempted to assess MPO activity by determining 3-chlorotyrosine by LC-MS/MS. This MPO-specific adduct could not be detected in unstable plaque of TS Apoe−/− mice as reported previously.14 Overall, the results of the MPO-Gd MRI and hydroethidium LC-MS/MS studies suggest that thresholds of MPO activity measured with non-invasive imaging may be able to delineate unstable from stable plaques.
Genetic deletion of myeloperoxidase increases cap thickness of unstable atherosclerotic plaque
To assess whether MPO contributes to the development of an unstable plaque phenotype, TS surgery was performed in Mpo−/−Apoe−/− mice. MPO-Gd MRI in such TS Mpo−/−Apoe−/− mice showed attenuated enhancement of the right carotid artery in the region of unstable plaque formation compared with Apoe−/− mice at 60 min (Figure 2A), confirming that activation of this tracer is MPO-dependent. We next determined fibrous cap thickness, the best morphological discriminator of plaque vulnerability to rupture.15 We evaluated the thickness of the cap based on its collagen content determined after staining sections with picrosirius red. We also quantified the cap: lesion height ratio as described in Supplementary material online, Figure S2A. Genetic deletion of MPO resulted in a significant 2- and 1.4-fold increase in mean cap thickness and cap: lesion height ratio, respectively, compared with control TS Mpo +/+ Apoe−/− mice (Figure 2 B–D), suggesting that MPO plays a role in cap thinning.
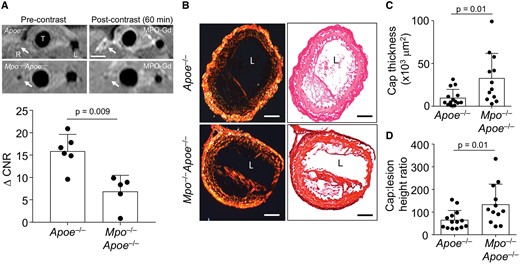
Deletion of myeloperoxidase (MPO) increases fibrous cap thickness in unstable plaque. (A) T1-TSE images of the right (R) and left (L) carotid arteries before and 60 min post-administration of MPO-Gd (0.3 mmol/kg) to TS Apoe− / − and TS Mpo− / −Apoe− / − mice with corresponding ΔCNR data. (B) Representative picrosirius red stained sections of unstable plaque in the right carotid artery viewed under polarised light (left) and bright field (right) from TS Apoe− / − and TS Mpo− / −Apoe− / − mice fed WD for 13 weeks. Scale bar = 100 μm. (C) Average fibrous cap thickness and (D) cap: lesion height ratio in TS Apoe− / − vs. TS Mpo− / −Apoe− / − mice at the end of the 13 weeks study. Data show individual values with mean + standard deviation analysed by Mann–Whitney rank sum test. L, lumen; T, trachea.
Oral administration of AZM198 decreases myeloperoxidase activity and increases cap thickness in unstable atherosclerotic plaque
We next investigated if pharmacological inhibition of MPO enzymatic activity increases cap thickness by administering AZM198, a MPO inhibitor with high potency in cell-free MPO activity assays (IC50 = 0.015 µM) (Supplementary material online, Figure S3),16 to TS Apoe− / − mice. Treatment with AZM198 for the entire 13 weeks duration of intervention had no significant effect on body weight of TS Apoe−/− mice (Supplementary material online, Figure S4A), and at termination resulted in a plasma drug concentration of 2.1 ± 0.25 µM (Supplementary material online, Figure S4B). This concentration is in the range aimed at and required to inhibit extracellular MPO activity by ∼98%.16 AZM198 had no significant effect on circulating total white blood cell, neutrophil, monocyte, eosinophil, or lymphocyte counts (Supplementary material online, Figure S5A–E), nor did it affect plasma lipid concentrations (Supplementary material online, Figure S6A–D). These results suggest that at the dose used, the MPO inhibitor did not have systemic immune-modulatory or lipid-lowering effects. Importantly, AZM198 decreased in vivo MPO activity in unstable plaque five-fold compared with corresponding arterial segments in untreated TS Apoe − / − mice (Figure 3A). AZM198 also significantly decreased MPO activity in unstable plaque assessed ex vivo (Figure 3B). AZM198 had no significant effect on the arterial concentrations of superoxide radical anion (the immediate precursor of H2O2), determined in vivo and ex vivo by the conversion of hydroethidine to 2-hydroxyethidium (Figure 3 C and D). Additionally, AZM198 did not significantly affect the abundance of MPO protein in unstable lesions, as assessed by immunohistochemistry (Figure 3 E and F). However, AZM198 significantly decreased MPO-Gd-mediated enhancement of unstable plaque compared with controls (ΔCNR 18.7 ± 6.1 vs. 6.9 ± 3.1, P = 0.03) (Figure 3G). Together, the data suggest that AZM198 decreases MPO specific activity in unstable plaque.
