





Vulnerable plaques
The majority of coronary thrombi (∼75%) is caused by plaque rupture.1,2 Prototype of the rupture-prone plaque contains a large, soft, lipid-rich necrotic core with a thin and inflamed fibrous cap, so-called thin-cap fibroatheroma (TCFA) (Figure 1).3,4 Other common features include expansive remodelling, large plaque size, plaque haemorrhage, neovascularization, adventitial inflammation, and ‘spotty’ calcifications.4 Thin-cap fibroatheroma caps are usually <65 µm thick.4Figure 2 summarizes factors contributing to the formation of vulnerable plaques. No distinct morphological features have been identified for the erosion-prone plaques, but they are usually rarely associated with expansive remodelling, scarcely calcified, and contain only limited inflammation.2,5
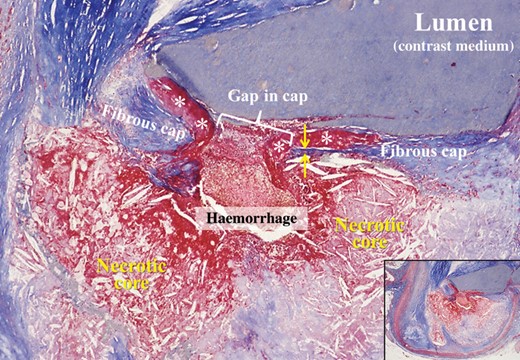
Cross-sectioned coronary artery showing a ruptured thin-cap fibroatheroma. The fibrous cap is very thin near the rupture site (between arrows) and a non-obstructive mural thrombus (asterisks) is bordering the gap in the disrupted cap. A haemorrhage has penetrated from the lumen through the gap into the lipid-rich necrotic core in which the characteristic cholesterol clefts are clearly seen. The lumen contains contrast medium injected postmortem. Overview in inset. Trichrome, staining collagen blue, and thrombus and haemorrhage red.
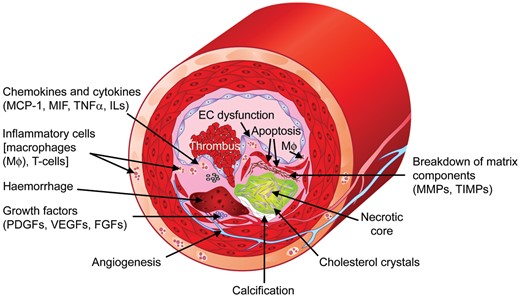
Factors contributing to the formation of vulnerable plaques. MCP-1, monocyte chemotactic protein-1; MIF, migration inhibitory factor; TNFα, tumour necrosis factor-α; ILs, interleukins; MMPs, matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinases; PDGFs, platelet-derived growth factors; VEGFs, vascular endothelial growth factors; FGFs, fibroblast growth factors; Mφ, macrophages.
Inflammatory cells, cytokines, chemokines, and growth factors
Vulnerable plaques contain monocytes, macrophages, and T-cells. Of the T-cells, CD4+ T-helper (Th) cells are the most prominent.6 T-cells can differentiate into a Th1 phenotype, which secretes and responds to IFN-γ or a Th2 phenotype, which secretes and responds to IL-4, IL-10, and IL-13 (Figure 3). T-cells promote the vulnerability of plaques through their effects on macrophages. Similarly, there are two main plaque macrophage phenotypes: pro-inflammatory M1 macrophages (IFN-γ-induced) and anti-inflammatory or regulatory M2 macrophages (IL-4/IL-13-induced).7
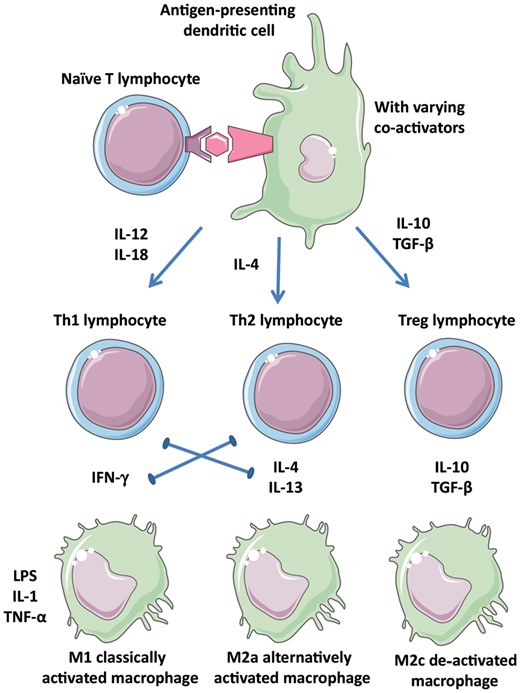
Diverse leucocyte populations in atherosclerotic plaques. Depending on the context of antigen presentation including the cytokine environment, naïve T-cells and macrophages may adopt several phenotypes that are more or less inflammatory. Polarization of the environment is promoted by the positive and negative interactions indicated. Th1, Th2, Treg: T-cell subtypes; M1, M2a, M2c: macrophage subtypes.
