





The current role of neutrophils in atherosclerosis, thrombosis and acute coronary syndromes (ACS)
Neutrophils—basic functions
Neutrophils are polymorphonuclear leucocytes that are distinguished and named by the special shape of their multi-lobed nucleus. They function as an early defence against pathogens and fulfil important functions in inflammatory and innate immune responses to protect the host. Neutrophils developed several strategies to accomplish these tasks, including degranulation, phagocytosis, apoptosis, release of reactive oxygen species (ROS), and the formation of neutrophil extracellular traps (NETs). These mechanisms are triggered by pathogens (e.g. bacterial lipopolysaccharides), but can also be activated by other leucocytes, locally produced cytokines, or other inflammatory stimuli.
Neutrophils produce cytoplasmic granules containing enzymes that can be released upon stimulation, including myeloperoxidase (MPO), matrix metalloproteinase 8 (MMP8), cathelicidin antimicrobial peptide (CAMP, also known as LL-37), azurocidin 1 (AZU1), cathepsin G (CTSG), and elastase 2 (ELA2). These enzymes serve to directly combat a pathogen or to enhance an inflammatory response. Despite their important function in innate immunity, the outcome of neutrophil activation is not always beneficial. Many studies are showing an involvement of neutrophils in the progression of cardiovascular diseases (CVD) including atherosclerosis, thrombosis, and ACS.
Neutrophils in atherosclerosis
In the past, the cellular focus of most studies investigating cellular mechanisms of atherosclerosis was set on the role of monocytes/macrophages and T cells. However, there are now many studies highlighting the contribution of neutrophils in the context of this disease.
Animal models have demonstrated that activated macrophages in atherosclerotic plaques produce chemokines that recruit neutrophils to the lesion site.1–4 In vitro experiments suggest that subsequent neutrophil transmigration through the endothelial layer could be mediated by oxidized low density lipoprotein (oxLDL), via an increase of endothelial contractility and upregulation of intracellular adhesion molecule-1 (ICAM-1).5 Another study showed that depletion of neutrophils reduced atherogenesis, likely by diminishing apoptosis and inflammation within the plaques.4 Of note, neutrophils and their mediators have also been found in atherosclerotic human lesions.6
Once inside a lesion, neutrophils release granular proteins: CAMP and AZU1 stimulate recruitment of inflammatory monocytes.7 Azurocidin 1 is preferentially deposited on the endothelium and triggers monocyte arrest and upregulation of endothelial adhesion molecules.8,9 Matrix metalloproteinase 8 is breaking down collagen and thus, is implicated in plaque instability.10 Myeloperoxidase catalyses the conversion of hydrogen peroxide to hypochlorous acid, which oxidises amines to toxic chloramines, consequently altering lipoprotein function.11
In addition to granular proteins, neutrophils release ROS, which can activate the endothelium, recruit additional neutrophils, oxidize LDL and contribute to plaque vulnerability.12 In atheromata, neutrophils are readily undergoing apoptosis and thereby release a cocktail of peptides that augment monocytes/macrophages migration into the lesion in order to phagocytose apoptotic cells.13,14
Neutrophils in thrombosis
Neutrophils are critically involved in thrombotic processes. In a mouse study of laser-induced endothelial damage, neutrophils have been shown to be the first cells at the site of damage, even preceding platelets.15 Notably, inhibition of neutrophil binding to the endothelium at the site of damage reduced the presence of tissue factor (TF, or FIII), the main initiator of the extrinsic coagulation cascade. This event decreased the final products of the coagulation cascade, fibrin generation, and platelet activation. Moreover, the neutrophil-derived proteinases cathepsin G and elastase degrade tissue factor pathway inhibitor (TFPI), the main inhibitor of TF, and thereby enhance coagulation.16 Additionally, neutrophils are able to activate platelets via production of ROS.17
Neutrophils in acute coronary syndromes
As described above, neutrophils promote atherothrombotic processes and thus may enhance pathways leading to acute myocardial infarction (MI). Early reperfusion therapy 3–6 h after onset of complete coronary occlusion with associated myocardial ischaemia and chest pain is the standard treatment in clinics. A short time window is central to salvage myocardial tissue. However, reperfusion can also damage the myocardium and increase infarct size. By re-perfusing ischaemic myocardium in dogs with whole blood or blood lacking neutrophils, Litt et al. have shown that reperfusion without neutrophils reduces infarct size, identifying neutrophils as key mediators of reperfusion injury.18 In fact, once re-perfused, the dying myocardium and damaged extracellular matrix release signals that attract and activate neutrophils.19 Initially, this helps the myocardium to heal, because the neutrophils phagocytose necrotic tissue and recruit monocytes that further degrade damaged tissue.7,20
However, degranulated proteins and ROS released by activated neutrophils are also damaging intact cells and extracellular matrix, activating endothelial cells, and recruiting more neutrophils, all of which may increase infarct size. In addition, the neutrophil-mediated recruitment of monocytes is thought to be detrimental in the long term, since it has been shown that the increased recruitment of monocytes after MI is accelerating atherosclerosis in mice.21 This finding is supported by the clinical observations of increased events in patients surviving a MI.22,23
