





Abstract
Aims Recent data from the COMPANION trial have documented that cardiac resynchronization therapy (CRT) with biventricular (BiV) pacing reduces mortality and hospitalization in patients with advanced CHF, but little is known regarding the cellular and molecular mechanisms of CRT. Our aim is to evaluate interstitial remodelling, tumor necrosis factor-α (TNF-α) expression, and apoptosis in patients with advanced CHF treated with CRT.
Methods and results We performed endomyocardial biopsies in 10 patients, aged 62, with dilated cardiomyopathy before and 6 months after the implantation of a BiV pacing device. Clinical status and left ventricular (LV) architecture and function were assessed as well as myocardial histology, TNF-α expression, and apoptotic index. CRT improved clinical status, as shown by a significant reduction of the Minnesota living with heart failure questionnaire (MLHFQ) score (from 53 to 40) and 6-min walked distance (from 290 to 330 m) (all P<0.05 vs. baseline). This was associated with reverse LV remodelling substantiated by significant reductions of LV volumes and end-systolic circumferential wall stress. Examination of myocardial tissue revealed a significant decrease of collagen volume fraction (CVF) (from 25.16 to 18.0%), TNF-α expression (from 9.5 to 3.6 pixel×103), and apoptotic index (from 2030 to 1408 apoptotic nuclei/106), with increased capillary density (from 1801 to 2011 capillary/mm2) after 6 months of CRT (all P<0.05 vs. baseline). Moreover, changes in TNF-α expression were positively correlated with both CVF and end-systolic circumferential wall stress (r=0.80 and 0.70, respectively).
Conclusion We provide the first evidence that CRT reduces interstitial remodelling, TNF-α expression, and apoptosis. The data may explain the beneficial effects of CRT on CHF progression and survival.
Cardiac resynchronization therapy (CRT) through biventricular (BiV) pacing represents a novel tool in the armamentarium of therapies available to patients with chronic heart failure (CHF) who remain symptomatic despite optimized standard therapies.1 Several studies have demonstrated that, by correcting the intra-, inter-, and atrioventricular dyssynchrony, CRT improves a wide array of clinical parameters, including maximal oxygen uptake, 6-min walk test, New York Heart Associstion (NYHA) class, and quality of life indexes, and partially reverses left ventricular (LV) remodelling.2–6 Very recently, data from the COMPANION trial have documented that CRT plus implantable defibrillator reduce major clinical endpoints including mortality and mortality plus hospitalization in a heart failure population.7 Despite such evidence may lead to a widespread use of implantable devices, the cellular and molecular mechanisms of BiV pacing are largely unknown.
In this regard, researchers have identified several molecular and cellular markers of heart failure progression.8,9 Chief among these are interstitial fibrosis, probably the best marker of heart failure, activation of the cytokine system, particularly tumor necrosis factor-α (TNF-α), and cell loss due to apoptotic death.8,9
Accordingly, the current study was aimed at evaluating interstitial remodelling, TNF-α expression, and apoptotic death in myocardial biopsies from 10 patients at baseline and 6 months after the implantation of a BiV pacing device, as well as indexes of clinical status and LV chamber remodelling. We herein demonstrate that BiV pacing reduces interstitial remodelling, TNF-α expression, and apoptotic death in CHF.
Methods
Study protocol
Ten patients with dilated cardiomyopathy were enrolled from March 2001 to December 2003. These patients were consecutively collected from a series of 28 patients, the inclusion criteria were as follows: symptomatic CHF NYHA class III or IV despite optimized therapy; stable medications for at least 1 month, β-blockade was started at least 5 months before entering the study; LV ejection fraction ≤35% and LV end-diastolic dimension ≥60; QRS duration of ≥150 ms and left bundle branch block in sinus rhythm; and informed consent. Exclusion criteria were unstable angina or recent MI, second or third-degree atrioventricular block, systolic blood pressure (BP) >170 and <80, heart rate >140, and serum creatinine levels >2.5 mg/dL.
During the enrolment period, we have implanted a total of 28 patients. The main reasons why not all implanted patients entered the study were unwillingness to participate in an experimental protocol (n=7), failure of implantation (n=1), atrial fibrillation (n=4), chronic renal failure (n=2), recent MI (n=4).
Written informed consent was obtained from all patients, and the study protocol was approved by the Ethics Committee of the Federico II University.
Patients were evaluated with the following tests: 12-lead scalar electrocardiogram, NYHA class, the peak VO2 by cardiopulmonary stress test, body weight, systolic and diastolic BPs, Doppler echocardiography, quality of life, before (pre-CRT) and 6 months after (post-CRT) the implantation of a BiV pacing device. Heart rate was calculated from the ECG, and systolic and diastolic BPs were measured by a cuff manometer after 15 min of supine rest. The QRS duration was measured during BiV pacing and during spontaneous rhythm. Quality of life was evaluated with the MLHFQ, whereas the 6-min walk test was carried out according to Guyatt's recommendation.
