





ABSTRACT
The Virus-X—Viral Metagenomics for Innovation Value—project was a scientific expedition to explore and exploit uncharted territory of genetic diversity in extreme natural environments such as geothermal hot springs and deep-sea ocean ecosystems. Specifically, the project was set to analyse and exploit viral metagenomes with the ultimate goal of developing new gene products with high innovation value for applications in biotechnology, pharmaceutical, medical, and the life science sectors. Viral gene pool analysis is also essential to obtain fundamental insight into ecosystem dynamics and to investigate how viruses influence the evolution of microbes and multicellular organisms. The Virus-X Consortium, established in 2016, included experts from eight European countries. The unique approach based on high throughput bioinformatics technologies combined with structural and functional studies resulted in the development of a biodiscovery pipeline of significant capacity and scale. The activities within the Virus-X consortium cover the entire range from bioprospecting and methods development in bioinformatics to protein production and characterisation, with the final goal of translating our results into new products for the bioeconomy. The significant impact the consortium made in all of these areas was possible due to the successful cooperation between expert teams that worked together to solve a complex scientific problem using state-of-the-art technologies as well as developing novel tools to explore the virosphere, widely considered as the last great frontier of life.
INTRODUCTION
The virosphere comprises the largest reservoir of unknown genetic diversity in the whole biosphere and is considered to be the last great frontier of life. It is constituted by viruses, which are sophisticated acellular infectious entities that typically consist of genetic material (RNA or DNA) enclosed in a protein shell called a capsid. Infection of a host organism, either prokaryotic or eukaryotic, is necessary to activate their genetic blueprints. Viruses replicate by hijacking the host metabolic machinery, and the viral life cycle is completed when newly assembled viral particles are released into their environment, ready to infect new host organisms and repeat the cycle of infection and replication. The number of virus particles on Earth has been estimated to be in the order of 1031, which makes them the most abundant entities in the whole biosphere (Hendrix et al. 1999; Mushegian 2020). By virtue of their biology, viruses are ubiquitous and can be found in all conceivable ecosystems, including those with extremes of temperature, pH, salinity, and pressure. Such ecosystems include hot springs in Iceland with temperatures close to the boiling point of water to ice-cold deep-sea habitats of the Arctic Ocean at water pressures of several hundred atmospheres (Romancer et al. 2007; Harrison et al. 2013). The abundance and diversity of viruses that flourish in extreme habitats is truly striking. Metagenomic analyses of water in hot springs, for example, reveal that these geochemical hot spots are rich in viruses with a reported titer ranging from 105 to 106 ml−1 and a production rate of approximately 109 viral particles per liter per day (Breitbart et al. 2004). While the overall diversity of the viral community is extremely high, only a small fraction of it (<1%) has been explored so far (Forterre and Prangishvili 2009; Mokili, Rohwer and Dutilh 2012; Krishnamurthy and Wang 2017). Therefore, the viral gene pool is critical to study functional dynamics of ecosystems, to investigate how viruses influence evolution of microbes and multicellular organisms (Rodriguez-Valera et al. 2009; Clokie et al. 2011; Sandaa and Bratbak 2018). As viruses seem to be a driving force in shaping biogeochemical cycles on a global scale (Middelboe et al. 1996; Gobler et al. 1997; Thingstad 2000; Danovaro et al. 2016), they may also provide the key to understanding biodiversity and biosphere functioning at the level of individual species and genomes (Thingstad 2000; Urich et al. 2014; Harrison and Brockhurst 2017; Hreggvidsson et al. 2017; Tuttle and Buchan 2020). Much remains to be investigated regarding virus-host interplay in nature and in an exploration of the relatively unexploited global virome as an immense genetic resource for innovation potential (Rinke et al. 2013; Paez-Espino et al. 2016). The virosphere thus offers enormous promise for the development of unique technologies for life science, industrial, medical and diagnostic applications.
