





ABSTRACT
In the rhizosphere, complex and dynamic interactions occur between plants and microbial networks that are primarily mediated by root exudation. Plants exude various metabolites that may influence the rhizosphere microbiota. However, few studies have sought to understand the role of root exudation in shaping the functional capacities of the microbiota. In this study, we aimed to determine the impact of plants on the diversity of active microbiota and their ability to denitrify via root exudates. For that purpose, we grew four plant species, Triticum aestivum, Brassica napus, Medicago truncatula and Arabidopsis thaliana separately in the same soil. We extracted RNA from the root-adhering soil and the root tissues, and we analysed the bacterial diversity by using 16S rRNA metabarcoding. We measured denitrification activity and denitrification gene expression (nirK and nirS) from each root-adhering soil sample and the root tissues using gas chromatography and quantitative PCR, respectively. We demonstrated that plant species shape denitrification activity and modulate the diversity of the active microbiota through root exudation. We observed a positive effect of T. aestivum and A. thaliana on denitrification activity and nirK gene expression on the root systems. Together, our results underscore the potential power of host plants in controlling microbial activities.
INTRODUCTION
The ability to secrete a wide range of compounds into the rhizosphere is one of the most remarkable metabolic features of plant roots with ~5–21% of the total photosynthetically fixed carbon being transferred into the rhizosphere through root exudates (Whipps, 1990; Marschner, 1995; Nguyen, 2003). These compounds are metabolized by soil-borne microorganisms as carbon and energy sources providing the basis for the establishment of plant-microorganism interactions that benefit plant growth by increasing the availability of mineral nutrients, production of phytohormones, degradation of phytotoxic compounds and suppression of soil-borne pathogens (Bais et al.2006; Philippot et al. 2013; Haichar et al.2014). This demonstrates the importance of studying the functional properties of the soil microbiota and the manner in which plant species influence bacterial diversity and microbial activities.
Denitrification is one of the microbial activities that occurs in the plant rhizosphere. Denitrification is the microbial process of the nitrogen cycle that allows the return of fixed nitrogen (reduction of atmospheric N2) to the atmosphere. The reduction of soluble NO3− or NO2− into gas (NO, N2O or N2) by denitrification is catalysed by metalloenzymes: nitrate reductases (Nar and Nap), nitrite reductases (NirK and NirS), nitric oxide reductases (cNor and qNor) and nitrous oxide reductase (Nos) (Zumft, 1997; Philippot, 2006). This stepwise reduction of nitrate is an alternative respiration pathway occurring in the case of oxygen depletion by phylogenetically diverse microorganisms in a wide range of ecosystems (Zumft, 1997). Most denitrifiers belong to various subclasses of bacteria, although the ability to denitrify has also been found in some archaea and fungi (Philippot, 2002). In addition, co-occurrences of denitrification genes do not appear to be randomly distributed among taxonomic groups (Graf et al.2014).
Denitrification represents a significant loss of nitrogen in soils (25–90%) (van der Salm et al.2007; Radersma and Smit, 2011), and this microbial process therefore affects the availability of nitrogen to plants. This process contributes to N2O emission, a powerful greenhouse gas (300-fold more heat-trapping capacity than CO2 per molecule) and the single most important ozone-depleting agent known (Ravishankara, Daniel and Portmann 2009; Coskun et al.2017).
The major factors regulating denitrification can be modulated in the rhizosphere: nitrate concentration (via absorption by plants) and oxygen partial pressure (via root respiration and the surrounding root moisture) are decreased, while the C availability (via rhizodeposition) is generally increased (Tiedje, 1988; Mounier et al.2004;). The impact of the rhizosphere on denitrifying activity has already been reported (Mahmood et al.1997; Mounier et al.2004; Henry et al.2008). For example, Philippot et al. (2002) observed that the presence of maize roots led to a change in the structure of the nitrate-reducing community. However, little is known about how denitrification is regulated in the rhizosphere. Determining the role of host plant on denitrification process modulation is of real importance in understanding the regulation of denitrification in the rhizosphere.
The aim of this study was to determine the impact of four plant species, wheat (Triticum aestivum), rape (Brassica napus), barrel clover (Medicago truncatula) and Arabidopsis thaliana (ecotype Colombia), cultivated in the same soil on (i) the diversity of the bacterial community by metabarcoding the 16S rRNA gene genes, and on (ii) the activity of the denitrifying community in the root-adhering soil (RAS) and on the root system (RS) by evaluating the denitrification activity and nirK and nirS gene expression.
MATERIALS AND METHODS
Plant growth
A laboratory experiment was performed with winter wheat (T. aestivum L. cv. Taldor), rape (B. napus cv. Drakkar), A. thaliana (ecotype Colombia) and barrel clover (M. truncatula, ecotype A17) on a calcareous silty-clay soil collected from the upper 20 cm layer of the agricultural site located in the Aix en Provence region. The pH was 8.2 and it contained 5.7% sand, 46.7% silt, 47.6% clay, 1.0% CaCO3, 1.8% organic C and 0.18% organic N. The soil was sieved (1-mm mesh size), air-dried, and the water-holding capacity (WHC) measured according to Bouyoucos (1929) based on the pulling out of water from soil by suction or vacuum forces. The WHC was determined in triplicates and was ~20%. Next, 150 g dry soil was placed into polypropylene cylindrical pots. Soil moisture was maintained at 75% of WHC by adding the necessary amount of water. Seeds were sterilised according to Achouak et al. (2004) and one seed was planted per pot. Sixteen pots of each plant were grown in a growth chamber. Four pots (four repetitions) per plant were dedicated to denitrification activity measurements (in the RAS and the RS), three pots (three repetitions) per plant were dedicated to molecular analysis (16S rRNA diversity from the RAS and the RS) and nine pots per plant were dedicated to isotope labelling experiments. Sixteen pots with soil but without plants (bulk soil treatment) were used: four pots (four repetitions) were dedicated to denitrification activity measurements, three pots (three repetitions) were dedicated to molecular analysis (16S rRNA diversity) and nine pots were dedicated to isotope labelling experiments. Plants and bulk soil pots were incubated in a growth chamber with a day–night period ~12/12, respectively; light intensity was 400 mmol photon m−2.s−1 and maximum daily temperature ranged from 20–22°C. Soil moisture was manually controlled.