Echocardiography
An ultrasound system equipped with a 2.5 MHz transducer (5500 Agilent Technologies, Andover, MA, USA) was used for complete M-mode and two-dimensional echocardiographic analysis. M-mode and two-dimensional recordings were made with the patients in the lateral recumbent position, according to the American Society of Echocardiography recommendations.10 The investigator reading the echoes was blinded to the study protocol. The methods are described in detail elsewhere.11 Measures of LV architecture and function were assessed according to the standard formulae. Biplane LV end-systolic and end-diastolic volumes were calculated from apical views, according to the modified Simpson's rule.12 Circumferential wall stress was calculated by two-dimensional echocardiography according to a previously validated formula.13 Area of mitral regurgitant jet was measured as previously described.2–6 As inter- and intra-ventricular dyssynchrony indexes, the following parameters were assessed: time from the onset of QRS to the onset of aortic and pulmonary flow (Q-Ao and Q-Pulm, respectively), the difference between Q-Ao and Q-Ap or the interventricular electromechanical delay (IMD), the delay between the initial inward septal and the inferolateral motion was identified by M-mode echocardiography.6
Cardiopulmonary exercise test
Symptom-limited cardiopulmonary exercise testing (Treadmills ‘Rammill Series’, Morgan Italia, Bologna, Italy) was performed, according to the Cornell-modified multistage treadmill protocol (2 min step increments) with a commercially available equipment (Benchmark Exercise Test System, Morgan Italia, Bologna, Italy). Measurements of oxygen consumption (VO2), were taken at rest and during exercise using a moving average of eight breaths. During each stage of exercise, data on heart rate and rhythm and BP were collected. All patients were encouraged to exercise until they felt unable to continue because of dyspnoea and/or fatigue. The maximum VO2 was defined as the highest VO2 value measured (peak VO2).
Pacemaker implantation
Patients were implanted with a BiV pacemaker (Contak TR or TR2, Guidant Inc.), and five received a BiV cardioverter-defibrillator (Contak Renewal 1 or 2, Guidant Inc.). LV pacing was obtained transvenously. Briefly, after coronary sinus angiography, a unipolar or bipolar lead with an over-the-wire system (Easytrak 1 or 2, Guidant Inc.) was advanced into the lateral or posterolateral cardiac vein. The final position was chosen on the basis of visual inspection and validated on the basis of both the best LV ventricular stimulation threshold and the best LV amplitude signal. The BiV pacing mode was programmed in DDD, and the lower rate was set at 40 b.p.m. The atrioventricular interval was optimized for maximal diastolic filling by Doppler echocardiography.
Myocardial tissue examination
In all patients, a myocardial perfusion scintigram at rest was obtained in order to ascertain whether perfusion defects were present at the septal–apical region of the left ventricle. Three to four endomyocardial biopsies ≈3 mm3 were collected from the septal–apical region of the left ventricle with a Pilling Weck bioptome (2.2 mm) under fluoroscopic and echocardiographic guidance. Biopsies were immediately immersed in buffered formalin 4% and embedded in paraffin, as assessed in previous pilot experiments on papillary muscle samples (unpublished results). Sections were observed with a Nikon Eclipse 1000 photomicroscope and analysed with Jandel SigmaScan Pro software. The following parameters were evaluated: cardiomyocyte diameter, collagen volume fraction (CVF), capillary density, immunodetection of TNF-α expression, and apoptotic index.
For cellular diameter determination, sections were stained with haematoxylin and eosin method, and only myocytes with a round nuclear profile were measured. Thirty myocytes were counted per slide.14,15 Sections were stained with Picrosirius Red method and total extracellular collagen content was determined by a semiquantitative analysis of the tissue area stained. CVF was expressed as percentage of connective tissue areas divided by total tissue area in the same field. Six low-power microscopic fields were analysed.16–18 Capillary density was taken as the number of capillaries in a given microscopic field (expressed as number of capillaries/mm2).
Sections were then immunostained with polyclonal anti TNF-α antibody (Endogen, Woburn, MA, USA) at a 1:50 dilution enhanced with a peroxidase conjugated avidin biotin kit (Pierce, Rockford IL, USA). Sections were also counterstained with haematoxylin. Four fields were counted for a semiquantitative evaluation of stained areas.19,20
Apoptosis was measured with in situ ligation of hairpin probe with single-base 3′ overhangs method (Apoptag ISOL kit, Intergen Co.),14–16 utilizing diaminobenzidine as cromogen. Sections were immediately processed for an anti-activated caspase-3 immunostaining. Polyclonal anti-caspase-3 (M30, Boheringer) was utilized at 1:300 dilution, enhanced with a phosphatase conjugated avidin biotin kit and revealed with fast red (Pierce). Positive and negative controls were performed, and apoptotic index was calculated as previously described.16 The number of myocyte nuclei labelled by hairpin probe divided by the numerical density of myocyte nuclei represents the apoptotic index.