Cytokines and chemokines important for regulating inflammatory and immune responses are listed in Supplementary material online, Table S1.7–11 The concept that Th1-related pro-inflammatory cytokines drive progression whereas Th2- and regulatory T-cell-related cytokines exert anti-atherogenic effects provides a useful theoretical framework (Figure 3). For an unstable phenotype, IFN-γ, IL-12, and IL-18 seem to be important factors. CCR5 drives Th1-type pro-inflammatory responses and contributes to plaque formation.12 Important players for plaque destabilization are macrophage migration inhibitory factor (MIF)13 and monocyte chemotactic protein-1.14 Of the plaque-stabilizing factors, IL-10 and TGF-β are of the greatest significance.11,15
Extracellular proteases and platelets
Extracellular proteases correlate with changes associated with plaque vulnerability,16 such as macrophage ingress and apoptosis, and loss of collagen and elastin.17,18 Apoptosis of SMCs contributes to weakening of plaques. Knockout models supported a role for matrix metalloproteinases (MMPs)19 and cathepsins20 in plaque rupture, although the effects of MMP inhibitors on plaque stability have been mixed. Recent studies with more selective drugs21 provide new hope that inhibiting proteases or preventing their secretion19 may lead to plaque stabilization.
Platelets contribute to atherogenesis through secretory functions and as modulators of inflammatory responses. Antiplatelet drugs might, therefore, serve as plaque-stabilizing compounds. However, experimental data on antiplatelet drugs are contradictory.22–25 Although antiplatelet drugs have proven benefits in the secondary prevention of CVD, their direct role in plaque stabilization remains unclear.
Endothelial dysfunction, wall stress, and shear stress
Impairment in endothelium-dependent vasodilatation is the clinical hallmark of endothelial dysfunction.26 In atherosclerosis, there is coexistence of segments with normal vasodilatory and abnormal vasoconstrictive responses to acetylcholine. In virtual histology (VH)-intravascular ultrasound (IVUS), segments with endothelial dysfunction have larger plaques and necrotic core areas.27 Under sympathetic activity, dysfunctional endothelium will respond with paradoxical vasoconstriction. The release from platelets of peptides, in particular serotonin and thrombin, leads to further vasoconstriction and perpetuation of the situation.28–31
Shear stress plays a key role during initiation of atherosclerosis.32,33 Recent studies indicate that it also predicts location of advanced lesions.34 Repeated measurements indicate that low shear stress is predictive of not only plaque location, but also of plaque growth.35,36 Role of wall stress in plaque rupture has been increasingly recognized, since it seems to predict plaque rupture better than shear stress.37 The levels of these forces inducing rupture are in the order of ∼150 kPa or ∼1100 mmHg, which have been shown to occur at the shoulder regions.37 In the PREDICTION trial, large plaque burden and low local endothelial shear stress provided independent and additive prediction to identify plaques that develop progressive enlargement and lumen narrowing.38
Angiogenesis
Microvessels increase with plaque progression, are abundant in vulnerable plaques,39 and can contribute to plaque inflammation.40 Fragile microvessels allow extravasation of lipoproteins and red blood cells,41 which contribute to plaque lipids. Whether haemorrhage from neovessels triggers plaque rupture (or vice versa) remains to be demonstrated. Mechanisms regulating plaque angiogenesis involve hypoxia-inducible factor and growth factors, such as VEGFs, PlGF, PDGFs, and FGFs.42 However, the net effect of all these regulators remains unclear. While VEGF is expressed in atherosclerotic lesions,43 patients receiving anti-VEGF antibodies for cancer show increased CVD complications. This finding is compatible with vasculoprotective effect of VEGF, and transient treatment of mice with VEGF has not increased atherosclerosis.44 Whether therapeutic manipulation of angiogenesis can stabilize plaques remains to be investigated.45
Progenitor cells
Clinical studies show correlations of endothelial progenitor cells (EPCs) with atherogenesis,46 suggesting that EPCs may provide protection against atherosclerosis. Recent data, however, question these findings: putative EPCs measured in the clinics have generated mostly inflammatory cells rather than endothelial cells.47 Also, mouse studies tracking endothelial origin in atherogenesis have found rare, if any, contributions from the blood.48
Smoking
Smoking is a major CVD risk factor causing endothelial damages, disturbances in coagulation, and inflammation.49 Stopping smoking is beneficial for plaque stabilization.