Plant harvesting
After five weeks of plant growth, plants were collected as follows: the RS of three replicates of each plant were separated from its RAS and 2 g of RAS from each plant were frozen in liquid N2 immediately and stored at -80°C for molecular analysis. The RSs were washed with water to remove adhering soil particles, frozen in liquid N2 and stored at -80°C. The bulk soil microcosms (four replicates) were also collected for microbial activities measurements and 2 g were frozen in liquid N2 immediately and stored at -80°C for molecular analysis.
The entire plant from four replicates for each plant was separated from the RAS and both, the whole plant and the RAS of the planted microcosm and soil of the unplanted microcosm, were used to measure denitrification activity.
The remaining nine replicates for each plant and the bulk soil treatment were used for isotope labelling experiments.
Measurements of microbial denitrification activities
Denitrification Enzyme Assay without carbon addition
We measured the denitrification activity of microbial communities inhabiting the RS in each plant rhizosphere in four replicates according to Guyonnet et al. (2017). The entire plant with the RS, rinsed with water to remove soil particles, was placed in a 150 ml airtight plasma-flask sealed by a rubber stopper. In each flask, air was removed and replaced with a He/C2H2 mixture (90/10 v/v) to create anoxic conditions and inhibit N2O-reductase. Potassium nitrate (50 µg of N-KNO3 g−1 of fresh root or dried soil) was added to each vial to provide microbial communities with a nitrate source. The amount of N2O during incubation at 28°C was measured each 4 h for 48 h. The slope of the linear regression was used to estimate anaerobic respiration (denitrification enzyme assay without the addition of carbon) to estimate the N2O (g−1.h−1) produced. N2O was measured with a gas chromatograph coupled to a micro-catharometer detector (µGC-R3000, SRA Instruments, Marcy l'Etoile, France).
Denitrification enzyme activity of plant roots and soils
We measured the denitrification enzyme activity (DEA) in soil, according to Bardon et al. (2014). Ten grams of fresh soil sample (RAS from each plant and bulk soil) were placed in a 150 ml airtight plasma-vial sealed with a rubber stopper. In each flask, air was removed and replaced with a mixture of He/C2H2 (90/10 v/v) to create anoxic conditions and inhibit N2O-reductase. A nutritive solution (0.5 ml) containing glucose (0.5 mg of C-glucose.g−1 of dried soil), glutamic acid (0.5 mg of C-glutamic acid g−1 of dried soil) and potassium nitrate (50 µg of N-KNO3 g−1 of dried soil) was added to the soil. The amount of N2O during incubation at 28°C was measured each hour for 5 h. The slope of the linear regression was used to estimate anaerobic respiration (denitrification) by measuring the production of N2O (g−1.h−1) using a gas chromatograph coupled to a micro-catharometer detector (µGC-R3000).
RNA extraction and cDNA synthesis
RNA was extracted from the RAS and RS from each plant rhizosphere in triplicate using an RNA ‘Power Soil isolation’ kit (MO BIO USA) that produced total non-degraded RNA according to the manufacturer's recommendations. DNA was removed from RNA extracted from RAS and root tissues by using RNeasy Mini Kit (Qiagen, Courtaboeuf, France) according to the manufacturer's recommendations. Next, 16S rRNA gene amplifications (300 bp) were carried out using the treated RNA as a template to ensure that DNA was completely removed. One µg of RNA from each RAS and RS sample retrieved from each plant was transcribed into complementary DNA (cDNA) using the SuperScript III Reverse Transcriptase (Life Technologies, Regensburg, Germany) according to the manufacturer's protocol and stored at -20°C.
nirK and nirS genes expression
Denitrifier abundance was estimated by real-time quantitative reverse transcription PCR (qRT-PCR) targeting the nirK and nirS genes encoding the copper and cd1 nitrite reductases, respectively. For nirK, the amplification was performed using the primers nirK876 (5’-ATYGGCGGVCAYGGCGA-3’) and nirK1040 (5’-GCCTCGATCAGRTTRTGGTT-3’) (Henry et al. 2004). The 20 µl final reaction volume contained SYBRgreen PCR Master Mix (QuantiTect SYBRgreen PCR kit, Qiagen), 1 µM of each primer, 400 ng of T4gp32 (MPbiomedicals, Illkvich, France) and from 1 to 3 µl of cDNA according to 16S rRNA gene normalisation. Thermal cycling was as follows: 15 min at 95°C; six cycles of 95°C for 15 s, 63°C for 30 s with a touchdown of -1°C by cycle, 72°C for 30 s; 40 cycles of 95°C for 15 s, 58°C for 30 s and 72°C for 30 s. For nirS, the amplification was performed using the primers nirSCd3aF (5’-AACGYSAAGGARACSGG-3’) and nirSR3cd (5’-GASTTCGGRTGSGTCTTSAYGAA-3’) (Throbäck et al. 2004). The 25 µl final reaction volume contained SYBRgreen PCR Master Mix (QuantiTect SYBRgreen PCR kit, Qiagen), 1 µM of each primer, 400 ng of T4gp32 (MPbiomedicals) and from 1 to 3 µl of cDNA according to 16S rRNA gene normalisation. Thermal cycling was as follows: 15 min at 95°C; six cycles of 95°C for 15 s, 59°C for 30 s with a touchdown of -1°C by cycle, 72°C for 30 s and 80°C for 30 s; 40 cycles of 95°C for 15 s, 54°C for 30 s and 72°C for 30 s and 80°C for 30 s. The standard curves for nirK and nirS qPCR were generated by amplifying 10-fold dilutions (107–102) of a linearised plasmid containing the nirK gene of Sinorhizobium meliloti 1021 and nirS gene of Pseudomonas stutzeri Zobell DNA (GenArt, Invitrogen, Life Technologies). Melting curves analysis confirmed that the specificity of amplification and amplification efficiencies for nirK and nirS genes were higher than 90%.