Statistical analysis
Data normality was evaluated through the Kolmogorov–Smirnov test. Because not all the variables assume a normal distribution, comparisons before and after CRT were performed by using the two-sided Wilcoxon matched pair test. Type 1 error was estimated by 95% confidence intervals test. Spearmann correlation was used as appropriate. All values are given as median and range. Sample size calculation was not performed in current pilot study. A value of P<0.05 was considered significant. Statistical analysis was performed using the Sigma Stat 3.1 package.
Results
Data reported were obtained from 10 patients, whose demographics are shown in Table 1. Medication was not changed over the follow-up period. The infarct location, in four patients with post-ischaemic heart failure, was inferolateral (two) and infero-apical (two). Clinical status improved after pacemaker implantation as substantiated by a variety of indexes (Table 2). One patient passed form 4° NYHA class to 3° and five patients passed from 3° class to 2° NYHA class. As showed in detail in Table 2, quality of life index significantly decreased from 53 to 40 (P=0.002) and this was paralleled by an increased walk distance in 6 min (from 290 to 330 m, P=0.002) and total exercise time (from 530 to 621 s, P=0.002). Evidence of wall motion resynchronization was provided by a significant decrease of interventricular electromechanical and septal-to-posterior wall motion delays from 82 to 19 ms (P=0.005) and from 181 to 16 ms (P=0.002), respectively. Echocardiographic data revealed a significant decrease of end-systolic volume index from 97 to 80 mL/m2 (P=0.006), an increase of ejection fraction (from 22 to 25%, P=0.002), and a reduction of mitral regurgitant jet (from 7 to 4.7 cm2, P=0.002).
Histological examination of myocardial tissue (Figure 1) showed a significant decrease of CVF (from 25.16 to 18.0%, P=0.002), an increase of capillary density (from 1801 to 2011 capillary/mm2, P=0.004), and no significant changes of myocyte diameter, compared with baseline values. Moreover, apoptotic death significantly decreased (from 2030 to 1408 apoptotic nuclei/106P=0.001) and also TNF-α expression decreased from 9.5 to 3.6 pixel number×103 (P=0.001) (Figure 2). A significant correlation was found between CVF and per cent reduction of TNF-α expression (r=0.73; P=0.014). Moreover, the per cent reduction of TNF-α expression was also correlated with the per cent reduction of end-systolic stress (r=0.68; P=0.046). All histological parameters, except TNF-α values, were also normally distributed. Two patients were considered non-responders according to an end-systolic volume decrease <15% from baseline value; however, they were included in the study.
Discussion
The current study provides the first evidence that CRT reduces interstitial remodelling, apoptotic death, and TNF-α expression. Therefore, in addition to the well-described improvements of clinical status, CRT impacts on fundamental cellular and molecular determinants of heart failure progression, which may in turn underlie the recently reported survival benefit.
In our patient population, CRT induced a remarkable attenuation of the typical interstitial remodelling observed in CHF with a significant reduction of CVF and a significant increase of capillary density. Myocardial fibrosis, which qualifies an increased collagen concentration, represents a hallmark and a major prognostic determinant of cardiac decompensation.8 It is associated with enhanced stiffness and electrical heterogeneity with attendant diastolic dysfunction and arrhythmias, both contributing to systolic impairment.Previous data obtained in animal models of heart failure have shown that a two to three-fold increase of CVF in the left ventricle is associated with diastolic dysfunction, whereas a further increase leads to combined diastolic and systolic dysfunction with clinical signs of congestive heart failure.21 Thus, our findings of reduced CVF and increased capillary density may lead to improved LV filling dynamics and systolic function, reduced arrhythmias, and improved oxygen diffusion capacity.