Biomarkers, genetic testing, and imaging in the detection of unstable plaques
Biomarkers and genetic testing
Single nucleotide polymorphism and GWAS studies have identified approximately 160 genetic loci that are associated with CVD, MI, and restenosis.50,51 However, there are no data to pinpoint specific genetic signatures to the vulnerable plaque. Although a genetic test to identify patients who carry vulnerable plaques is the ultimate goal, this seems currently unlikely. It seems clear that part of the gene–environment interactions are regulated by epigenetics.52 As chromatin alterations are reversible, epigenetic modifications are amendable to pharmacological interventions, which may provide new treatments for CVD.
Detection of unstable plaques
Plaque burden correlates well with calcification, but is not an indicator of stability. Computerised tomography shows that lesion area in ruptured plaques is larger than in stable lesions.53 Using IVUS, it was found that patients with acute MI had larger plaque area compared with patients with unstable or stable angina. VH-IVUS allows classification of lesions as fibrous, fibrocalcific, fibroatheroma, and TCFAs.54 The definition of IVUS-derived TCFA is a lesion with plaque burden ≥40% and confluent necrotic core ≥10% in direct contact with lumen. VH-IVUS identified the following characteristics as predictors of clinical events5: TCFA, plaque burden ≥70%, and minimum lumen area ≤4 mm2. However, even combining these characteristics resulted in only 18% event rate during 3 years, which illustrates current limitations of imaging techniques.
Optical coherence tomography (OCT) gives a spatial resolution of ≤20 µm allowing more accurate assessment of cap thickness (Figure 4).55 Optical coherence tomography has potential to assess plaque macrophage content. In non-flow-limiting coronary lesions, high-risk plaque characteristics (such as thin fibrous cap, large lipid pools, and microchannels) were associated with plaque progression.56 NIR spectroscopy is another technique designed to identify lipid-containing plaques.57 Emerging imaging techniques utilize MRI markers homing to rupture-prone plaques and markers of macrophage metabolic activity.58 Developing such techniques remains a challenge for the future.59
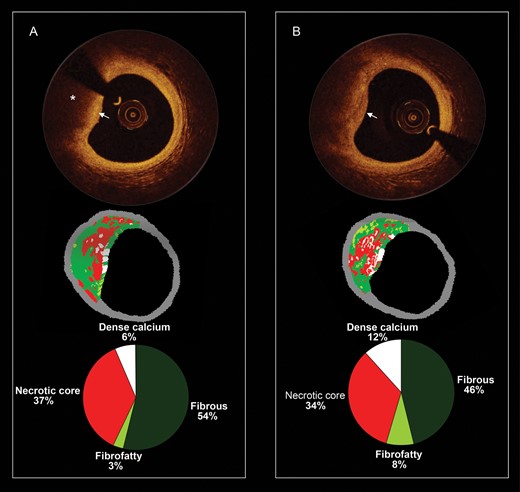
Serial and corresponding virtual histology and optical coherence tomography. (A) The baseline optical coherence tomography frame (top), the corresponding virtual histology frame (mid), and the quantification of the virtual histology tissue types (bottom). The optical coherence tomography frame shows from 8 to 12 o'clock a fibroatheroma which is composed of a fibrous cap (white arrow) and a necrotic core (asterisk). The corresponding virtual histology frames show also a fibroatheroma (necrotic core-rich plaque in red colour). In (B), the patient was reimaged at 1 year and some plaque changes have been observed. In the optical coherence tomography frame, the fibrous cap became thinner (please note that at baseline, the signal rich area overlying the necrotic core was wider).