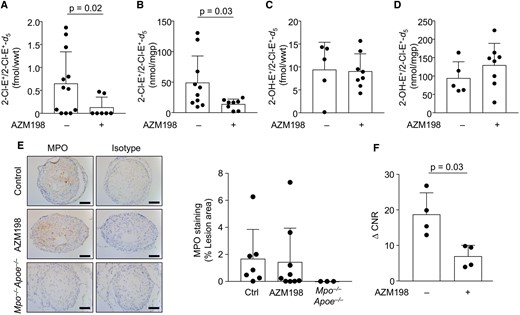
AZM198 treatment decreases myeloperoxidase (MPO) activity in unstable lesions. Unstable plaque from TS Apoe− / − mice fed WD ± AZM198 for 13 weeks was analysed for in vivo (A) and ex vivo 2-chloroethidium (2-Cl-E+) (B) as well as in vivo (C) and ex vivo 2-hydroxyethidium (2-OH-E+) (D) using LC/MS-MS as described in Methods section. (E) Representative IHC of MPO in unstable plaque from TS mice ± AZM198. IHC was performed as described in Methods section, using rabbit IgG isotype and sections from Mpo− / −Apoe− / − mice as controls, with corresponding quantitative data. Scale bar = 100 μm. (F) ΔCNR (CNR60 min − CNRprecontrast) following MPO-Gd T1-TSE. Quantitative data show individual data with mean + standard deviation analysed by Mann–Whitney rank sum test.
AZM198 significantly increased cap thickness and cap: lesion height ratio in unstable plaque compared with untreated controls (Figure 4A–C). Specifically, TS Apoe − / − mice treated with AZM198 had a 65% and 50% increase in fibrous cap thickness and cap: lesion height ratio in unstable plaque, respectively, compared with untreated controls (Figure 4 B and C), without concomitant increases in smooth muscle cells as assessed by smooth muscle actin immunohistochemistry (Supplementary material online, Figure S7). Together, these results suggest that inhibiting MPO in TS Apoe−/− mice promotes a more stable plaque phenotype.
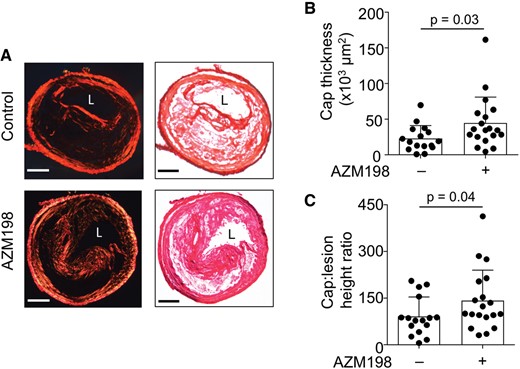
Pharmacological inhibition of myeloperoxidase (MPO) increases fibrous cap thickness in unstable plaque. (A) Representative picrosirius red stained sections of unstable plaque viewed under polarised light (left) and bright field (right) in TS Apoe− / − mice fed WD ± AZM198 for 13 weeks. Scale bar = 100 μm. L, lumen. (B) Fibrous cap thickness and (C) cap: lesion height ratio in TS mice fed WD ± AZM198. Quantitative data show individual data with mean + standard deviation analysed by Mann–Whitney rank sum test.
Genetic deletion and pharmacological inhibition of myeloperoxidase decreases fibrin and haemosiderin in unstable atherosclerotic plaque
An increase in atherosclerotic plaque vascularity and proliferation of the adventitial vasa vasorum are markers of atherosclerotic plaque vulnerability,9 where microrupture and extravasation can cause lesional accumulation of erythrocytes. We did not observe any immunohistochemical evidence for differences in lesion haemoglobin or Ter119, a marker of red blood cells, between control Apoe−/− and either Mpo−/−Apoe−/− or drug-treated Apoe− / − mice 6 weeks after TS surgery (Supplementary material online, Figure S8). However, extravasated haemoglobin is phagocytosed by local macrophages with haemoglobin-derived iron eventually being deposited as haemosiderin. Deletion of the Mpo gene or AZM198 treatment significantly decreased haemosiderin in unstable plaque as assessed by Perl’s Prussian Blue staining compared with control Apoe−/− mice (Figure 5 A and B). Furthermore, deletion of the Mpo gene or AZM198 treatment significantly decreased fibrin in unstable plaque assessed by Martius Scarlett Blue staining compared with control Mpo +/+ Apoe−/− mice (Figure 5 C and D) indicative of reduced fibrin deposition secondary to coagulation and clotting following plaque rupture events. Together, these results suggest that MPO inhibition decreases indicators of intraplaque haemorrhage or prior cap disruption.