Several lines of experimental evidence suggest that the TNF system is part of a portfolio of biologically active molecules that contribute to the progression of heart failure independent of the haemodynamic status by virtue of direct toxic effects on the heart and the vasculature.22 In fact, TNF-α, which is not constitutively present in the normal myocardium but is activated by mechanical stretch,23 provokes a hypertrophic growth response in cultured adult cardiac myocyte, participates in the extracellular matrix remodelling, and triggers myocyte apoptosis.22,23 Moreover, transgenes overexpressing myocardial TNF-α or rats submitted to high dose of exogenous TNF-α develop cardiac hypertrophy, fibrosis, and manifest the clinical phenotype of CHF.24
In addition to replacement and reactive fibrosis and the activation of the cytokine system, cardiomyocyte loss has been suggested as an important causative factor in the development of heart failure.25 Although controversy still exists as to the true incidence of apoptosis, particularly in patients with moderate-to-severe heart failure,8,9,25 recent evidence has proven that myocyte apoptosis may be a casual mechanism of heart failure, contributing to ventricular remodelling and CHF progression.26 Apoptotic death could indeed result in a suboptimal chamber thickness-to-volume ratio contributing to continued increase in wall stress and LV dilation during the progression of CHF. Thus, our findings of a significant reduction of ISOL and caspase-3 positive cardiomyocyte point to an overall reduction of cardiomyocyte loss that may have contributed to the reverse remodelling.
Taken together, our patients exhibited evidence of improved clinical status and LV reverse remodelling which were associated with significant attenuation of interstitial fibrosis and remarkable reduction of apoptotic death and TNF-α expression. Considering that heart failure is a syndrome characterized by multiple anatomic, functional, and biological alterations interacting in a complex manner,9 it is difficult to speculate on the precise pathophysiological mechanisms underlying the observed findings. However, the hypothesis can be put forward that the rapid reduction of LV dyssynchrony due to CRT decreases the overall mechanical stretch on the LV myocardium and in turn reduces the stretch-induced up-regulation of signalling pathways favouring LV remodelling including the TNF and the apoptotic systems. This partial LV unloading may lead to reduced collagen deposition in the interstitial space and increased capillary density with beneficial architectural and functional changes. This interpretation is supported by the significant direct correlation found between TNF-α expression and circumferential end-systolic stress reduction.
In this regard, several lines of experimental evidence support the concept that the mechanical stress is a major trigger of myocardial growth and remodelling and is indeed capable of modifying the expression of a plethora of genes involved in myocardial growth and remodelling (e.g. TNF-α, apoptotic cascade, etc.), either directly or indirectly through activation of myocardial autocrine/paracrine mechanisms.8 Also well documented is that patients with contractile discoordination in addition to the elevated LV parietal stress typical of dilated, poorly contracting ventricles, have a further increase of mechanical stretch secondary to internal sloshing from early to late-activated LV regions resulting in a rightward shift of the end-systolic pressure–volume point, higher end-systolic stress, and worsened function.1 In this regard, the Kass and coworkers27 have recently demonstrated that the late-activated high-stress lateral endocardium with LV dyssynchrony generates protein dysregulation with transmural and transchamber expression gradients of calcium handling and gap junction proteins that impair function and increase arrhythmia susceptibility. However, because in the current study the biopsies were taken from the unloaded septo-apical region and not from the reloaded lateral wall, although contract at high stress, our findings of reduced fibrosis may depend upon a global reduction of LV volume and stretch. Alternatively, one may argue that the passive stretch of the septum during the late systole may also precipitate fibrosis and that this is reversed by CTR.
Very few studies have addressed the reversibility of defects in myocardial biology in the setting of CHF. The most widely employed clinical setting includes patients waiting for heart transplants and supported by an LV-assist device. Such unloaded hearts exhibit a dramatic reduction of fibrosis, TNF-α myocardial content, cellular hypertrophy, and improvements of myocyte contractile properties, and β-adrenergic responsiveness.18,20 Also relevant to our investigations are the classic studies by Krayenbuehl et al.28 in patients with aortic valve disease and by Brilla et al.17 in hypertensive heart disease, both performed using LV biopsies. Specifically, the regression of fibrosis averaged 25% in nine patients with aortic stenosis 70 months after valve replacement,28 whereas in a group of hypertensive patients, 6 months of therapy with lisinopril reduced CVF by ∼9%.17 This is congruent with our findings of more marked reduction of TNF-α expression than CVF, in keeping also with the half-life of fibrillar collagen.
An obvious limitation of the current study is the small sample size and the lack of a control group, essentially due to its invasive nature. LV endomyocardial biopsies represent only a small part of the LV. However, we tried to avoid areas of gross reparative fibrosis by identifying scar tissue in the septal–apical region with echocardiography and perfusion scintigrams. Furthermore, scar tissue presents with a typical histological appearance characterized by a three-layered structure composed by high percentage, ≈40%, of collagen type I, whereas reactive fibrosis represents a diffuse accumulation of fibrillar collagen within the cardiac interstitium.8 The limited amount of myocardial tissue per biopsy hampered a more extensive assessment of myocardial fibrosis and apoptotic death using biochemical and/or molecular markers including hydroxyproline concentration or DNA laddering. However, several studies have shown an excellent correlation between morphometrically and biochemically detected CVF in several model systems and that changes in hydroxyproline concentration are even more marked than changes in Picrosirius Red staining.16 Other study limitations include (i) the site where the biopsies were taken; in fact, the study consider only specimens from apical septum, as LV-free wall biopsies are not routinely taken as the risk of wall rupture; (ii) the presence of four post-ischaemic patients. Furthermore, it must be stressed that this is one of the few clinical investigations examining tissue samples from patients with moderate-to-severe heart failure, whereas most previous data were obtained from patients with end-stage disease candidate to heart transplant, generally submitted to intravenous inotropic drugs which possess an intrinsinc myocardial toxicity and may therefore directly alter tissue architecture.