Sequencing of 16S rDNA gene
For each plant, 10 µL of cDNA obtained from RNA extracted from the RS and the RAS from each plant in triplicate were sent to FASTERIS (Switzerland) for sequencing using MiSeq Illumina technology. The V3-V4 domain of 16S rRNA was amplified with tagged primers 16S Fwd primer 3’-CCTACGGGNGGCWGCAG-5’ and 16S Rev primer 3’-GACTACHVGGGTATCTAATCC-5’. The tagged primers' structure was 5’- N2–4X6Pn -3’, with ‘N2–4’ random bases, ‘X6’ 6-bases tag and ‘Pn’ was the specific primer. Amplification conditions were 3 min at 95°C, 35 cycles of 30 s at 95°C, 30 s at 55°C, 90 s at 72°C and 5 min at 72°C.
Isotope labelling and analysis of plant and soil material
After five weeks of plant growth, all pots (nine pots per plant and nine pots of bulk soil) received the same amount of two forms of nitrogen: ammonium (NH4+) and nitrate (NO3−), and where the nitrogen was either 14N or 15N. Three replicates from each plant and bulk soil treatment received 15N labelled ammonium and unlabelled nitrate (15NH4+ + NO3−) and three other replicates received unlabelled ammonium and 15N labelled nitrate (NH4+ + 15NO3−). The remaining three replicates per plant and bulk soil treatment received unlabelled nitrogen (NH4+ + NO3−) in order to measure the natural abundance of 15N. The four plant species and the unplanted soil underwent three treatments in triplicate resulting in 45 pots.
In total, 0.24 µg N.g−1 dry soil was added with 50% of each nitrogen form. Five millilitres of each solution were added to each pot in 1 ml aliquots with a single injection homogeneously distributed over the soil surface. Each aliquot was injected with a syringe with 21-gauge needle that was slowly removed to ensure uniform distribution throughout the profile. After 12 h of incubation, the shoots of all pots were cut off and stored at -80°C. The RSs and their RAS fractions were subsequently separated. The roots were rinsed with tap water. All roots and RAS samples were stored at -80°C. The 12 h incubation time was used in accordance with previous studies, which showed maximal uptake of label around this time (Streeter, Bol and Bardgett 2000; Bardgett, Streeter and Bol 2003; Weigelt et al.2003).
Values of atom fraction and concentration of N (in %) were used to calculate uptake of 15N on a per unit mass basis (mg excess of 15N per gram). Mean values of 15N abundance of the unlabelled control plants were used as reference for 15N excess.
Bioinformatics analysis
Sequence processing and data analysis
Sequence data were processed and analysis of high-throughput community sequencing data was performed with QIIME version 1.8 (Caporaso et al.2010). Sequences were trimmed of barcodes and primers, and then short sequences (<200 bp), sequences with ambiguous base calls, and sequences with homopolymer runs >6 bp were removed. Operational Taxonomic Units (OTUs) were then defined by clustering at 97% similarity. Final OTUs were taxonomically classified using Blast (Altschul et al.1990) and compiled at each taxonomic level into both ‘counts’ and ‘percentage’ files. OTU tables were rarefied at the lowest sequencing depth to control for differences in sequencing depth. A total of 7185 297 valid reads and 392 892 OTUs were obtained from the 27 samples through sequencing analysis [three root samples and three root-associated soil samples per plant species (four species) and three bulk soil samples].
Alpha diversity analysis was performed using the Phyloseq R package version 1.20.0 (McMurdie and Holmes 2013). Species richness (observed richness) and species evenness (Inverse Simpson index) were estimated by multiple subsampling with replacement (n = 103) to the minimum libraries' sizes to standardise the sequencing effort.
Core microbiota
The core microbiota was identified using QIIME (Caporaso et al.2010) and was determined by plotting OTU abundance in the core at 5% intervals (from 50% to 100% of samples). We defined the core microbiota of each plant as the OTUs present in 100% of samples. Determination of a core microbiota was accomplished by comparing all samples from each plant across the two compartments (RAS and root). Any taxa found to be ubiquitous across all samples were then defined to be part of the core microbiota of the compartment. From these data Venn diagrams were constructed using the R package: VennDiagram, to show common and unique OTUs within the four plant species (T. aestivum, B. napus, M. truncatulaandA. thaliana).
Network analysis
Network inference was made by computing all Spearman's rank correlations between all OTUs of the ‘family’ taxonomic level. P-value was adjusted using the procedure of Benjamini and Hochberg (1995) to finally consider only significant correlation (Spearman correlation coefficient |r| >0.8 and P-adj <0.05). The same procedure was used to compute the correlation between each OTU and each compartment (RS and RAS). Each network was visualised, analysed and different metrics (Table S1) (i.e. degree, diameter, average path length, average clustering coefficient, modularity) were calculated using Gephi open-source software (Bastian, Heymann and Jacomy 2009).
We analysed OTUs' interactions by comparing the high and low DEA graphs for the RS and RAS compartments. By computing intersection, union or difference of these graphs, we can retrieve edges present or not in our DEA condition of interest. The graphical analysis was performed using the igraph R package.
Statistical analysis
All results are presented as means (± standard error). For each plant, a one-way analysis of variance (ANOVA) and post-hoc Tukey HSD were performed to test the effect of root exudation on measured variables. Before analysis, Shapiro and Bartlett tests were performed to ensure conformity with the assumptions of normality and homogeneity of variances. Effects with p <0.05 are referred to as significant.
In order to test the significance between microbiota inhabiting the RAS and those colonising the root tissues in all plants a non-parametric permutation-based multivariate analysis of variance (PERMANOVA, vegan R package)) on abundance-based (Bray-Curtis) dissimilarity matrix was performed. All statistical analyses were carried out using R statistical software 3.1.0. ).
Data deposition
The sequence data generated in this study was deposited at the EMBL-ENA public database (http://www.ebi.ac.uk/ena/data/view/PRJEB25281).