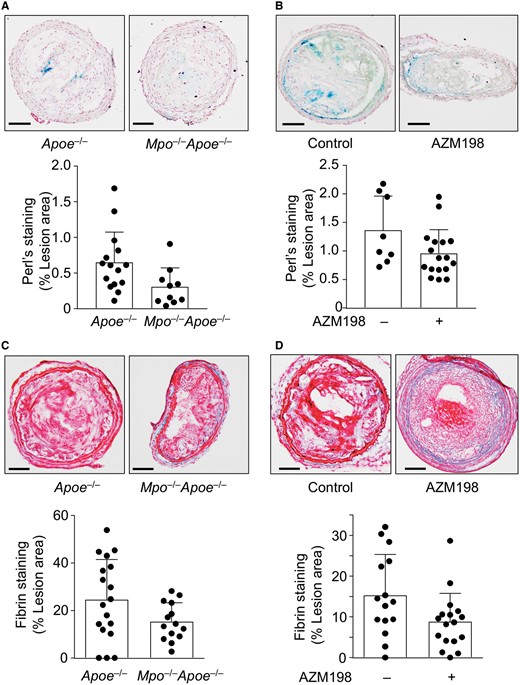
Effect of AZM198 on intraplaque fibrin and haemosiderin. Unstable plaque was assessed by Perl’s Prussian Blue and Martius Scarlet Blue (MSB) staining in TS Mpo− / −Apoe− / − and TS Apoe− / − mice ± AZM198 to detect haemosiderin and fibrin, respectively. (A and B) Representative images of Perl’s staining and quantification of haemosiderin+ area expressed as percentage per total lesion area. (C and D) Representative images of lesion fibrin (bright red stain) by MSB staining with quantification showing percentage fibrin+ area per total lesion area. Quantitative data show individual data with mean + standard deviation analysed by Mann–Whitney rank sum test (A, B, D) and unpaired t-test (C). Scale bar = 100 μm.
AZM198 does not affect the inflammatory cell content or lipid composition in unstable atherosclerotic plaque
Compared with TS Apoe−/− control mice, there was no significant difference in macrophage (Supplementary material online, Figure S9A and C) or monocyte chemoattractant protein-1 (MCP-1) content (Supplementary material online, Figure S9B and D) in TS Mpo−/−Apoe−/− or drug-treated Apoe− / − mice. Similarly, genetic deletion of MPO or its pharmacological inhibition did not change the lesion lipid content as assessed by Oil Red O staining (Supplementary material online, Figure S9E and F). These results indicate that the plaque stabilising effects of AZM198 may not be mediated by a reduction in lipid content or macrophage numbers, although functional modulation of plaque macrophages cannot be excluded.
Discussion
We investigated the role of the inflammatory enzyme MPO in the TS mouse model of atherosclerotic plaque instability. Our data show that MPO activity is significantly increased in unstable compared with stable plaque, and that genetic deletion or pharmacological inhibition of MPO increases fibrous cap thickness and decreases markers of intraplaque haemorrhage, indicative of a more stable plaque phenotype. We also demonstrate that elevated MPO activity in unstable plaque can be detected non-invasively with MPO-Gd MRI and distinguished from stable plaque, highlighting the translational potential of this strategy to improve identification of high-risk disease. Our results implicate elevated MPO activity in experimental unstable atherosclerotic plaque and build upon previous clinical studies showing a strong association between circulating or plaque-specific MPO and adverse prognosis.4
We used three separate experimental approaches to determine MPO activity. The activatable MRI probe, MPO-Gd, has been used previously in preclinical studies to non-invasively demonstrate MPO activity in vivo, including atherosclerotic plaque in the aortic root of Apoe− / − mice.13 Characterization of MPO-Gd has established its specificity for MPO.11 We previously reported that formation of 2-chloroethidium from hydroethidine is specific for the MPO-product hypochlorite12 and has greater sensitivity for MPO activity than measurement of 3-chlorotyrosine for the evaluation of mouse atherosclerotic lesions in vivo and ex vivo.14 The latter can be explained readily on kinetic grounds.12 , 17 Together, the combination of these distinct experimental approaches provide unambiguous evidence for active MPO being present at elevated levels in unstable compared with stable plaque.