Conclusion
In conclusion, this study demonstrates that CRT not only reverses chamber LV remodelling, but also has a profound impact on the molecular and cellular biology of the failing myocardium, similar to that showed by beta-blockers and converting enzyme inhibitors.
Conflict of interest: none declared.
These authors contributed equally to the study.
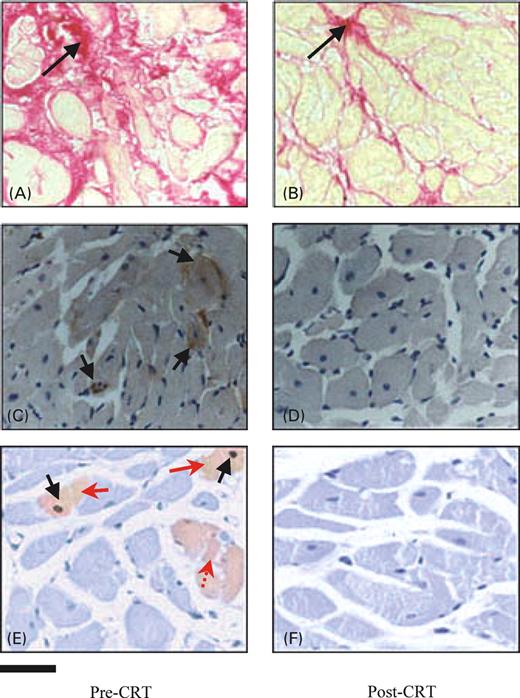
Figure 1 Representative histological images of myocardial tissue sampled before (A, C, and E) and after (B, D, and F) 6 months of CRT. (A) and (B) depict CVF stained with Picrosirius Red method. Black arrows indicate collagen deposition (in magenta or red). (C) and (D) show TNF-α immunoreaction before (pre-CRT) and after (post-CRT) 6 months of CRT, respectively. Note that immunohistochemical signal (brownish cells with black arrows) is present only before CRT. After 6 months, there are no or few scattered cells TNF-α immunopositive. (E) and (F) represent a double reaction for apoptosis detection. Black arrows show apoptotic nuclei marked with DAB by hairpin probe, whereas red arrows show immunoreaction for activated caspase-3, marked in light red by fast red. Note that in (E), there is a cell (red arrow, dotted) immunopositive only for caspase-3. Bar=20 µm.
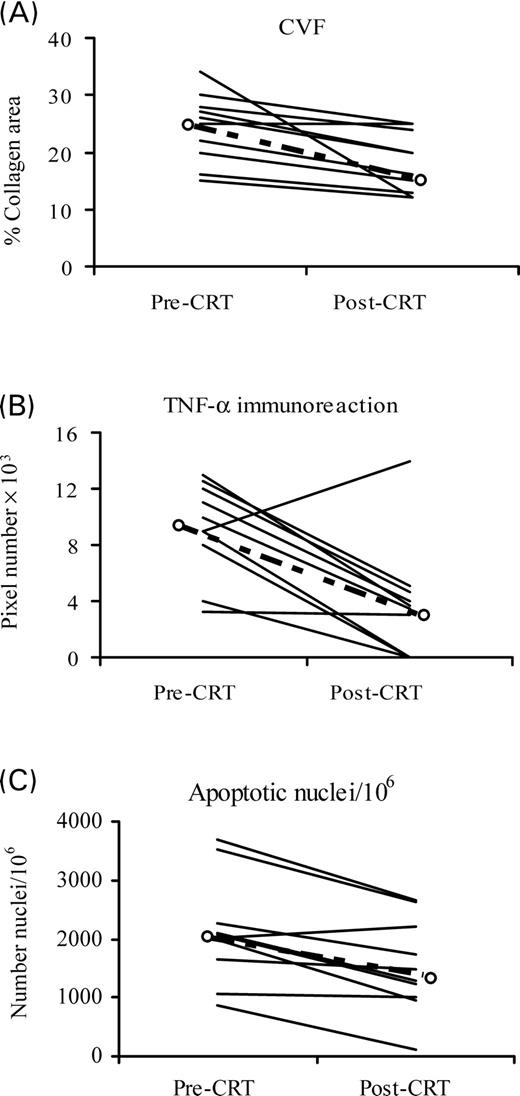
Figure 2 Individual data related to CVF (A), TNF-α immunoreaction (B), and apoptosis (C) before (pre-CRT) and after (post-CRT) 6 months of CRT. Bold dashed lines represent the median.