Current treatments and future perspectives of plaque stabilization
Statin therapy
Patients receiving pravastatin 3 months before carotid endarterectomy showed significantly less inflammation and a higher collagen content in carotid plaques, suggesting plaque stabilization.60 The ATROCAP study randomized two-step bilateral carotid endatherectomy patients to atorvastatin or placebo for 4–6 months after the first procedure. Plaques from treated patients showed a trend towards fewer inflammatory cells, whereas no change was observed in controls.61 Results are consistent with pleiotropic, anti-inflammatory effects of statins, which may contribute to the stabilization of plaques.62 Recent experimental finding of plaque stabilization with ezetimide, which lacks pleiotropic effects, lends support to the lipid-lowering therapy per se.63 However, it is not yet possible to discern the contribution of each mechanism to clinical results.
The German Atorvastatin Study demonstrated that hyperechogenicity of plaques significantly increased after 12 months compared with non-statin-based lipid lowering.64 In the ASTEROID65 and SATURN studies,66 aggressive lipid lowering regressed atheroma volume in IVUS. Data from several prospective IVUS trials confirmed significant atheroma regression after LDL reduction67 and the PROVE-IT trial showed lower CVD endpoints after 24 months under intense lipid lowering in ACS patients.68 In addition, the JUPITER study in healthy subjects with LDL <3.4 mmol/L and hs-C-reactive protein above 2 mg/L showed that 20 mg of rosuvastatin significantly reduced major CVD events in this low-risk group.69
Antiplatelet and antihypertensive therapies
Aspirin is effective for CVD secondary prevention, and a major reduction in CVD events was found in the CURE trial, where clopidogrel was added to aspirin in ACS patients.70 However, these agents mostly reduce complications of plaque rupture and may not be plaque-stabilizing agents per se. Whether new platelet inhibitors ticagrelor and prasugrel and platelet thrombin receptor inhibitor vorapaxar have plaque-stabilizing properties remains unknown, although recent meta-analyses have suggested the same benefits in patients with recent ACS.71
Four recent IVUS trials have shown that β-blockers slow the progression of CVD.72 Endothelial function can be improved by renin–angiotensin inhibitors, and HOPE73 and ONTARGET74 trials have shown a larger reduction in CVD events that could be predicted from the reduction in blood pressure, supporting plaque-stabilizing effects.
Other anti-atherosclerotic therapies
HDL-raising therapies
ApoA1-Milano and other HDL-like apoA1 complexes have been shown to regress atherosclerosis possibly via several HDL-related protective mechanisms like reverse cholesterol transport, anti-oxidative activity, endothelial vasoprotection, and reduction of platelet activation.75 HDL also inhibits coagulation cascade. Cholesterol ester transfer protein (CETP) is a plasma protein that catalyses exchange of cholesteryl esters and triglycerides between lipoproteins. Reduction in CETP activity is associated with cholesterol reduction in VLDL and LDL and enrichment of HDL. However, the ILLUMINATE trial with a CETP inhibitor torcetrapib failed due to toxicity.76 New trials with novel CETP inhibitors such as anacetrapib (Supplementary material online, Table S2) are underway, but dal-OUTCOMES trial assessing dalcetrapib has been stopped due to the lack of efficacy.77 Thus, these recent trials and a large Mendelian randomization study78 question the usefulness of HDL-raising therapies, if no simultaneous beneficial changes can be achieved in VLDL or LDL levels.