Importantly, the MPO inhibitor, AZM198, had no significant effect on tissue concentrations of 2-hydroxyethidium, a specific oxidation product of superoxide,18 which in turn is the common precursor of H2O2. This, together with the absence of overt differences in MPO protein in plaque of drug-treated vs. control mice, implies that the observed decrease in MPO activity in drug-treated mice was likely due to a decrease in the specific activity of MPO rather than the availability of its substrate (H2O2) or expression of MPO protein.
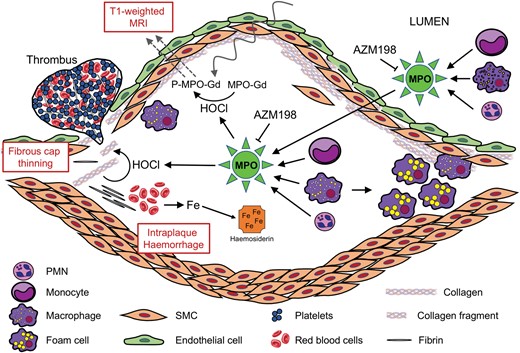
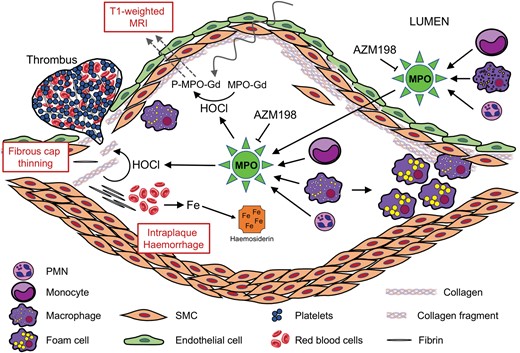
Proposed role of myeloperoxidase (MPO) as a molecular imaging and therapeutic target to identify and stabilize high-risk atherosclerotic plaque. Unstable plaques contain elevated levels of MPO activity and its product, hypochlorous acid (HOCl). Plaque MPO may be derived from MPO released by circulating and/or plaque phagocytes. Increased MPO activity/HOCl can be utilized for non-invasive, T1-weighted magnetic resonance imaging (MRI) using the MPO sensor MPO-Gd, due to its increased arterial retention via linkage to proteins (P-MPO-Gd). MPO/HOCl destabilize plaque by fibrous cap thinning, thereby contributing to increased intraplaque haemorrhage (leading to increased deposition of haemosiderin iron) and thrombotic events. Inhibition of MPO activity by the 2-thioxanthine AZM198 results in plaque stabilization via inhibition of plaque destabilizing activities of MPO/HOCl.
We assessed the impact of genetic deletion or pharmacological inhibition of MPO on atherosclerotic plaque characteristics and used fibrous cap thickness as the primary determinant of plaque stability. This approach has been used previously to demonstrate the plaque-stabilizing effect of statins in humans.19 Our data demonstrate that genetic deletion of MPO in TS mice results in a doubling of fibrous cap, and that partial inhibition of MPO activity with AZM198 increased the size of the fibrous cap by ∼65%. The comparable extents of benefit observed with MPO deficiency and pharmacological MPO inhibition suggest that the plaque stabilising activity of AZM198 represents an on-target effect. Based on previous studies, the observed increase in cap thickness represents a significant shift towards a more stable plaque phenotype.15 The exact mechanisms linking MPO to a decrease in fibrous cap thickness are yet to be fully elucidated. Previous studies have demonstrated that MPO-derived oxidants degrade collagen via activation of latent collagenase20 and metalloproteinase-7, as well as by inactivation of tissue inhibitor of metalloproteinase-1.21 , 22 Therefore, the increase in collagen content in the fibrous cap produced by genetic or pharmacological blockade of MPO may be due to inhibition of collagen degradation at sites of active inflammation. As humans have five-fold higher neutrophil concentrations, a five-fold higher neutrophil MPO content,23 and significantly higher concentrations of circulating and plaque-specific MPO,24 the impact of MPO inhibition on plaque phenotype may be of greater relevance in the clinical setting than in TS mice. Furthermore, as MPO inhibition has been shown to improve myocardial remodelling and function after experimental infarction,25 this therapeutic strategy may be particularly effective in the secondary prevention setting.