Baseline clinical characteristics of the patient population
| Age | 62(54–70)a |
| Gender | 10 males |
| Atiology | six idiopathic, four post-ischaemic |
| NYHA functional class | IV(n=2); III (n=8) |
| Concomitant therapy | |
| ACE-inhibitors or AT1 receptor blockers | 9/10 |
| Beta-blockers | 7/10 |
| Digitalis | 6/10 |
| Diuretics | 10/10 |
| Aldosterone receptor blockers | 6/10 |
| Amiodarone | 4/10 |
| Age | 62(54–70)a |
| Gender | 10 males |
| Atiology | six idiopathic, four post-ischaemic |
| NYHA functional class | IV(n=2); III (n=8) |
| Concomitant therapy | |
| ACE-inhibitors or AT1 receptor blockers | 9/10 |
| Beta-blockers | 7/10 |
| Digitalis | 6/10 |
| Diuretics | 10/10 |
| Aldosterone receptor blockers | 6/10 |
| Amiodarone | 4/10 |
a Data are expressed as median and range.
Baseline clinical characteristics of the patient population
| Age | 62(54–70)a |
| Gender | 10 males |
| Atiology | six idiopathic, four post-ischaemic |
| NYHA functional class | IV(n=2); III (n=8) |
| Concomitant therapy | |
| ACE-inhibitors or AT1 receptor blockers | 9/10 |
| Beta-blockers | 7/10 |
| Digitalis | 6/10 |
| Diuretics | 10/10 |
| Aldosterone receptor blockers | 6/10 |
| Amiodarone | 4/10 |
| Age | 62(54–70)a |
| Gender | 10 males |
| Atiology | six idiopathic, four post-ischaemic |
| NYHA functional class | IV(n=2); III (n=8) |
| Concomitant therapy | |
| ACE-inhibitors or AT1 receptor blockers | 9/10 |
| Beta-blockers | 7/10 |
| Digitalis | 6/10 |
| Diuretics | 10/10 |
| Aldosterone receptor blockers | 6/10 |
| Amiodarone | 4/10 |
a Data are expressed as median and range.
Clinical status, electrocardiographic, and echocardiographic parameters before and after 6 months of CRT
| Baseline | 6 months after CRT | P-Value | |
|---|---|---|---|
| NYHA class | IV(n=2); III (n=8) | IV(n=1); III (n=4); II (n=5) | — |
| Distance walked in 6 min (m) | 290 (187–420) | 330 (210–495) | 0.002 |
| Total exercise time (s) | 530 (160–690) | 621 (187–797) | 0.002 |
| Peak oxygen consumption (mL/kg/min) | 12.5 (10–20) | 14.5 (13–21) | 0.002 |
| Score on the MLHFQ | 53 (31–93) | 40 (24–71) | 0.002 |
| Heart rate (b.p.m.) | 70 (67–95) | 65 (63–82) | 0.006 |
| QRS duration (ms) | 164 (139–199) | 130 (103–153) | 0.004 |
| IMD (ms) | 82 (67–132) | 19 (9–59) | 0.005 |
| Septal-to-posterior wall motion delay (ms) | 181 (116–383) | 16 (−23.8–169.2) | 0.002 |
| LV end-diastolic volume index (mL/m2) | 108 (84–194) | 77 (58–196) | 0.037 |
| LV end-systolic volume index (mL/m2) | 97 (68–154) | 80 (45–130) | 0.006 |
| Relative wall thickness | 0.21 (0.12–0.30) | 0.26 (0.16–0.34) | 0.005 |
| Ejection fraction (%) | 22 (17–28) | 25 (20–31) | 0.002 |
| Circumferential wall stress (kdynes/cm2) | 408 (212–647) | 382 (122–522) | 0.020 |
| Area of mitral regurgitant jet (cm2) | 7 (2.85–12.6) | 4.7 (1.72–8.40) | 0.002 |
| Baseline | 6 months after CRT | P-Value | |
|---|---|---|---|
| NYHA class | IV(n=2); III (n=8) | IV(n=1); III (n=4); II (n=5) | — |
| Distance walked in 6 min (m) | 290 (187–420) | 330 (210–495) | 0.002 |
| Total exercise time (s) | 530 (160–690) | 621 (187–797) | 0.002 |
| Peak oxygen consumption (mL/kg/min) | 12.5 (10–20) | 14.5 (13–21) | 0.002 |
| Score on the MLHFQ | 53 (31–93) | 40 (24–71) | 0.002 |
| Heart rate (b.p.m.) | 70 (67–95) | 65 (63–82) | 0.006 |
| QRS duration (ms) | 164 (139–199) | 130 (103–153) | 0.004 |
| IMD (ms) | 82 (67–132) | 19 (9–59) | 0.005 |
| Septal-to-posterior wall motion delay (ms) | 181 (116–383) | 16 (−23.8–169.2) | 0.002 |
| LV end-diastolic volume index (mL/m2) | 108 (84–194) | 77 (58–196) | 0.037 |
| LV end-systolic volume index (mL/m2) | 97 (68–154) | 80 (45–130) | 0.006 |
| Relative wall thickness | 0.21 (0.12–0.30) | 0.26 (0.16–0.34) | 0.005 |
| Ejection fraction (%) | 22 (17–28) | 25 (20–31) | 0.002 |
| Circumferential wall stress (kdynes/cm2) | 408 (212–647) | 382 (122–522) | 0.020 |
| Area of mitral regurgitant jet (cm2) | 7 (2.85–12.6) | 4.7 (1.72–8.40) | 0.002 |
Data are presented as median and range.
