





Abstract
Ambient air pollution is a leading cause of non-communicable disease globally. The largest proportion of deaths and morbidity due to air pollution is now known to be due to cardiovascular disorders. Several particulate and gaseous air pollutants can trigger acute events (e.g. myocardial infarction, stroke, heart failure). While the mechanisms by which air pollutants cause cardiovascular events is undergoing continual refinement, the preponderant evidence support rapid effects of a diversity of pollutants including all particulate pollutants (e.g. course, fine, ultrafine particles) and gaseous pollutants such as ozone, on vascular function. Indeed alterations in endothelial function seem to be critically important in transducing signals and eventually promoting cardiovascular disorders such as hypertension, diabetes, and atherosclerosis. Here, we provide an updated overview of the impact of particulate and gaseous pollutants on endothelial function from human and animal studies. The evidence for causal mechanistic pathways from both animal and human studies that support various hypothesized general pathways and their individual and collective impact on vascular function is highlighted. We also discuss current gaps in knowledge and evidence from trials evaluating the impact of personal-level strategies to reduce exposure to fine particulate matter (PM2.5) and impact on vascular function, given the current lack of definitive randomized evidence using hard endpoints. We conclude by an exhortation for formal inclusion of air pollution as a major risk factor in societal guidelines and provision of formal recommendations to prevent adverse cardiovascular effects attributable to air pollution.
Introduction
Environmental stressors represent important risk factors for the development and progression of cardiovascular disease (CVD).1–5 Recently, the Global Burden of Disease (GBD) Study established that ambient outdoor air pollution due to particulate matter <2.5 μm (PM2.5), was the fifth ranking global risk factor in 2015, causing 4.2 million deaths annually, with cardiovascular deaths accounting for most of these deaths (Figure 1A and B).6 Importantly, this statistic does not include other occupational and environmental stressors including smoking, household air pollution, or noise which taken together, arguably could outrank the global impact of many classical risk factors in combination. Scientific understanding of the scale and scope of pollution and its association with a much wider range of diseases, particularly non-communicable diseases have provided a better framework of understanding of pollution’s true global impact.7 Importantly, while chemicals in the air may initiate or potentiate cardiometabolic disease, non-chemical pollutants such as temperature, noise exposure, socioeconomic factors, or mental stress caused by work strain, grief, or social isolation may co-segregate with air pollution and potentially amplify associations with disease.8–12 Alterations in vascular function were the earliest pathophysiological mechanism described in response to air pollution exposure in humans, and indeed disturbances in endothelial function are a critical initiating event that is widely relevant to almost all classical risk factors.13 , 14 In the present review, we focus on the evidence supporting the impact of air pollution and, its particulate and gaseous constituents on vascular function. We address to what extent alterations in vascular function are responsible for mediating systemic diseases in response to air pollutants. Finally, we examine the impact of mitigation strategies to reduce air pollution exposure on vascular function and biochemical pathways known to modulate vascular reactivity such as reduction in redox stress and/or markers of endothelial health.
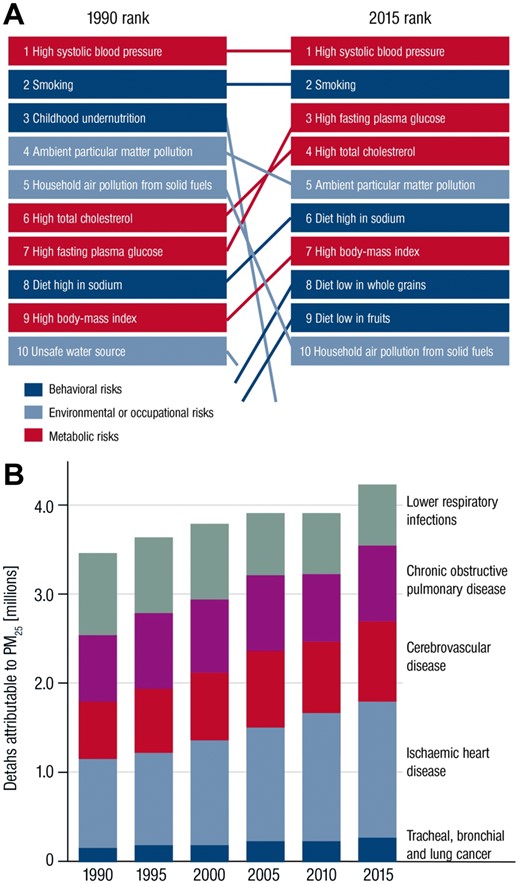
(A) Global Burden of Diseases global risk factors for deaths 1990 compared with 2015. (B) Deaths attributable to ambient particulate matter pollution by year and cause. PM2.5 = particles with aerodynamic diameter less than 2.5 µm. Reused from doi: 10.1016/S0140-6736(17)30505-66 according to the terms and conditions of the Creative Commons Attribution License (CC BY, see https://creativecommons.org/licenses/by/4.0/ for details).