RESULTS
Denitrifying enzyme activity in plant root systems
To investigate the impact of the root exudates produced by each plant species on denitrifying activity, we measured the emission of N2O after nitrate amendment only in each plant RS, without adding any carbon source (Fig. 1A). The denitrification activity of the microbiota colonising the RS of each plant species differs significantly from one another. The denitrification activity was higher in the RS of T. aestivum and A. thaliana (5.2 and 4.6 g N-N2O h−1.g−1 dried roots, respectively) and very low in the RS of M. truncatula and B. napus (0.6 and 0.08 g N-N2O h−1.g−1 dried roots, respectively). Interestingly, B. napus root exudates appear to inhibit denitrification activity in the RS or counterselect denitrifying microorganisms.
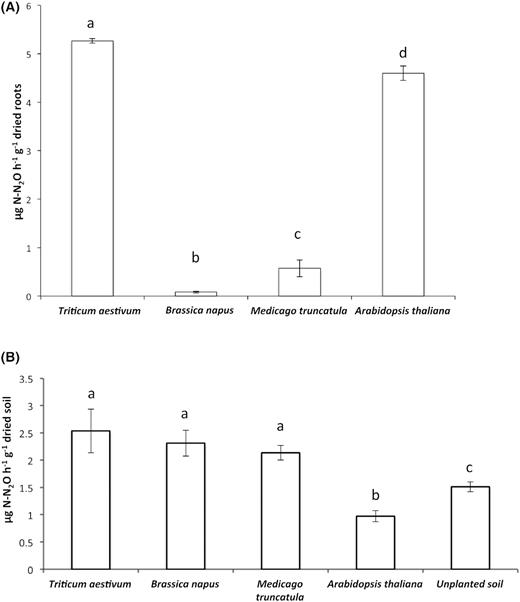
Denitrification activities of microbiota colonising the root system and inhabiting the root-adhering soil of wheat (Triticum aestivum), rapeseed (Brassica napus), barrel clover (Medicago truncatula) and Arabidopsis thaliana plantlets. (A) Impact of root exudates on denitrifying activity of microbial community colonising the root system. Denitrification activity (g N-N2O h−1.g−1 dried roots) was measured in triplicate after the addition of nitrate source only. (B) Denitrification enzyme activity (DEA) of soil samples amended with nitrate (50 µg of N-KNO3 g−1 of dried soil) and carbon sources (0.5 mg of C-glucose and 0.5 mg of C-glutamic acid g−1 of dried soil) was measured in triplicate. Letters show which means differed between treatments (Tukey's test; α = 0.05). Vertical bars: means ± standard errors.
Denitrifying enzyme activity in the root-adhering soil
To determine the potential rate of the denitrification of microorganisms present in the RAS, the DEA of the RAS fractions retrieved from each plant rhizosphere was measured. The DEA of T. aestivum, B. napus and M. truncatula are significantly different from the unplanted soil (Fig. 1B), indicating a rhizosphere effect. However, the DEA of A. thaliana is significantly lower than that of the unplanted soil and the rhizosphere of the other plants. In addition, T. aestivum, B. napus and M. truncatula do not differ significantly from each other (Fig. 1B).
Preferential uptake of soil nitrogen forms by plant species: NO3− and/or NH4+
In shoot tissues, the 15N content after the separate addition of NO3− or NH4+ for each plant species reveals a significant preferential uptake of NO3− by T. aestivum, B. napus and A. thaliana plantlets (Fig. 2A). Conversely, none of the forms of N tested are preferentially absorbed by M. truncatula under our experimental conditions. B. napus had a significantly greater uptake capacity for NO3− than the other plant species. The patterns of the root tissue contents in 15N show the same trend as the shoot tissues with a significant preferential uptake of NO3− by T. aestivum and B. napus (Fig. 2B). As with the shoot tissues, M. truncatula uptakes NO3− or NH4+ forms of nitrogen.
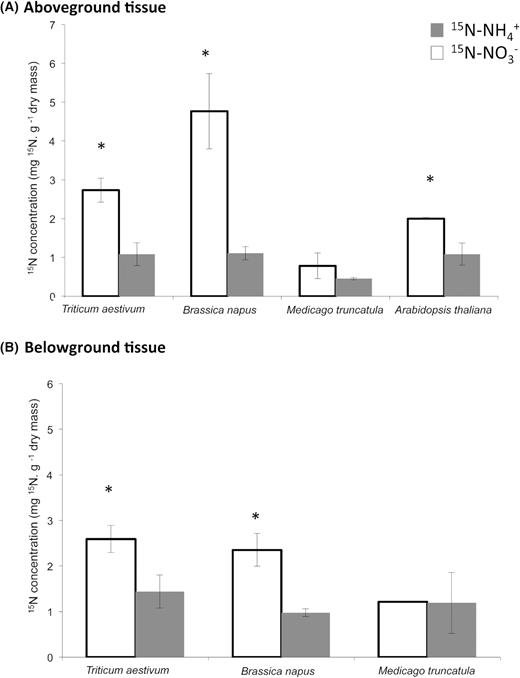
Nitrogen uptake of ammonium and nitrate for (A) aboveground and (B) belowground tissues 12 h after 15N labelling (NO3− or NH4+) of wheat (Triticum aestivum), rapeseed (Brassica napus), barrel clover (Medicago truncatula) and Arabidopsis thaliana plantlets. Vertical bars: means ± standard errors. Means significantly different from 15N-NO3−: *, P <0.05. Nitrogen uptake was not measured belowground for A. thaliana due to low amount of roots.
Diversity of active microbiota inhabiting each plant rhizosphere
To investigate the impact of the root exudates produced by each plant species on the selection of active microbiota, the RNA extracted from the RAS and the RS was reverse transcribed and analysed using 16S rRNA gene sequencing. An average of 7185 297 sequences were generated and utilised for analysis. The rarefaction curves, displaying the observed OTUs' richness as a function of the sequencing effort, indicated that the sequencing depth had almost been reached to completely capture the diversity present in the bulk soil, RAS and root tissues of all the plants (Fig. S1). The richness and diversity of species (Inverted Simpson) were significantly increased in the RAS compared with the bulk soil for each plant, except for M. truncatula, and decreased in the root compartment compared with the RAS fraction, especially for A. thaliana and B. napus (Fig. S2).