Neutrophil extracellular traps and cardiovascular diseases
A more recently discovered mechanism of defending the host against pathogens is the formation of NETs. Neutrophil extracellular traps involve the release of DNA and granular proteins from the cell.24 It is currently debated whether this DNA is of mitochondrial or nuclear origin and whether NET formation is a death sentence for the neutrophil (for recent reviews see25,26). It is clear however, that NETs confer antimicrobial properties and are implicated in pathophysiological processes. Their formation may be triggered by bacterial signals such as LPS, but also by P-selectin expressed by activated platelets or the activated endothelium.27,28 Indeed, recent clinical studies confirmed the presence of NETs in atherosclerotic lesions, which is correlated with a severe atherothrombotic state.29,30 In a lesion, NETs may promote atherogenesis by exposing granular proteins and ROS to their environment as described above.
Neutrophil extracellular trap formation in the bloodstream exerts strong effects on thrombosis. The released DNA acts as a scaffold for binding neutrophil granular proteins and soluble proteins from the blood such as von Willebrand factor and fibronectin (FN1), which both promote platelet binding.27,31 Neutrophil extracellular traps have also been shown to bind active coagulation factor XII16 and contain TF,32,33 both of which are triggering the coagulation cascade and lead to generation of the clot stabiliser FIa and the platelet activator thrombin (FII). Furthermore, NETs bind fibrinogen (FI), which serves as a substrate for FII and stabilises the clot once it is cleaved to fibrin (FIa).31 After these studies showed an involvement of neutrophils in experimental venous and arterial thrombosis in animals, the presence of NETs was also confirmed in human coronary thrombi and in stent thrombi.34–36
Recently, NETs were reported in ischaemia-reperfusion injury in rats.37 Moreover, adopting strategies from some pathogens that produce nucleases to cleave NETs,38–40 in vivo studies in rodents could show that the use of the nuclease DNAse I protects from thrombosis, ischaemic stroke, and MI.37,41–44
Prognostic value of neutrophils
Given their role in atherothrombosis and their occurrence in human CVD, neutrophils emerge as a surrogate of cardiovascular risk. Several smaller studies showed that plasma neutrophil elastase levels correlate with severity of CAD45–50 Assessing plasma levels of MPO in coronary angiography patients could predict cardiovascular mortality and measuring MPO together with C-reactive protein further improved risk prediction.51 Moreover, MPO levels correlate with the extent of CAD, neutrophil counts, and severity of ACS.52–55
A large number of clinical studies investigated the number of neutrophils in the blood and the ratio of neutrophils to total leukocytes: they found that neutrophil counts correlate with severity46,47,56 and complexity of CAD,57 recurrent ischaemic events,58 mortality and acute MI,59,60 larger MI,61 and arterial stiffness.62 Moreover, recent studies demonstrated that NETs are associated with severe coronary atherosclerosis and a prothrombotic state29,32
Summary and conclusions
The immune reactions of neutrophils are important to fight infections. Yet, they can also inflict serious damage on healthy tissues. Especially in non-infectious inflammatory diseases such as atherothrombosis, neutrophil activation may induce disease progression (Figure 1). Clinical studies show that neutrophil numbers and markers improve cardiovascular risk prediction. This notion raises the intriguing question whether inhibiting neutrophils could be beneficial in these disease contexts. One approach could be to target degranulation proteins using antibodies. Another option is to target NETs.
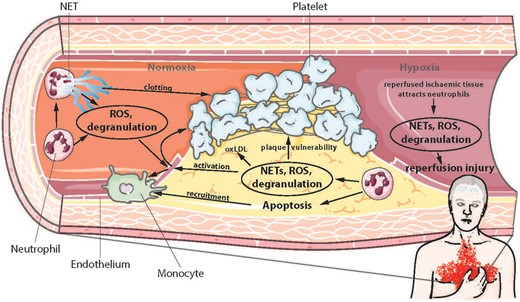
Effects of neutrophils in atherosclerosis, thrombosis, and ischaemia-reperfusion injury. NETs, neutrophil extracellular traps; ROS, reactive oxygen species; oxLDL, oxidised low-density lipoprotein.
Recent studies showed that NETs aggravate acute CVD. Thus, in the past, scientists may have missed to identify NETs as an important component of neutrophil function and defence. Beyond their diagnostic and prognostic value in CVD, NETs appear worthwhile to be tested for a therapeutic intervention: NETs can be inhibited targeting key pathway molecules involved in NET release. Along these lines, the use of DNAse 1 to digest the DNA may be the most promising method to minimize neutrophil-mediated damaging effects. However, targeting neutrophils using this strategy could be delicate, as it is likely to interfere with the host’s ability to fight acute infections.
To address this problem, further research to determine the differences of neutrophil activation by pathogens vs. activation by sterile inflammation is warranted. Understanding these differences may guide us to identify opportunities to specifically inhibit activation of neutrophils in non-infectious chronic inflammatory diseases such as atherothrombosis.



Acknowledgements
The work related to this article was funded by the Swiss National Science Foundation to S.S. and C.M.M., by Novartis Consumer Health Foundation to S.S. and by the University Research Priority Program Integrative Human Physiology at the University of Zurich to D.S.G. and C.M.M. The Figure was designed using Servier Medical Art by Servier under a Creative Commons Attribution 3.0 Unported License.
Conflict of interest: none declared.
References
References are available as supplementary material at European Heart Journal online.


{kind=link}