Physiology and pathophysiology of endothelial function
The vascular endothelium represents a biologically active tissue regulating vascular tone, modulating vascular inflammation, thrombosis, and vascular injury mainly via the radical nitric oxide (•NO).14 It is also well established that vascular (endothelial) dysfunction develops in the setting of traditional cardiovascular risk factors including smoking, hypercholesterolaemia, diabetes, and arterial hypertension but also in response to recently recognized environmental stressors such as noise and is often the first manifest abnormality.8 , 14 The alteration in vascular function has its genesis in the abnormal generation of excess reactive oxygen species (ROS). This is of great importance, since the half-life of •NO, and therefore, its biological activity and breakdown, respectively, is decisively determined by ROS such as superoxide. •NO reacts in a facile fashion with superoxide to form the highly reactive intermediate peroxynitrite (ONOO−) in a reaction that is about 10 times faster than the dismutation of superoxide by the superoxide dismutase (rate constant 5–10 × 109/M/s).15 ONOO− may act as a vasoconstrictor and most importantly, as a cytotoxic molecule that causes oxidative damage to proteins, lipids, and DNA. It can cause eNOS uncoupling and tyrosine nitration of prostacyclin synthase and manganese superoxide dismutase, thereby oxidatively inactivating these enzymes and greatly limiting the power of endothelial mediators with protective function. A number of superoxide sources have been identified such as nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), mitochondria, and uncoupled •NO Synthase, that depending on the context, may contribute to excess ROS. Given the fact that •NO release is diffuse, a category of more targeted alterations in proteins such as S-nitrosylation, commonly referred to as S-nitrosylation, has been proposed as a stable post-translational modification that directly regulates vascular and cellular function.16 Defective nitrosylation has been linked to endothelial barrier dysfunction and may represent an important as yet poorly investigated link between risk factors such as air pollution and CVD.16
Air pollution mixtures, chemistry, sources and exposure assessment
While air pollutants have been reported since antiquity, their sources and composition have changed, with anthropogenic (combustion-derived) air pollutants being the current major concern from a public health perspective.17 Air pollution results from the complex interaction of multiple emissions and chemical reactions, and the traditional classification of fine particles based on size or mass may provide an incomplete picture.5 Over 90% of the pollutant mass in the mixture in urban settings is from gases or vapour-phase compounds and secondary pollutants, including ozone (O3), nitrogen dioxide (•NO2), volatile organic compounds (including benzene), carbon monoxide (CO), and sulfur dioxide (SO2). Combustion particulates that contain ultrafines or PM0.1 (PM <0.1 μm in diameter) demonstrate enhanced cardiovascular toxicity owing to features such as high particle counts, high surface area to mass ratio, oxidative stress potential, high solubility and charge, resulting in facile distal airway penetration and likely systemic penetration.5 Air pollution is largely influenced by climate conditions (some of the most aggressive pollutants being generated during hot periods with high UV index)18 but also contributes directly to global warming, which in turn can affect cardiovascular health.19
Air pollution concentration ranges and exposure assessment