Several phyla were present in different amounts between the two compartments studied (RAS and RS) and between the plants (Fig. 3A). In addition, globally in all plants, significant differences (p-value = 0.001) were observed between microbiota inhabiting the RAS and those colonising the RS. The general trend is a very low difference between microbial diversity in the RAS fraction of all the plantlets compared with the one of the reservoir (bulk soil) at the phyla level. Another general trend is an important increase in the Bacteroidetes abundance (5-fold in the rhizosphere of T. aestivum, B. napus and M. truncatula) and of the OD1 phylum (from x15 to x150 in the four rhizosphere samples) (Additional File 1). In the root tissues, Chloroflexi was more abundant in the RS of T. aestivum (8%) than the other plants (3–5%), while Bacteroidetes was more abundant in the RS of B. napus (15%) and T. aestivum (13%) compared with the other plants (9–10%) (Fig. 3A, Additional File 1). Verrucomicrobia was highly enriched in the RS of B. napus (8%) compared with the other plants (1–3%). Firmicutes and Acidobacteria were less abundant in the plant RSs (1–2% and 1–3%, respectively), except in the root tissues of A. thaliana where they were slightly more abundant (3% and 5%, respectively) (Fig. 3A, Additional File 1). The relative abundance of Crenarchaeota was increased in the RS of A. thaliana (0.3%) compared with that of the other plants (<0.07%). For the RAS fractions, few differences in phyla abundance were observed between the plant species. However, Bacteroidetes was more abundant in the RAS of A. thaliana (6%) than in those of T. aestivum, B. napus and M. truncatula (2–3%). Planctomycetes was counter-selected in the RAS fraction of A. thaliana (9%) compared with other plant rhizospheres and bulk soil (15–17%) (Fig. 3A, Additional File 1).
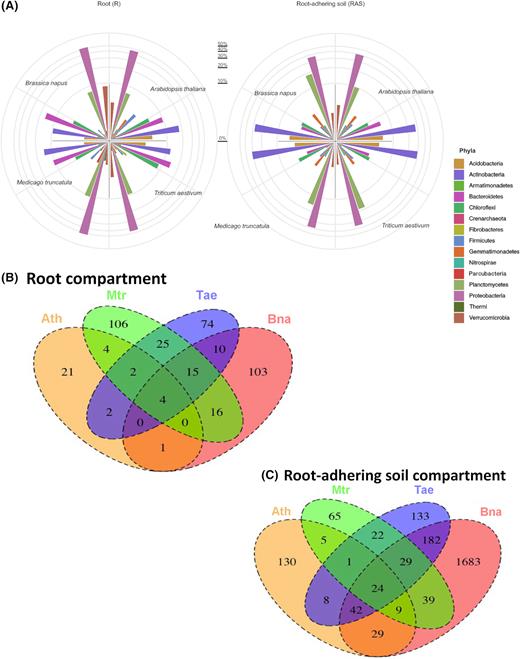
Analysis of bacterial diversity. (A) Distribution of the 15th major bacterial phyla (abundance in %) among the root system and the root-adhering soil of wheat (Triticum aestivum), rapeseed (Brassica napus), barrel clover (Medicago truncatula) and Arabidopsis thaliana plantlets. The Venn diagrams show shared and unique bacterial OTUs at 100% identity among bacterial community (B) colonising the root system and (C) those inhabiting the root-adhering soil retrieved from the rhizosphere of wheat (Triticum aestivum), rapeseed (Brassica napus), barrel clover (Medicago truncatula) and Arabidopsis thaliana.
Some phyla have been more enriched in the RAS than in the root tissues and vice versa, independently of the plant species (Fig. 3A, Additional File 1). For example, this is the case for Actinobacteria, Gemmatimonadetes, Plantomycetes and Crenarchaeota, which were more abundant in the RAS of B. napus, T. aestivum and M. truncatula compared with their RSs (from 2-fold to 10-fold increases). Despite the high abundance of Proteobacteria in all the plant rhizospheres, they were more abundant in the root tissues (44–52%) than in the RAS (34–39%).
To examine a potential role of the plant species in the root-associating bacterial assemblage, we compared the bacterial profiles obtained from T. aestivum, B. napus, M. truncatula and A. thaliana root compartments (Fig. 3B). Within the root OTUs (rOTUs) community, only four OTUs were enriched in the RSs of all the plants studied, while 21, 106, 74 and 103 OTUs were significantly enriched in the RSs of A. thaliana, M. truncatula, T. aestivum and B. napus, respectively (Fig. 3B, Additional File 2). The common and unique OTUs retrieved from the RS of each plant are shown in Additional File 2.
To examine the impact of the plant species on shaping the bacterial community inhabiting the RAS, we compared the bacterial profiles obtained in the RAS of T. aestivum, B. napus, M. truncatula and A. thaliana (Fig. 3C). We identified 24 RAS OTUs (rasOTUs) shared by the four plant species. These shared rasOTUs can be regarded as the core microbiome of the four rhizospheres. Unique rasOTUs corresponding to the bacterial community specifically inhabiting each plant species were also observed with 130, 65, 133 and 1683 OTUs found in the rhizospheres of A. thaliana, M. truncatula, T. aestivum and B. napus, respectively. Common and unique OTUs retrieved from the plant RAS are shown in Additional File 3.
Network description
To explore co-occurrences among the bacterial phyla colonising the RSs and inhabiting the rhizospheres (RAS fractions) of T. aestivum, B. napus, M. truncatula and A. thaliana, we used a network inference based on strong and significant correlations (using non-parametric Spearman's) (Fig. 4, Table S1). The number of positive correlations (co-occurrences) was higher than the number of negative correlations (co-exclusions) in the rhizospheres of B. napus and A. thaliana and equal for the two others (T. aestivum andM. truncatula) (Table S1).