Niacin/nicotinic acid
With interest in HDL-raising therapies, niacin has been recently re-investigated. Two trials in statin-treated patients with low HDL have shown that modified-release nicotinic acid significantly reduced carotid atherosclerosis,79 and that the use of slow-release niacin significantly reduced carotid intima-media thickness in comparison with statin.80 However, AIM-HIGH and HPS-2-THRIVE trials failed to show any added benefits (Supplementary material online, Table S2), which raises doubts about the usefulness of this approach.
Phospholipase inhibitors
Another approach to reduce plaque inflammation is to inhibit lipoprotein-associated phospholipase A2, which has prevented increase in necrotic core when compared with placebo.81 Two major trials testing this approach are now ongoing (STABILITY and SOLID-TIMI 52 trials).
New approaches
Antagonists against pro-atherogenic chemokine receptor CCR5 and its ligand CCL5 have been developed.82 Migration inhibitory factor is involved in atheroprogression and its inhibition with biologicals or small molecules may be useful for plaque stabilization.13
PCSK9 is involved in hypercholesterolaemia by favouring degradation of LDL receptor.83 Some natural PCSK9 mutations increase its function and cause hypercholesterolaemia, whereas loss-of-function mutations cause hypocholesterolaemia. Therefore, PCSK9 is an attractive target for lowering plasma LDL with potential plaque-stabilizing features. The ODYSSEY OUTCOMES trial is now testing the efficacy of PCSK9 inhibitor in CVD (Supplementary material online, Table S2).
siRNAs against apoB100 have been used to reduce LDL levels.84 Whether this technology will be useful in the prevention of plaque ruptures remains unknown. Innate and adaptive immunity regulates pro-atherogenic inflammation. Immunization of hyperlipidaemic animals with LDL preparations or apoB100 fragments reduces atherosclerosis, suggesting that vaccination may become a potential strategy for the prevention of CVD.85,86
Key points (adapted from Ylä-Herttuala et al.1)
Vulnerable plaques are prone to rupture and thrombosis. Two types of vulnerable plaques are rupture-prone and erosion-prone. Prototype of the rupture-prone plaque contains large and soft lipid-rich necrotic core covered by thin and inflamed fibrous cap.
Thin-cap fibroatheroma: If the fibrous cap is thin, the plaque is called TCFA. Thin fibrous caps are usually heavily inflamed.
Plaque stabilization can be achieved by increasing thickness of fibrous cap, reducing inflammation in the fibrous cap, and reducing size of atheromatous core. Plaques may be stabilized against thrombosis independent of changes in plaque size and luminal obstruction.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This study was supported by ESC Working Group of Atherosclerosis and Vascular Biology.
Conflict of interest: I.H. has received research grants from NWO, CTMM, BMM, and TI Pharma. J.W.J. has received grants from Astellas, Astra-Zeneca, Biotronik, Boston Scientific, Daiichi Sankyo, Lilly, Genzyme, Medtronic, MSD, Pfizer, Orbus Neich, Novartis, Roche, Servier, Sanofi-Aventis, the Netherlands Heart Foundation, the Interuniversity Cardiology Institute of the Netherlands, and the European Community Framework FP7 Programme. R.K. has received grants from the Netherlands and British Heart Foundations, the CTMM, the BBSRC and EPSRC, the FP7 Programme, and Johnson & Johnson. B.R.K. has received grants from the Swiss National Science Foundation, Fondation Leenaards, and the Fondation Prevot. N.M. has received research grants from Boehringer Ingelheim, GSK, MSD, and Takeda. A.N. has received grants from the British Heart Foundation and the UK National Institute for Health Research. He is also consultant for PlaqueTec. G.P. is a co-founder of Cavadis. C.W. is a shareholder of Carolus Therapeutics, Inc. S.Y.-H has received grants from the Academy of Finland, the Finnish Heart Foundation, and EU FP7 programme grants CliniGene, Baculogenes, BIOMAGSCAR, and BAMI.
Acknowledgements
The invaluable help of Ms Marja Poikolainen in preparing the manuscript is greatly acknowledged.
References
Author notes
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.


{kind=link}
{kind=link}
{kind=link}
{kind=link}