The typical range of ambient concentrations for several air pollutants, including the latest U.S. NAAQS and the European Commission regulatory criteria, have been previously reviewed.1–3 Since health effects of air pollution across large populations cannot be studied using individual monitoring, models are needed to predict personal level exposure. Satellite based techniques use the optical properties of the aerosol column (Aerosol Optical Depth, AOD) to produce indirect estimates of ground-level PM2.5 concentrations, which were the basis of GBD estimates for air pollution. Studies have shown high correlations between satellite and ground-level measurements for PM10 (PM < 10 μm), PM2.5, and nitrogen dioxide (•NO2) and are available for most locations on earth at approximately 1 × 1 km resolution.20 These measures of air pollution are typically combined with data on land use characteristics and emissions sources (e.g. roads, population density, energy use, land use), to model fine-scale spatiotemporal air pollution patterns.21 Figure 2A presents an update of the global estimate of CVD deaths from PM2.5,21 based on public health statistics of the World Health Organization (WHO) for the year 2015. The integrated exposure-response functions have been adopted from Cohen et al.,6 used for the GBD of 2015 (Figure 1). The total mortality rate from CVD is estimated at 2.43 million per year, with a 95% confidence interval of 1.70–3.08 million per year. These attributable deaths are based on WHO data for ischaemic heart disease and cerebrovascular disease (ischaemic and haemorrhagic strokes). Since the global total mortality attributable to PM2.5 is about 4.2 million annually, the contribution by CVD is about 57%. Note that in Europe this fraction is considerably higher than the global mean, being 74%. The breadth and scope of personalized monitoring devices have also expanded dramatically and many of these devices can provide unprecedented personal exposure information, in addition to biologic variables such as vascular function, refining our ability to discern the true impact of the environment on health (Figure 2B).22 Of note, the choice of exposure-response functions, the methods behind air pollution measurement and the study populations are major confounders explaining the variations observed among the different large population studies.23 These differences in health impact assessments in large part can help explain differences in estimates (deaths or life years lost) attributable to air pollution. For instance, when estimates from the European Study of Cohorts for Air Pollution Effects (ESCAPE) cohort instead of the American Cancer Society (ACS) cohort study was used, the attributable burden of ambient air pollution nearly doubles.23
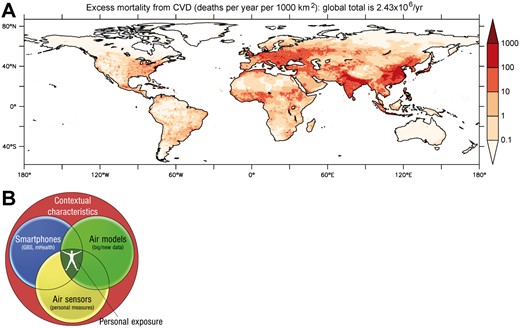
(A) Estimated excess deaths from cardiovascular disease induced by PM2.5 during the year 2015; updated from Lelieveld et al. 21 (B) Integration of personalized biometric data on vascular function with atmospheric models using big data and personalized air sensors.
Air pollution exposure and cardiovascular events
The association between air pollution exposure, in particular for PM2.5, and cardiovascular events including cardiovascular mortality, myocardial infarction (MI), stroke, heart failure including hospitalization is strong and is the subject of extensive reviews.1–3 , 5 , 24 Just to mention a few examples, a systematic review and meta-analysis of studies of short-term air pollution exposures and MI showed that PM2.5, along with •NO2, and SO2 and CO were associated with increased risk of MI. The ESCAPE project (n = 100 166 from multiple cohorts), showed a significant 13% increase in non-fatal acute coronary events, with a 5 µg/m3 elevation in long-term exposure to PM2.5. Patients with underlying coronary artery disease (CAD) may be at particularly high risk. The best recent evidence for acute coronary syndrome (ACS) risk with PM2.5 comes from Utah (Intermountain Health Care, n = 16 314 patients), where concurrent-day PM2.5 was associated with an increase for ACS. Excess risk was observed only among individuals with angiographic CAD, leading to an increase in ST-segment elevation MI. Long-term survival following ACS is also reduced by chronic PM2.5 exposure.