The exploitation of the virosphere and its genetic diversity was a central objective of the Virus-X project (Viral Metagenomics for Innovation Value; http://virus-x.eu). This EU Horizon 2020 endeavour launched in 2016 and continued through 2020, engaged in a collaborative effort of four biotech companies representing Small and Medium-sized Enterprises (SME partners) and 10 research groups from 8 European countries. The project acronym Virus‐X refers to the unknown virus and thus to the uncharted territory of viral genomics. The research activities of the consortium particularly focused on bacterial and archaeal viruses from extreme habitats, their impact on microbial dynamics, and viruses as a source of novel enzymes for biotech applications. The latter was crucial as, due to their highly unusual biology and lifecycles, viruses have developed specialized replication machineries employing proteins with unique properties often very different from cellular host enzymes (Kazlauskas, Krupovic and Venclovas 2016; Zhou et al. 2017; Kazlauskas et al. 2018). Many milestones of modern molecular biology and biotechnology were accomplished using viral proteins as their genomes are packed with genes encoding unusual nucleic acid processing enzymes with remarkable properties (Ofir and Sorek 2018). Today, key tools in biotechnology originate from only very few bacterial viruses (e.g. λ, T4 or T7), mostly infecting the model organism E. coli (Murray and Gann 2007). Furthermore, many new applications are based on old enzymes put to new uses, albeit sometimes following structural/functional modification (Kaczorowski and Szybalski 1998; Wilson et al. 2013; Lu et al. 2020). Further technological progress in the field depends on the discovery and development of enzymes from novel sources (van den Burg 2003; Castelan-Sanchez et al. 2019; Jin et al. 2019). Until now, this reservoir has been near inaccessible since methodologies for infecting and propagating viruses in a laboratory environment are cumbersome for many microbes, and only a small fraction of microorganisms can be cultivated. The advent of metagenomics that exploits Next Generation Sequencing technologies (NGS) has finally made the virome genetic content accessible for exploration (Angly et al. 2006; Kristensen et al. 2010; Schoenfeld et al. 2010; Rosario and Breitbart 2011; Beerenwinkel et al. 2012). However, exploring the virosphere remains a challenging task of emerging metagenomic methodologies for a number of reasons. Sampling in extreme environments is difficult and grueling, the concentration of viral genetic material is often very low, and up to 70% of the genes in novel environmental viruses bear little or no similarity to known nucleotide sequences deposited in the GenBank non-redundant database (Breitbart et al. 2002; Edwards and Rowher 2005; Hjörleifsdottir et al. 2014; Gil et al. 2021). Thus, this enormous genetic resource, largely unexplored, can be considered as the dark matter of life. Consequently, the Virus-X project was set to analyse and exploit the virome of extreme natural habitats with the ultimate goal of developing new gene products with high innovation value for applications in the bioeconomy. For this objective, enzyme discovery and development in the Virus‐X project focused on specific non-structural viral proteins such as proteins participating in nucleic acids metabolism, e.g. polynucleotide kinase (Blöndal et al. 2005a) and RNA ligase (Blöndal et al. 2005b); viral recombination machinery (Stefanska et al. 2014, Stefanska et al. 2016); nucleotide metabolism; transcription; replication, including DNA polymerases (Hjorleifsdottir et al. 2014), DNA helicases, single‐strand DNA binding proteins, and other DNA and RNA processing enzymes. Also of particular interest were enzymes involved in bacterial cell lysis as potential antimicrobials (Plotka et al. 2014; Plotka et al. 2015) and components of antiviral mechanisms, including the CRISPR system (Nordberg Karlsson et al. 2020). In addition, the consortium focussed on the unknown—gene products with not yet determined properties and the potential of completely new functionalities.
Project overview
All activities within the project were divided into four platforms that cover metagenome retrieval (P1), bioinformatics (P2), protein characterisation (P3), and invention to innovation (P4). While each platform was led by one partner, all partners contributed to at least two platforms, creating a highly intertwined network of collaboration and ensuring the overall success of the project (Fig. 1).