Clinical status, electrocardiographic, and echocardiographic parameters before and after 6 months of CRT
| Baseline | 6 months after CRT | P-Value | |
|---|---|---|---|
| NYHA class | IV(n=2); III (n=8) | IV(n=1); III (n=4); II (n=5) | — |
| Distance walked in 6 min (m) | 290 (187–420) | 330 (210–495) | 0.002 |
| Total exercise time (s) | 530 (160–690) | 621 (187–797) | 0.002 |
| Peak oxygen consumption (mL/kg/min) | 12.5 (10–20) | 14.5 (13–21) | 0.002 |
| Score on the MLHFQ | 53 (31–93) | 40 (24–71) | 0.002 |
| Heart rate (b.p.m.) | 70 (67–95) | 65 (63–82) | 0.006 |
| QRS duration (ms) | 164 (139–199) | 130 (103–153) | 0.004 |
| IMD (ms) | 82 (67–132) | 19 (9–59) | 0.005 |
| Septal-to-posterior wall motion delay (ms) | 181 (116–383) | 16 (−23.8–169.2) | 0.002 |
| LV end-diastolic volume index (mL/m2) | 108 (84–194) | 77 (58–196) | 0.037 |
| LV end-systolic volume index (mL/m2) | 97 (68–154) | 80 (45–130) | 0.006 |
| Relative wall thickness | 0.21 (0.12–0.30) | 0.26 (0.16–0.34) | 0.005 |
| Ejection fraction (%) | 22 (17–28) | 25 (20–31) | 0.002 |
| Circumferential wall stress (kdynes/cm2) | 408 (212–647) | 382 (122–522) | 0.020 |
| Area of mitral regurgitant jet (cm2) | 7 (2.85–12.6) | 4.7 (1.72–8.40) | 0.002 |
| Baseline | 6 months after CRT | P-Value | |
|---|---|---|---|
| NYHA class | IV(n=2); III (n=8) | IV(n=1); III (n=4); II (n=5) | — |
| Distance walked in 6 min (m) | 290 (187–420) | 330 (210–495) | 0.002 |
| Total exercise time (s) | 530 (160–690) | 621 (187–797) | 0.002 |
| Peak oxygen consumption (mL/kg/min) | 12.5 (10–20) | 14.5 (13–21) | 0.002 |
| Score on the MLHFQ | 53 (31–93) | 40 (24–71) | 0.002 |
| Heart rate (b.p.m.) | 70 (67–95) | 65 (63–82) | 0.006 |
| QRS duration (ms) | 164 (139–199) | 130 (103–153) | 0.004 |
| IMD (ms) | 82 (67–132) | 19 (9–59) | 0.005 |
| Septal-to-posterior wall motion delay (ms) | 181 (116–383) | 16 (−23.8–169.2) | 0.002 |
| LV end-diastolic volume index (mL/m2) | 108 (84–194) | 77 (58–196) | 0.037 |
| LV end-systolic volume index (mL/m2) | 97 (68–154) | 80 (45–130) | 0.006 |
| Relative wall thickness | 0.21 (0.12–0.30) | 0.26 (0.16–0.34) | 0.005 |
| Ejection fraction (%) | 22 (17–28) | 25 (20–31) | 0.002 |
| Circumferential wall stress (kdynes/cm2) | 408 (212–647) | 382 (122–522) | 0.020 |
| Area of mitral regurgitant jet (cm2) | 7 (2.85–12.6) | 4.7 (1.72–8.40) | 0.002 |
Data are presented as median and range.
References
Kass DA. Ventricular resynchronization: pathophysiology and identification of responders.