The following aspects of air pollution exposure and cardiovascular events are widely acknowledged: (i) The chronic effects are much larger than acute effects. (ii) Elderly and individuals with prior CVD or risk factors such as obesity are at higher risk. (iii) The relationship between exposure levels and cardiovascular events suggest a ‘no lower threshold’ limit, with recent studies continuing to demonstrate a strong relationship at levels below current regulatory limits.25
Pathophysiological insights into mechanisms of air pollution-mediated vascular dysfunction
Oxidative stress as a common denominator of the vascular effects of air pollution
Oxidative stress is the pre-eminent pathophysiological factor for the adverse vascular health effects of air pollution, is well documented in the lungs and almost certainly occurs with immediate exposure.26 The lungs’ endogenous defenses in the form of a protective surfactant material with a complex mixture of proteins and phospholipids (which are continually replenished) and alveolar macrophages, prevent the systemic penetration of particulates and reactive gases such as ozone. Depletion of low molecular weight antioxidants such as ascorbate, glutathione, and tocopherol with subsequent depletion of reduced cofactors such as NADPH may result in potentiation of oxidative stress.18 Certain types of ultrafine particles may demonstrate direct penetration.27 Likewise, surface-bound reactive co-pollutants such as transition metals, endotoxins, and reactive quinones/aldehydes (partially formed by photochemical reactions in the atmosphere) may be carried by larger particles and released to the lung tissue and circulation leading to secondary toxicity.18 Alternatively, with continual exposure over long periods and/or in the presence of additional pre-disposing factors, chemical transformation of endogenous molecules may overwhelm endogenous defenses resulting in systemic exposure.26 Chemical transformation of thiols and fatty acids (•NO2 and •NO); lipid peroxidation, generation of reactive aldehyde, ketone and endoperoxide fatty acid products; oxidation of thiols to disulfides or sulfoxides and DNA base oxidation can all occur. The proposed mechanism(s) of air pollution-induced endothelial dysfunction and how vascular dysfunction may translate to cardiovascular events is presented in Figure 3. The model is supported by extensive evidence from in vitro cell culture and animal studies, human panel and interventional studies suggesting that both solid and gaseous constituents of air pollution may increase oxidative stress, endothelial dysfunction, and promote acute and chronic cardiovascular sequelae (Supplementary material online, Tables S1–S3). The demonstration of systemic oxidative stress in human studies is somewhat inconsistent and although there are now many studies, they have been only in plasma or airway fluid and are dependent on the types of assays which are sometimes simplistic. The assays used to assess the footprint of systemic ‘oxidative stress’ or damage may also significantly influence the results.26
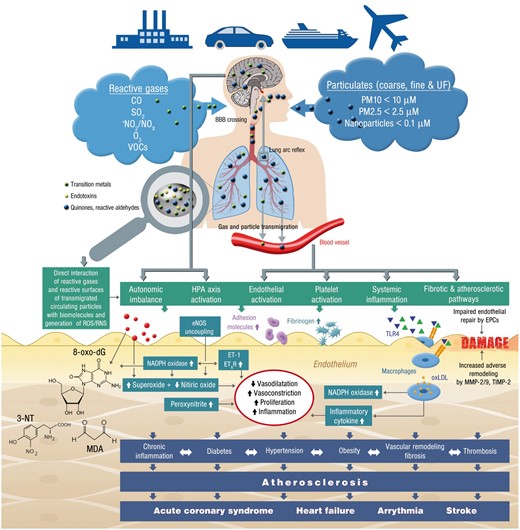
Summary of pathophysiological mechanisms by which air pollution components such as reactive gases and particulates causes endothelial dysfunction, increased oxidative stress, inflammation and subsequently cardiovascular disease. Green and blue triangles are damage- and pathogen-associated molecular patterns (e.g. free DNA fragments, hyaluronan, 7-ketocholesterol, oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine, lipopolysaccharide) as well as soluble heavy/transition metals. EPCs, endothelial progenitor cells; ET-1, endothelin-1; ETAR, endothelin type A receptor; HPA axis, hypothalamic–pituitary–adrenal axis; MDA, malondialdehyde; MMP, metalloproteinase; 3-NT, 3-nitrotyrosine; 8-oxo-dG, 8-oxo-deoxyguanosine; TIMP, tissue inhibitor of metalloproteinases; TLR4, toll-like receptor 4; VOCs, volatile organic compounds.