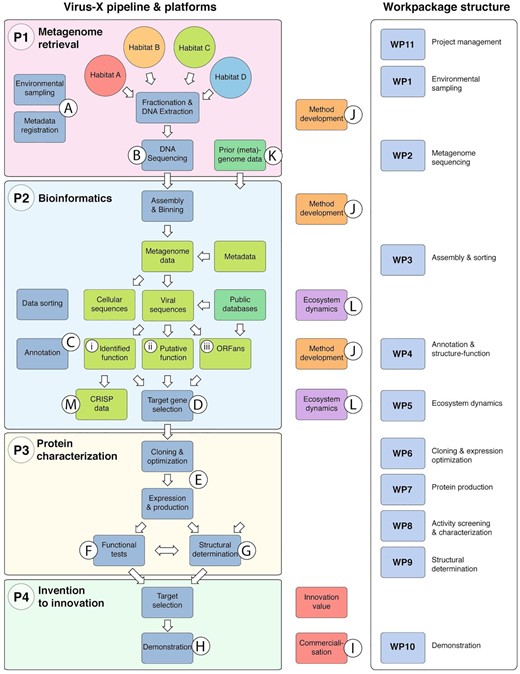
The Virus-X discovery pipeline was designed to bring viral gene products from environmental sampling (A) via sequencing (B), bioinformatics and annotation (C), selection of gene targets (D), cloning, expression and production (E), functional (F) and structural characterisation (G), to demonstration (H) and ultimately to commercialization (I). Although industrially driven, the project was designed to be a vehicle for method development (J) in the field of metagenomics as well as to address some specific challenges that set viral metagenomics apart from metagenomics of cellular genomes. The viral metagenome approach brought the need for improvements and innovative approaches in the functional assignment of genes as the focus was on exploring the outer realm of sequence diversity where many gene sequences lack significant similarity to known sequences (Uchiyama and Miyazaki 2009). Therefore, particular emphasis was put on bioinformatics approaches to gene annotation. Data from previous bacteriophage metagenome studies and genome data were used as reference sequences and to initiate bioinformatics and the downstream workflow in the project at an early stage in the project timeline (K). In silico annotation was complemented with functional and structural studies to establish the function of targeted gene products. Functional studies (F) required cloning and expression of selected genes and functional assays in a high‐throughput set‐up. Structure determination by X‐ray crystallography (G) complemented bioinformatics and biochemical characterisation for selected targets in all categories. In addition to the research and development of selected gene products, the work program involved the study of specific aspects of the functional dynamics of microbial communities (L) and the interplay between prokaryotic cellular population and the associated viral population, such as by the study of viral diversity, bacterial immunity and the CRISPR system (M). Sequence data were thus also acquired from the cellular members of the sampled communities.
One key objective of the Virus-X project was the development of new technologies for metagenomics ranging from bioprospecting of complex viromes in extreme environments to the analysis and annotation of large-scale metagenomic data sets. For this purpose, new algorithms were developed, which formed part of the Virus-X metagenomics toolbox, many of which are now implemented in freely available webservers or as free, open-source software (Table 1).
The Virus-X metagenomics toolbox includes freely available databases except for EMGB, free, open-source software and a user-friendly webserver.
| PHROGs | database of deeply clustered and well-annotated viral protein groups (Terzian et al. 2021 submitted) https://phrogs.lmge.uca.fr |
| Uniclust | database providing functionally homogenous clusters of sequences at clustering depths of 90%, 50%, and 30% sequence identity (Mirdita et al. 2016); https://uniclust.mmseqs.com |
| Soil | Marine Reference Catalogues | catalogues of 2 billion proteins from soil metagenomes (SRC) and 300 million proteins from marine metatranscriptomes assembled (MERC) using Plass http://wwwuser.gwdg.de/∼compbiol/plass |
| BFD | database of over 65 million protein profiles based on combined and clustered 2.5 billion protein sequences from SRC, MERC, Metaclust, and Uniprot https://bfd.mmseqs.com |
| WIsH | prediction of the prokaryotic host of viral contigs as short as3 kb that runs several hundred times faster than standard bioinformatic implementations (Galiez et al. 2017); https://github.com/soedinglab/wish |
| Linclust | first protein clustering algorithm that scales linearly with the input, clusters billion of protein sequences within a single day on a single server (Steinegger and Söding 2018); https://github.com/soedinglab/mmseqs2 |
| HH-suite3 | software suite for fast remote homology detection and deep protein annotation (Steinegger et al. 2019); https://github.com/soedinglab/hh-suite |
| Plass | assembles short-read sequencing data on a protein level. Plass is able to extract ten times more proteins from complex metagenomes (Steinegger et al. 2019); https://github.com/soedinglab/plass |