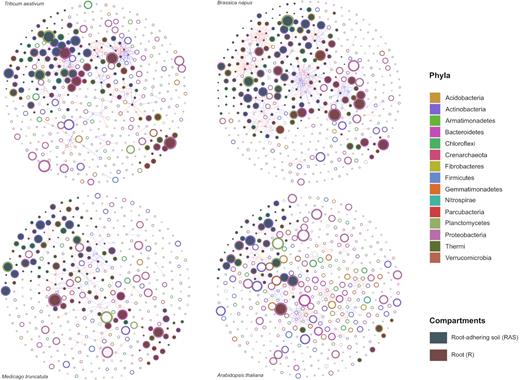
Significant co-occurrence and co-exclusion relationships among the microbiota inhabiting the root-adhering soil (in green) and colonising the root system (in brown) of wheat (Triticum aestivum), rapeseed (Brassica napus), barrel clover (Medicago truncatula) and Arabidopsis thaliana plantlets. The colour of nodes (rings of the circles) corresponds to the phylum, while the size of the nodes is proportional to their abundance. The red and blue lines specify significant positive and negative correlations (Spearman correlations ≥0.8 or ≤-0.8, P.adj < 0.05) between two nodes, respectively. Full circles of a green or brown colour correspond to OTUs more abundant in the root system and the root-adhering soil, respectively. Empty circles correspond to OTUs with the same abundance between the root system and the root-adhering soil compartments.
The bacterial network of OTUs from the RS (rOTUs) and the RAS (rasOTUs) compartments of B. napus is highly connected (Fig. 4). The structure of the network demonstrates densely connected groups of nodes, forming a clustered topology. Comparing the properties of the calculated networks, we observed that the B. napus network contains a significantly higher percentage of negative (220) and positive (171) edges than the other plants (Table S1). OTUs considered to be keystone species belonged primarily to different genera within the phyla of Proteobacteria, Planctomycetes and Actinobacteria (Fig. 4).
The T. aestivum bacterial network that associates with its rOTUs and its rasOTUs is also highly connected (Fig. 4) with 105 positive edges and 110 negative edges (Table S1). The OTUs considered to be keystone species (depicted as nodes with larger sizes in the network) belonged primarily to different genera within the phyla of Proteobacteria, Bacteroidetes and Actinobacteria (Fig. 4).
For the bacterial networks of M. truncatula and A. thaliana, we noticed a clear separation between the rOTUs and rasOTUs with a very weak connection between them. More bacterial phyla colonising RAS co-occur in the rhizosphere of M. truncatula compared with the RS (Fig. 4). For A. thaliana, the co-occurring bacterial phyla display an equal distribution in the RAS and the RS, and very few co-occur on the root tissues.
Finally, co-exclusions and co-occurrences found between certain rOTUs and/or rasOTUs retrieved from each plant are shown in Additional File 4. Overall, regardless of the plant studied, we noticed more co-exclusion and less co-occurrence relationships among the significant interactions (Fig. 4 and Table S1). Among several positive interactions observed, we noted strong co-occurrence relationships for some members of Firmicutes (Paenibacillaceae) with Acidobacteria and Actinobacteria (Frankiaceae) in the RAS of B. napus (Additional File 4). A positive correlation was also observed between Nitrospiraceae and Actinobacteria in the RAS of B. napus and A. thaliana and between Nitrospiraceae and Planctomycetes in the RAS of T. aestivum, B. napus and A. thaliana. A positive correlation was also observed between Rhizobiaceae and Chloroflexi and between Chlamydiales and Planctomycetales in the RSs of T. aestivum and M. truncatula, respectively (Additional File 4).
Among several negative interactions observed, we noted a co-exclusion relationship for A. thaliana for the OTUs of the Acidobacteria (Chloracidobacteria) found in the RAS with the OTUs of the Firmicutes (Paenibacillaceae) found in the RS (Additional File 4). In the rhizosphere of T. aestivum, the OTUs from Gemmatimonadetes were negatively correlated with those of the Opitutales (Verrucomicrobia). For example, in the rhizosphere of B. napus, the OTUs of the Chloroflexi correlate negatively with the OTUs of the Comamonadaceae.
By comparing OTUs' interactions under high and low DEA conditions in the RS and RAS compartments of the studied plant, we have identified a few OTUs that co-occur only in the RAS of T. aestivum and A. thaliana, with high DEA, while other OTUs co-occur only in the RS of T. aestivum and B. napus with high DEA (Additional File 5), suggesting a potential role for these OTUs in denitrification. This is the case, for example, of the co-occurrence in the RAS of T. aestivum and B. napus between OTUs of Nitrospira and Solirubrobacterales and Rhodospirillales and Candidatus nitrososphaera or between OTUs of Acidobacteria and Verrucomicrobia in the RS of T. aestivum and A. thaliana (Additional File 5).
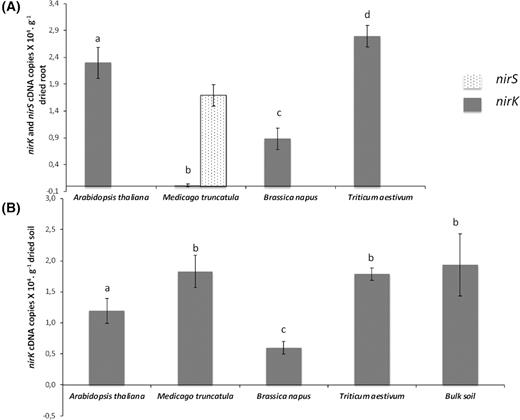
nirS and nirK transcript levels in the rhizosphere microbiota
To gain additional insights into the role of root exudates in regulating denitrification, we measured the expression of the nirK and nirS genes in the RS and in the RAS of each plant species (Fig. 5). The number of nirK (encoding a copper-containing nitrite reductase) transcripts from the denitrifiers was higher in the RS of A. thaliana and T. aestivum (2.3 × 104 and 2.8 × 104nirK cDNA copies per g of dried root, respectively), in contrast with a lower expression in the RS of B. napus (0.9 × 104nirK cDNA copies per g of dried root) and the almost complete absence of nirK expression in the RS of M. truncatula (Fig. 5A). The nirS gene that encodes a cytochrome cd1-containing nitrite reductase was detected only in the RS of M. truncatula (1.7 × 104nirS cDNA copies per g of dried root).
Predictive functional metagenome composition. Heatmap of the normalised relative abundances of the predicted functional categories (level 3) of the microbiota colonising the bulk soil, the root system and the root-adhering soil (RAS) of wheat (Triticum aestivum), rapeseed (Brassica napus), barrel clover (Medicago truncatula) and Arabidopsis thaliana plantlets.