However, there are several ongoing questions on the specifics of oxidative stress in response to PM and how it may modulate vascular function. These include the extent to which endogenous immune vs. non-immune cells contribute to ROS formation and vascular dysfunction, the role of pathways in the lung in transduction of air pollution effects, the contribution of direct translocation of particulate constituents through the lung, and finally, the role of secondary damage associated molecular patterns including oxidatively modified proteins and lipids, receptors such as Toll-like receptors (TLRs) and RAGE in response to air pollution exposure.26 , 28 While the intersection of oxidative stress with inflammation is well known,29 the role of systemic inflammation as mediator of air pollution effects in humans is far from clear. There is clear evidence that most environmental stressors initiate adverse genetic (e.g. epigenetic or microRNA) changes of signalling cascades leading to an imbalance between ROS production and detoxification.30 Also central effects have been described consisting of autonomic imbalance and hypothalamic–pituitary–adrenal axis activation that is partially based on the lung arc reflex as well as potential crossing of the blood–brain barrier by reactive gases, ultrafine particles and soluble, PM surface-bound compounds such as transition metals, endotoxins and reactive (photo-activated) quinones/aldehydes (Figure 3).26 , 31
Animal studies
A variety of different approaches have been employed to study the effects of air pollution in the experimental context. These may differ with regards to route/duration of exposure, strain/susceptibility of animals, source of pollutants, and compositional characteristics of particles.5 , 26 Supplementary material online, Table S1 summarizes selected studies to date that have provided insights on vascular effects of exposure to either direct installation of particles on intact vessels or vascular endothelial cells. In general, these studies have demonstrated an important effect on ROS pathways, while reduction of ROS sources has been shown to ameliorate endothelial function, •NO availability, endothelial cell activation (adhesion molecule expression), and inflammation. Supplementary material online, Table S2 summarizes important in vivo studies utilizing concentrated ambient particles (PM2.5, ultrafines, diesel, gases, or mixtures). These studies provide definitive evidence for rapid systemic effects of air pollution exposure, often within hours, with recent findings in humans suggesting that nanoparticles and their constituents inhaled into the lung could rapidly cross the alveolar membrane and appear systemically (Figure 3).27 Many studies have demonstrated heightened vasoconstriction of conduit vessels as well as microvasculature with acute inhalational exposure. In both mice and rats, sub-acute and chronic exposure to air pollution alone and/or in conjunction with agents such as angiotensin II, resulted in increased superoxide () and potentiation of vasoconstrictor responses. Several reports have highlighted amplified endothelin-1/ETA-receptor signalling upon exposure to diesel exhaust and reactive gases (Supplementary material online, Table S2, Figure 3), which is in line with NADPH oxidase-driven endothelin-1 promoter activation and vice versa conversely, activation of NADPH oxidase and production by endothelin-1.14 , 29 production in response to chronic PM2.5 exposure was abolished by the NAD(P)H oxidase inhibitor apocynin and the NOS inhibitor N-omega-nitro-L-arginine methyl ester (L-NAME), suggesting that -mediated reduction in nitric oxide bio-availability, due to NADPH oxidases and uncoupled NOS respectively may be an important mechanism inducing adverse vascular effects.32 , 33 Increased microvascular adhesion of inflammatory monocytes in the adipose microcirculation has been noted with concentrated PM2.5 exposure, together with perivascular deposition of mononuclear cells, with deficiency of Nox2 and Tlr4 improving vascular responsiveness.34 , 35 Deficiency of the p47phox sub-unit of NADPH oxidase also abolished production in the visceral adipose tissue from PM2.5-exposed animals and improves insulin resistance.36 While there are limited comparative studies of ultrafine particles relative to PM2.5, these generally show equal or in some cases larger effects for diesel exhaust exposures (Supplementary material online, Table S2). In at least one study, ultrafine exposure in ApoE− / − mice resulted in greater atherosclerosis compared to PM2.5. Besides the higher systemic penetration, exposure to ultrafine particles may result in inhibition of anti-inflammatory capacity of high-density lipoprotein and greater systemic oxidative stress as evidenced by increased hepatic malondialdehyde, up-regulation of Nrf2-regulated antioxidant genes and lipid peroxidation products in the plasma and liver.37 , 38
All ozone exposure studies to date are acute and are consistent in showing rapid (hours) endothelial dysfunction and enhanced vasoconstriction. The mechanisms appear to be related to rapid depletion of •NO, decrease in aortic eNOS levels, and reversibility with scavengers (Supplementary material online, Table S2). Some studies appear to suggest factors in the serum that induce endothelial dysfunction and neuronal inflammation. While an endothelial locus of generation of putative factors that then circulate to cause systemic vascular and neuronal injury has been theorized, these findings need to be confirmed.39 In the only primate study with ozone, acute exposure of primates to ozone (0.5 ppb) resulted in increased aortic mitochondrial damage. In one short-term 2-day study, there appeared to be no additive effects noted in ozone and diesel exhaust. In general, the concentration of ozone in rodent studies are 0.5–1 ppm, which is nowhere close to the concentration of doses used in human studies or for that matter the regulatory standards (current U.S. National Ambient Air Quality Standard is 0.075 ppm averaged for an 8-h period, while the European Commission has set a standard at 0.057 ppm). There are too few studies examining the differential impact of gases on vascular function and those that have been performed have examined a limited number of endpoints (Supplementary material online, Table S2).