The current study demonstrates that genetic deletion and pharmacological inhibition of MPO significantly decrease plaque fibrin content. This indicates either a reduction in contained plaque rupture events or leakage from neovessels that form a microvascular network throughout unstable atherosclerotic plaque. Although we did not observe a significant change in erythrocyte markers, MPO blockade decreased intraplaque haemosiderin, the long-term storage form of iron derived from erythrocytes/haemoglobin. Taken together, a reduction in intraplaque fibrin and haemosiderin provides further evidence that inhibition of MPO promotes stabilization of atherosclerotic plaque.
By investigating the impact of MPO on the development of unstable atherosclerotic plaque phenotypes, the present work significantly differs from previous studies examining the role of MPO on the size of stable atherosclerotic plaque.26 , 27 Brennan et al. 26 reported MPO deficiency to increase lesion size in the aortic root of Ldlr− / − mice but were unable to demonstrate the presence of MPO activity in such lesions. Keliher et al. 13 reported MPO activity in the aortic root of Apoe− / − mice, and we observed a modest decrease in atherosclerosis lesion size at that site in TS Mpo− / −Apoe− / − compared with TS Apoe− / − mice (Supplementary material online, Figure S10A). Therefore, differences in the mouse models used may explain the discordant results. Tiyerili et al. 27 reported 4-aminobenzoic acid hydrazide (ABAH) to decrease atherosclerosis in the aortic root of Apoe− / − mice, whereas we observed no significant effect of AZM198 in TS Apoe− / − mice (Supplementary material online, Figure S10B). In contrast to AZM198, however, ABAH is unlikely to effectively inhibit MPO activity in vivo 28 and Tiyerili et al. did not determine MPO activity. Overall therefore, MPO appears to affect the phenotype rather than the size of atherosclerotic lesions in mouse models of this disease.
The current study has some limitations. Firstly, although the TS model produces atherosclerotic plaque with an unstable phenotype that approximates human disease, some features are distinct, including the small vessel calibre. Secondly, although fibrous cap disruption is a definitive indicator of plaque vulnerability, we used cap thickness as our primary endpoint as it is quantified more reliably than cap disruption on histological analysis of these small calibre vessels. Finally, AZM198 was administered concurrently with Western Diet and, as such, MPO inhibition may prevent the development of unstable plaque rather than stabilising established advanced disease. Future studies will need to address if late inhibition of MPO promotes transformation of unstable to stable plaque phenotypes.
We show that molecular MRI with MPO-Gd can non-invasively discriminate increased MPO activity in unstable plaque and thus has the potential to be a non-invasive marker of plaque vulnerability. Previous studies have demonstrated that the presence of certain high-risk plaque characteristics on coronary imaging can be prognostically significant,29 , 30 however such evaluation is not currently used for the routine assessment of coronary artery disease. This may be due to the absence of a clinically validated, non-invasive diagnostic tool for the reliable identification of inflamed unstable plaque. An imaging strategy using MPO-Gd or a related MPO sensor has several potential advantages. Firstly, such a tracer would be effective for coronary imaging, as it is not constrained by background uptake in the surrounding myocardium. Secondly, as the tracer is enzymatically activated, it provides a direct index of disease activity and thus may have utility in identifying high-risk plaque. Finally, unlike other contrast agents used to image atherosclerosis, MPO-Gd allows for the direct localization and quantification of a potential therapeutic target, which could be used to guide administration of MPO inhibitors or other therapies that modulate inflammation.
In conclusion, our data implicate MPO in fibrous cap thinning and intraplaque haemorrhage, both features of plaque instability, and suggest it is a target for pharmacological intervention warranting further evaluation. Our data also indicate that non-invasive imaging of plaque MPO activity may be useful for both the identification of high-risk unstable plaque and for the evaluation of novel therapies targeting vascular inflammation. This may provide a novel approach to identify and treat patients at risk of developing acute coronary events.
Acknowledgements
We thank Prof RM Graham for critically reading the manuscript and Mr Brendan Lee for providing technical support for imaging experiments.
Funding
This work was supported by National Health and Medical Research Council (NHMRC) of Australia [1060804 to R.S. and A.J.K., Program 1052616 to R.S.], the Australian Research Council [Discovery Project 170101453 to R.S.], and a St Vincent’s Clinic Foundation to R.S., I.R., and A.J. Y.C.C. was supported by a fellowship of the National Heart Foundation of Australia. K.P. and R.S. are supported by NHMRC Fellowships. We also acknowledge infrastructure support from New South Wales Health, the Victorian State Government and the Australian National Imaging Facility.
Conflict of interest: none declared.
Footnotes
See page 3311 for the editorial comment on this article (doi: )
References
Author notes
Imran Rashid, Ghassan J. Maghzal and Yung-Chih Chen authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}