Given the level of expression of the nirK gene in the RAS, no significant difference was found between the RAS of T. aestivum and M. truncatula compared with the bulk soil. In contrast, the expression of nirK was significantly decreased in the RAS of A. thaliana and B. napus compared with the bulk soil (Fig. 5B). As in the RS, the expression of nirK is the lowest in the RAS of the B. napus plantlets (Figs 5A and 5B). No expression of nirS was detected in the RAS fraction of the four plant species and in the bulk soil. In summary, only nirK (not nirS) was expressed in the RS of T. aestivum and A. thaliana and, to a lesser extent, in the one of B. napus and repressed in the presence of M. truncatula. nirK was repressed in the RAS of A. thaliana and B. napus and unchanged in that of T. aestivum and M. truncatula. Surprisingly, nirS expression was detected only in the RS of M. truncatula.
DISCUSSION
Plant species shape denitrification activity through root exudation
The measurement of the denitrification activity of the RS without additional carbon (Fig. 1), as conducted in a previous study (Guyonnet et al.2017), likely displayed higher activity than in the RAS where carbon source was added. This result suggests the role of the root exudates of each plant species in the management of the denitrification activity. This plant intervention might be conducted by selecting beneficial microorganisms involved in plant nutrition and protection against pathogens, which may happen to denitrify. The host plant may also produce compounds that could control the expression of the genes involved in denitrification. Fig. 1 shows a higher denitrification activity in the RS of T. aestivum and A. thaliana, which is consistent with the high expression of the bacterial nirK gene for these two host species (Fig. 5). This suggests a potential positive effect of the root exudates from T. aestivum and A. thaliana to select bacterial populations able to denitrify or to activate the expression of denitrification genes. The very low level of denitrification activity in the RS of B. napus is consistent with a lower level of expression of the nirK gene compared with that of the RS of T. aestivum and A. thaliana. These results suggest that the plantlets of B. napus may not recruit bacterial populations able to denitrify or may alter the expression of denitrification genes in the RS probably via root exudation. In addition, by measuring the preferential uptake of B. napus for nitrate and ammonium, our results demonstrated a greater avidity of B. napus for nitrogen uptake primarily in the form of nitrate compared with the other plants (Fig. 2), suggesting potential competition for nitrate with denitrifiers as previously observed for other plants (Kuzyakov and Xu, 2013; Bardon et al.2014). Denitrification inhibition, named Biological Denitrification Inhibition (BDI), has already been observed in Fallopiasp. and has been defined as the ability of the plant to release secondary metabolites such as procyanidins that inhibit denitrifiers, and therefore enables the plant to recover nitrate for its growth (Bardon et al. 2014, 2016, 2017). B. napus may perform BDI because the presence of procyanidins has already been demonstrated in its roots (Wronka et al.1994, Nesi et al.2009).
In the RS of M. truncatula, the nitrogen uptake was very low regardless of its form, suggesting that unlike B. napus, competition for nitrogen with denitrifiers is not expected. However, the low denitrification rate primarily achieved by denitrifying bacteria harbouring the nirS gene indicates a counter-selection of the populations harbouring the nirK gene. However, we did find that a high level of denitrification activity in the legume RAS positively correlated with the nirK gene expression (Fig. 1). Several studies have investigated the effect of legume cultivation in nitrogen cycle processes, since legumes associated with nitrogen-fixing bacteria can be used as a substitute for mineral fertilisers (Philippot, Hallin and Schloter 2007). Researchers reported high denitrification rates with legume rhizospheres compared with other plants (Svensson et al.1991; Kilian and Werner, 1996), which is consistent with our data. Nevertheless, several studies have reported that denitrification is very common in rhizobia and that many strains can denitrify both as nodule bacteroids and as free-living bacteria (Philippot, Hallin and Schloter 2007). In our study, either M. truncatula root-associated Rhizobia (bacteroids or free-living bacteria) are not involved in the denitrification process, since nirK gene expression was not detected (Fig. 5, Additional File 2), or it can be assumed that only the nirS copy is transcribed, since it was recently suggested that certain rhizobia may possess Cu-type (nirK) and cd1-type (nirS) nitrite reductase genes (Sánchez and Minamisawa, 2018).
Our results provided evidence that the denitrification activity of the microbial communities inhabiting the rhizosphere (RAS) of T. aestivum was significantly higher than that of the unplanted soil, where less carbon is available (Fig. 1). Similarly, the measurement of the denitrification rate of cereals, such as barley grown in pots (Klemedtsson et al.1987, Metz et al.2003) and maize in field conditions (Mahmood et al.1997), also revealed high denitrification activity compared with the unplanted soil. However, even though denitrification is primarily conducted by bacteria, we cannot exclude the role of fungi and archaea as previous studies have already shown their capacity to denitrify (Philippot, 2002, Maeda et al.2015). In addition, the nirK and nirS primers used in this study are not universal enough to survey all the soil denitrifiers, since they were designed based on a limited number of sequences, primarily from laboratory strains. Further studies developing new nirK and nirS primers and targeting denitrification genes expression from fungi and Archaea are needed to confirm our hypothesis.
Core and specific active microbiota among plant species
Metabarcoding of 16S rRNA revealed the enrichment of members of Chloroflexi in the RS of T. aestivum, Bacteroidetes and Verrucomicrobia in that of B. napus, and Firmicute and Acidobacteria in that of A. thaliana at the RS level (Fig. 3A, Additional File 1). These results are consistent with previous data using a stable-isotope probing (SIP) approach on B. napus (Gkarmiri et al.2017), A. thaliana (Bulgarelli et al.2012; Lundberg et al.2012) and wheat (Wang et al.2016).
Comparison of the microbiota inhabiting the RAS revealed differences in the phyla abundance with high abundance, for example, Bacteroidetes in the rhizosphere of A. thaliana (Fig. 3A, Additional File 1), which is consistent with previous research by Bulgarelli et al. (2012) on this model plant. Remarkably, the rhizosphere of A. thaliana was enriched with the Crenarchaeota phylum, which is consistent with the results of Bressan et al. (2009). This phylum, considered to be the most abundant ammonia-oxidising phylum in soil ecosystems (Leininger et al.2006), has been found to be involved in root exudate assimilation in the rhizosphere of A. thaliana using a SIP approach (Bressan et al.2009).