There have been insufficient studies on the impact of chronic air pollution exposure on the surfactant milieu. The phospholipids and surfactant proteins, which are an important line of defence, are continually replenished and provide a potent barrier against air pollution; even those that manage to penetrate the distal airways. However, chronic ongoing exposure to air pollution may overcome surfactant defenses18 and increase oxidatively modified derivatives of 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (PAPC) and the major oxidation product of cholesterol, 7-ketocholesterol. These processes may participate in endothelial barrier dysfunction, inflammatory cell recruitment (Figure 3), and facilitate transition of air pollutants, chemokines, and other secondary mediator signals to the bone marrow and systemic circulation.34 These mediators may enhance oxidative stress in the vasculature via activation of TLR4 pathways in monocytes and macrophages.13 , 40 Deficiency of TLR4, NOX2, and p47phox have all been shown to attenuate ROS generation, reduce inflammatory monocyte infiltration into vasculature and improve vascular function in response to inhalational exposure to concentrated PM2.5.34 , 36 The formation of 7-ketocholesterol in response to chronic PM2.5 exposure and its accumulation within low-density lipoprotein and subsequent uptake by CD36, a scavenger receptor recognizing specific oxidized phospholipids and lipoproteins, facilitating entrapment within plaque macrophages, may represent a paradigm for air pollution mediated endothelial dysfunction and promotion of atherosclerosis.41 , 42 Impairment of endothelial regeneration owing to depletion of endothelial progenitor cells (EPCs) may also represent an important mechanism of sustained endothelial dysfunction (Supplementary material online, Table S2).
Two recent studies provide evidence that the pulmonary anti-oxidant barrier may be critical in modulation of systemic responses.43 , 44 In the first study, exposure to concentrated ambient PM2.5 (CAP) reduced insulin-stimulated Akt/eNOS activation in the aorta in 9 days, an effect reversed by the antioxidant TEMPOL or lung-specific overexpression of extracellular SOD.43 In the second study, a similar 9-day exposure to CAP reduced EPCs, VEGF (vascular endothelial growth factor)-stimulated aortic Akt phosphorylation, and plasma •NO levels in wild-type mice, but not in mice overexpressing extracellular superoxide dismutase (ecSOD-Tg) in the lungs. Further, EPCs from CAP-exposed wild-type mice failed to augment hindlimb perfusion when injected into unexposed mice subjected to hindlimb ischaemia. In contrast, EPCs derived from CAP-exposed ecSOD-Tg mice restored hind limb ischaemia.44 Adverse remodelling and fibrotic/atherosclerotic processes conferred by matrix metalloproteinases and TIMP-1 further contribute to endothelial and vascular damage (Supplementary material online, Table S2).
Activation of sympathetic nervous system and hypothalamic inflammation occurs in response to PM2.5 exposure with potentiation of blood pressure responses.45 Air pollutants can penetrate the central nervous system, inducing inflammation in areas of the central nervous system responsible for blood pressure regulation and metabolic control.1 , 26 , 46 , 47 Receptors such as the transient receptor potential cation channel, subfamily A, member 1 (TRPA1) receptors in airway sensory neurons can also sense the environmental toxicants and aerogenic oxidants, resulting in neurogenic inflammation.48 Inhibition of central IKKβ, prevented peripheral inflammation and abnormalities such as insulin resistance and whole body metabolism.40 , 45
Evidence of the association between particulate matter air pollution and oxidative stress/vascular dysfunction in humans
Evidence from panel studies and large epidemiological databases
Though not entirely consistent, the available studies demonstrate that acute exposure to air pollution may induce systemic oxidative stress in human subjects, under certain circumstances, that may depend on multiple factors, including chemical composition of the air pollutants, associated co-pollutants and host susceptibility.26 The evidence for changes in vascular function in panel studies where examined, has demonstrated associations across multiple acute time windows across a range of concentrations, including most recently with very high exposure levels in China (Supplementary material online, Table S3). Both the MESA-Air and the Framingham cohort have demonstrated that long-term exposures to ambient levels of PM2.5 are linked to chronic reductions in brachial endothelial function.49 , 50 Collectively, this supports the concept that air pollution exposure in humans through changes in oxidative stress causes vascular endothelial dysfunction, the hallmark physiological change underlying the initiation and progression of atherosclerosis. Platelet activation seems to be another hallmark of air pollution exposure in humans (Supplementary material online, Table S3).51
There have been insufficient studies on the impact of effect modifiers that may determine individual patient susceptibility. Polymorphisms in several oxidative stress-related genes such as GSTM1, GSTP1, GSTT1, HFE C282Y, CAT, and heme oxygenase have been found to be associated with the vulnerability to PM2.5.26
Controlled exposure studies