Abraham WT, Fisher WG, Smith AL, Delurgio DB, Leon AR, Loh E, Kocovic DZ, Packer M, Clavell AL, Hayes DL, Ellestad M, Trupp RJ, Underwood J, Pickering F, Truex C, McAtee P, Messenger J; MIRACLE Study Group. Multicenter InSync Randomized Clinical Evaluation. Cardiac resynchronization in chronic heart failure.
Cazeau S, Leclerq C, Lavergne T, Walker S, Varma C, Linde C, Garrigue S, Kappenberger L, Haywood GA, Santini M, Bailleul C, Daubert JC; Multisite Stimulation in Cardiomyopathies (MUSTIC) Study Investigators. Effects of multisite biventricular pacing in patients with heart failure and intraventricular conduction delay.
Auricchio A, Stellbrink C, Sack S, Block M, Vogt J, Bakker P, Huth C, Schondube F, Wolfhard U, Bocker D, Krahnefeld O, Kirkels H; Pacing Therapies in Congestive Heart Failure (PATH-CHF) Study Group. Long-term clinical effect of hemodynamically optimized cardiac resynchronization therapy in patients with heart failure and ventricular conduction delay.
Stellbrink C, Breithardt OA, Franke A, Sack S, Bakker P, Auricchio A, Pochet T, Salo R, Kramer A, Spinelli J; PATH-CHF (PAcing THerapies in Congestive Heart Failure) Investigators; CPI Guidant Congestive Heart Failure Research Group. Impact of cardiac resynchronization therapy using hemodynamically optimized pacing on left ventricular remodeling in patients with congestive hear failure and ventricular conduction disturbances.
Pitzalis MV, Iacoviello M, Romito R, Massari F, Rizzon B, Luzzi G, Guida P, Andriani A, Mastropasqua F, Rizzon P. Cardiac resynchronization therapy tailored by echocardiographic evaluation of ventricular asynchrony.
Bristow MR, Saxon LA, Bohemer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM; Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure (COMPANION) Investigators.Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure.
Mann DL. Mechanisms and models in heart failure. A combinatorial approach.
Sahn DJ, DeMaria A, Kisslo J, Weyman A. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements.
Fazio S, Sabatini D, Capaldo B, Vigorito C, Giordano A, Guida R, Pardo F, Biondi B, Sacca L. A preliminary study of growth hormone in the treatment of dilated cardiomyopathy.
Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I, Silverman NH, Tajik AJ. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography.
Douglas PS, Reichek N, Plappert T, Muhammad A, St John Sutton MG. Comparison of echocardiographic methods for assessment of left ventricular shortening and wall stress.
Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes.
Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart.
Cittadini A, Isgaard J, Monti MG, Casaburi C, Di Gianni A, Serpico R, Iaccarino G, Sacca L. Growth hormone prolongs survival in experimental heart failure.
Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease.
Bruckner BA, Stetson SJ, Farmer JA, Radovancevic B, Frazier OH, Noon GP, Entman ML, Torre-Amione G, Youker KA. The implications for cardiac recovery of left ventricular assist device support on myocardial collagen content.
Nagueh SF, Stetson SJ, Lakkis NM, Killip D, Perez-Verdia A, Entman ML, Spencer WH 3rd, Torre-Amione G. Decreased expression of tumor necrosis factor-α and regression of hypertrophy after nonsurgical septal reduction therapy for patients with hypetrophic obstructive cardiomyopathy.
Torre-Amione G, Stetson SJ, Youker KA, Durand JB, Radovancevic B, Delgado RM, Frazier OH, Entman ML, Noon GP. Decreased expression of tumor necrosis factor-α in failing human myocardium after mechanical circulatory support. A potential mechanism for cardiac recovery.
Capasso JM, Palackal T, Olivetti G, Anversa P. Left ventricular failure induced by long-term hypertension in rats.
Mann DL. Tumor necrosis factor-induced signal transduction and left ventricular remodeling.
Palmieri EA, Benincasa G, Di Rella F, Casaburi C, Monti MG, De Simone G, Chiariotti L, Palombini L, Bruni CB, Sacca L, Cittadini A. Differential expression of TNF-α, IL-6, and IGF-1 by graded mechanical stress in normal rat myocardium.
Sivasubramanian N, Coker ML, Kurrelmeyer KM, MacLellan WR, DeMayo FJ, Spinale FG, Mann DL. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor.
Kang PM, Izumo S. Apoptosis in heart: basic mechanisms and implications in cardiovascular diseases.
Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanicistic role for cardiac myocyte apoptosis in heart failure.
Spragg DD, Leclerq C, Loghmani M, Faris OP, Tunin RS, DiSilvestre D, McVeigh ER, Tomaselli GF, Kass DA. Regional alterations in protein expression in the dyssynchronous failing heart.


{kind=link}
{kind=link}