The comparison of the microbial assemblages revealed differences and similarities in the composition of the microbiota inhabiting the roots and the rhizosphere retrieved from the four plant species (Fig. 3B,C). First, we demonstrated that four rOTUs belonging to members of the families Caulobacteraceae, Comamonadaceae, Chitinophagaceae and Bacillaceae were enriched in the RSs of all the plants studied. These rOTUs can be considered generalists, since they are associated with the RS of all plants studied as determined by Haichar et al. (2008). Second, some clear differences were observed between the microbiota from the four plant RSs (Fig. 3B,C). For example, some members of the Cytophagaceae and the Chitinophagaceae family are specifically selected by A. thaliana (Additional File 3), while members of the families Opitutaceae, Verrucomicrobiacea, Solibacterales and Ellin 6075 were specifically associated with B. napus. In contrast, the enrichment of the members of the Microbacteriaceae family appears to be a distinct feature of the microbiota of the T. aestivum roots. In a recent study by Tomasek et al. (2017), a positive correlation between the abundance of Cytophagaceae, Chitinophagaceae and Microbacteriaceae and denitrification activity was observed, suggesting that these families potentially play an important role in denitrification at the root of A. thaliana and T. aestivum.
For microbiota colonising the RAS, we identified 24 rasOTUs common to all the plants studied (Additional File 3), suggesting that these OTUs represent generalist bacteria, while those retrieved only from a single plant species are considered to be specialist bacteria (Haichar et al.2008).
By comparing the root microbiota with the RAS microbiota, it appears that the root compartment is more selective than the soil surrounding the RS, indicating the specific recognition and nutritional selection of bacterial communities in the root tissues prior to the release of nutrients into the rhizosphere (Haichar et al.2008, Haichar, Roncato and Achouak 2012, Haichar, Fochesato and Achouak 2013).
As the bacterial community colonising the RS differs according to the plant species, the differences in denitrification activity could also be linked to the selection of certain denitrifying bacteria that are more or less effective for each plant through root exudates. Similarly, the diversity of the bacterial community inhabiting the RAS could also explain the denitrification activity profiles. Future studies targeting the diversity of denitrifiers using denitrification genes, such as nirK and nirS, are needed. As described above, the current challenge is to design optimal nirK and nirS universal primer pairs to target entire denitrifier communities in the environment, as already suggested by Bonilla-Rosso et al. (2016).
Microbial interaction networks
Positive correlations between microbial populations suggest the occurrence of a mutualistic interaction and co-existence, while negative correlations might suggest the presence of host-competitive exclusion or a predation relationship between the microorganisms (Steele et al.2011, Barbéran et al.2012, Ju and Zhang, 2015). A closer look at the bacterial networks shows that several associations confirm or reveal interesting ecological patterns for the taxa that have not been as well studied, as was also shown by Barbéran et al. (2012) (Fig. 4, Additional File 4). This is the case for the Crenarchaeotal OTU, which is particularly remarkable because of our poor understanding of the ecological niches occupied by this taxon, even though it has been proposed that related Crenarchaeota are ubiquitous in the soil (Bates et al.2011; Barbéran et al.2012) and that they may have an important role in the nitrogen cycle as ammonia oxidisers (Simon, Dodsworth and Goodman 2000; Leininger et al.2006; Barbéran et al.2012). In this study, taxa related to Crenarchaeota OTUs co-occurred with the Alphaproteobacteria class (Ellin329 order) and Acidobacteria in the RAS of B. napus and with Chloroflexi (Anaerollinea class) in the RAS of M. truncatula. The co-occurrence between the Crenarchaeota OTUs is probably involved in the nitrogen cycling with the OTUs of the families Acidobacteria, Alphaproteobacteria and Chloroflexi, which are probably also involved in carbon cycling (Ward et al.2009; Hug et al.2013; Brauer et al.2016), highlights the link between these two cycles and the importance of each for a better understanding of the other.
It is interesting to note that Nitrospirales and Solirubrobacterales co-occur only in the RAS of T. aestivum and B. napus displaying high DEA, suggesting a syntrophic relationship, in which ammonium released by Solirubrobacterales during the organic metabolism of N (Tu et al.2017 could serve as a substrate for Nitrospira to achieve nitrification (Daims et al.2015) and provide nitrate to denitrifiers. Similar interactions might also be suggested between Candidatus nitrosphaera participating in the nitrification process (Spang et al.2012) and Rhodospirillales known to perform denitrification, as described by Saarenheimo, Tiirola and Rissanen (2015).
CONCLUSION
Collectively our results have shown that plant species shape denitrification activity and modulate the diversity of active microbiota through root exudation. As expected, a positive effect of the root exudates on denitrification activity was observed in the RS of A. thaliana and T. aestivum, while B. napus appears to alter denitrification activity in the RS through root exudates. This denitrification inhibition is probably due to competition between B. napus and the denitrifiers for nitrate, since nitrate is preferentially utilised by B. napus. Some OTUs were associated with all the plants studied and are considered generalists, while others are specifically associated with plant species and are considered to be specialists. Network analysis indicated that Nitrospirales and Solirubrobacterales co-occur only in the RAS of T. aestivum and B. napus presenting high denitrification activity, while Acidobacteria and Verrucomicrobia co-occur at the roots of A. thaliana and T. aestivum presenting high denitrification level.
ACKNOWLEDGEMENTS
This work was supported by the French national programme EC2CO-MicrobiEn, CNRS, (RhizoDen Project). We thank Elise Lacroix for her help in plant culture and harvesting. Plant culture was performed at the greenhouse platform (FR41, University Lyon1). We thank J. Gervaix for his help on nitrogen form uptake experiments. The authors gratefully thank the editor Prof. Wietse de Boer and reviewers for improving our manuscript.
CONFLICT OF INTEREST. The authors declare no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}