Several controlled exposure studies demonstrate that acute exposure to PM2.5 and dilute diesel exhaust result in rapid vascular dysfunction that manifests as conduit or microvascular endothelial dysfunction, or transient constriction of a peripheral conduit vessel that is reversible (Supplementary material online, Table S3). In some of these studies, concentrated PM2.5 exposure diminished conduit artery endothelial-dependent vasodilatation in a delayed fashion, after 24 h (but not immediately).52 PM2.5 mass and TNF-α level post-exposure have both been associated with the degree of endothelial dysfunction, suggesting that systemic inflammation induced by particles and the degree of pollution are likely responsible.52 Concentrated PM2.5 has not always been shown to induce endothelial dysfunction, underscoring the importance of composition, individual propensity, and methods of determining endothelial function, among many factors that determine vascular response.53 Studies of ultrafine particles including inhalation of elemental carbon as well as diluted diesel exhaust have been shown to induce rapid endothelial dysfunction in the microcirculation.54 , 55 The blunted responses to acetylcholine often persists for 24 h in healthy adults following cessation of exposure.56 In a randomized controlled clinical trial, diesel exhaust (200 mg/m3 of fine particulate matter), or filtered air increased blood pressure rapidly with persistent effects.57 Exercise-induced ST-segment depression and ischaemic burden were significantly greater during diesel exhaust exposure compared with filtered air exposure.58 Further analyses revealed that diesel exhaust but not filtered diesel exhaust causes impaired vasodilation to the endothelium-dependent vasodilators bradykinin and acetylcholine.59 In addition, in vitro organ chamber experiments with isolated vessels demonstrated that diesel exhaust particles and not carbon nanoparticles induce endothelial dysfunction, an effect that was markedly improved by the administration of superoxide dismutase suggesting that reduced vascular •NO bioavailability is likely due to increased production of ,59 although vascular •NO production by the •NO-synthase actually seems to be increased.60
Ozone exposures have been shown to impair conduit vessel endothelial function in panel studies (Supplementary material online, Table S3). However, acute exposure at least in one study did not demonstrate abnormalities in endothelial function. Similarly, many panel studies have demonstrated an association between NO2 levels, while, acute exposure to •NO2 failed to cause endothelial dysfunction in man.61 There are currently insufficient controlled interventional studies on other gaseous co-pollutants such as •NO2 and sulfur dioxide although the data at least from animals seems to suggest that these by themselves are less harmful but rather it is the associated co-pollutants admixture that may exert toxic effects.17
In summary, the preponderance of evidence supports an effect of particulate air pollutants on endothelial function in both conduit and resistance vessels, including the coronary circulation. Endothelial dysfunction may result in changes in arterial stiffness and afterload, that may translate into persistent hypertension with chronic exposure. Air pollution exposure, indeed, is associated with incident hypertension at both low levels and high levels with associations with traffic related air pollutants being strong.24 , 62 Chronic alterations in endothelial function and blood pressure may indeed represent an important mechanism for accelerated atherosclerosis. In the largest study, linking chronic exposures to coronary artery calcium (MESA-Air cohort, n = 6795 across 6 U.S. regions), each 5 µg/m3 increase in long-term PM2.5 exposure was associated with a greater progression of CAC (4.1 Agatston units/year).63
Air pollutants and cardiovascular risk factors
PM2.5 inhalation promotes not only acute elevations in blood pressure over hours-to-days but also chronic development of hypertension per se. This has been the topic of multiple systematic reviews and meta-analyses. One study that assessed the chronic (lifelong exposure) over >50 weeks in an animal model noted evidence of left ventricular hypertrophy, diastolic dysfunction, abnormalities in flow reserve, and molecular changes in the myocardium including foetal gene expression and increase in SERCA2a levels.64 This finding is supported by epidemiological studies linking traffic air pollutants and PM2.5 with left and ventricular structural changes (MESA-Air and the Jackson Heart Study).24 Similarly, PM2.5 has been associated with the development of insulin resistance, a finding that has been corroborated by epidemiologic evidence.47 While the mechanisms are likely multifactorial, given the widely accepted link between insulin resistance and endothelial dysfunction and the broad recognition that insulin’s action to enhance its own vascular delivery (and that of its substrates) is integral to its overall action, it is likely that insulin resistance is a direct consequence of air pollution mediated endothelial dysfunction.65 , 66 Also obesity26 and thrombosis51 are consequences of air pollution and represent important triggers of atherosclerosis leading to manifest cardiovascular events such as stroke, ACS, arrhythmia and heart failure (Figure 3).
Effects of air pollution mitigation on cardiovascular intermediate outcomes
The significant improvements in air quality during the past few decades has now been shown to translate into an increase in overall life expectancy and do so with no increase in economic costs.1 , 7 Further, improvement in air quality besides, the health co-benefit may bring much needed focus on issues such as global warming. Unfortunately, population growth, increasing energy and transportation demands, and numerous geopolitical-economic factors continue to delay improvements in air quality in many parts of the world. Unless some actions are taken to protect the health of millions of at-risk individuals, the burden of air pollution-related cardiometabolic diseases will only further increase. In this regard, personal protection through the use of masks, filtration devices and other strategies to reduce personal exposure are the only bulwark to protect individuals at risk for air pollution mediated CVD (Supplementary material online, Table S4). Several ‘personal-level’ strategies including drugs have been shown to reduce exposures and to improve adverse effects including vascular function, blood pressure, heart rate variability, and other intermediate outcomes.2
Air pollution and cardiovascular risk: a role for the cardiovascular community and society at large
Despite the overwhelming evidence concerning the detrimental effects of air pollution on cardiovascular health including development of risk factors such as hypertension and diabetes, as well as cardiovascular events, there are no randomized controlled trials that have investigated the efficacy of pollution-directed interventions on cardiovascular outcomes such as stroke, ischaemic heart disease, heart failure, arrhythmia, and all-cause cardiovascular mortality.24 , 67 Further research is needed to identify interventions that reduce risk to pollution exposures such as improved ventilation and indoor air filtration systems, facemasks and medicines to reduce impact of air pollution and also harmonization of risk assessment models is urgently needed. There is little mention of this risk factor in the European and American guidelines. The ESC Guidelines for prevention only mention that air pollution can adversely affect cardiovascular health68 but to date do not provide concrete recommendations on what the individual can do to mitigate risk and what solutions can be provided to governmental and non-profit agencies to impact air pollution levels and to protect societal health. It would be important that legal PM2.5 thresholds worldwide adhere to the WHO air quality guidelines in the future since those in Europe are still high (25 µg/m3) when compared with those in Canada and the USA (10–12 µg/m3). It is only a matter of time that the continued drumbeat of evidence will have to persuade the cardiovascular community that concrete guidance on personal and societal mitigation of the threat of air pollution may need to be part of parcel of guidance on cardiovascular risk prevention. We have to acknowledge that air pollution is a cardiovascular risk factor, that governments and non-profit organizations may need to focus on and not simply health professionals. Given the multiple constraints faced by health care providers, identification of target patients most likely to benefit from intervention could be facilitated as an initial step to protect vulnerable patients. Quantitative estimates of at risk populations and translation of exposures into individual estimates of risk may be possible in the future with personalized approaches.69 Finally, broad societal based interventions are mandatory to tackle the risk posed by air pollution.
Funding
The present work was supported by a vascular biology research grant from the Boehringer Ingelheim Foundation for the collaborative research group, Novel and neglected cardiovascular risk factors: molecular mechanisms and therapeutic implications to study the effects of new cardiovascular risk factors (to A.D. and T.M.). T.M. and T.G. are PIs of the DZHK (German Center for Cardiovascular Research), Partner Site Rhine-Main, Mainz, Germany. This work was also supported by 5U01ES026721-03, R01ES026291 and R01ES015146 (to S.R.).
Conflict of interest: none declared.
References
Author notes
Andreas Daiber and Sanjay Rajagopalan authors contributed equally to this study.


{kind=link}
{kind=link}
{kind=link}