| MetaEuk | enables large-scale eukaryotic metagenomics through reference-based, sensitive taxonomic and functional annotation (Levy Karin, Mirdita and Söding 2020); https://metaeuk.soedinglab.org |
| SpacePHARER | sensitive identification of phages from CRISPR spacers in prokaryotic hosts (Zhang et al. 2021); https://spacepharer.soedinglab.org |
| MMseqs2 software suite | software suite to search and cluster huge protein and nucleotide sequence sets (Steinegger and Söding 2017), sensitive taxonomy assignment (Mirdita et al. 2021), and user-friendly webserver (Mirdita, Steinegger and Söding 2019); https://github.com/soedinglab/mmseqs2 https://search.mmseqs.com |
| EMGB | web-based metagenome annotation browser for large datasets (Henke et al. manuscript in preparation) https://github.com/metagenomics/EMGB2 |
| PHROGs | database of deeply clustered and well-annotated viral protein groups (Terzian et al. 2021 submitted) https://phrogs.lmge.uca.fr |
| Uniclust | database providing functionally homogenous clusters of sequences at clustering depths of 90%, 50%, and 30% sequence identity (Mirdita et al. 2016); https://uniclust.mmseqs.com |
| Soil | Marine Reference Catalogues | catalogues of 2 billion proteins from soil metagenomes (SRC) and 300 million proteins from marine metatranscriptomes assembled (MERC) using Plass http://wwwuser.gwdg.de/∼compbiol/plass |
| BFD | database of over 65 million protein profiles based on combined and clustered 2.5 billion protein sequences from SRC, MERC, Metaclust, and Uniprot https://bfd.mmseqs.com |
| WIsH | prediction of the prokaryotic host of viral contigs as short as3 kb that runs several hundred times faster than standard bioinformatic implementations (Galiez et al. 2017); https://github.com/soedinglab/wish |
| Linclust | first protein clustering algorithm that scales linearly with the input, clusters billion of protein sequences within a single day on a single server (Steinegger and Söding 2018); https://github.com/soedinglab/mmseqs2 |
| HH-suite3 | software suite for fast remote homology detection and deep protein annotation (Steinegger et al. 2019); https://github.com/soedinglab/hh-suite |
| Plass | assembles short-read sequencing data on a protein level. Plass is able to extract ten times more proteins from complex metagenomes (Steinegger et al. 2019); https://github.com/soedinglab/plass |
| MetaEuk | enables large-scale eukaryotic metagenomics through reference-based, sensitive taxonomic and functional annotation (Levy Karin, Mirdita and Söding 2020); https://metaeuk.soedinglab.org |
| SpacePHARER | sensitive identification of phages from CRISPR spacers in prokaryotic hosts (Zhang et al. 2021); https://spacepharer.soedinglab.org |
| MMseqs2 software suite | software suite to search and cluster huge protein and nucleotide sequence sets (Steinegger and Söding 2017), sensitive taxonomy assignment (Mirdita et al. 2021), and user-friendly webserver (Mirdita, Steinegger and Söding 2019); https://github.com/soedinglab/mmseqs2 https://search.mmseqs.com |
| EMGB | web-based metagenome annotation browser for large datasets (Henke et al. manuscript in preparation) https://github.com/metagenomics/EMGB2 |
The Virus-X metagenomics toolbox includes freely available databases except for EMGB, free, open-source software and a user-friendly webserver.
| PHROGs | database of deeply clustered and well-annotated viral protein groups (Terzian et al. 2021 submitted) https://phrogs.lmge.uca.fr |
| Uniclust | database providing functionally homogenous clusters of sequences at clustering depths of 90%, 50%, and 30% sequence identity (Mirdita et al. 2016); https://uniclust.mmseqs.com |
| Soil | Marine Reference Catalogues | catalogues of 2 billion proteins from soil metagenomes (SRC) and 300 million proteins from marine metatranscriptomes assembled (MERC) using Plass http://wwwuser.gwdg.de/∼compbiol/plass |
| BFD | database of over 65 million protein profiles based on combined and clustered 2.5 billion protein sequences from SRC, MERC, Metaclust, and Uniprot https://bfd.mmseqs.com |
| WIsH | prediction of the prokaryotic host of viral contigs as short as3 kb that runs several hundred times faster than standard bioinformatic implementations (Galiez et al. 2017); https://github.com/soedinglab/wish |
| Linclust | first protein clustering algorithm that scales linearly with the input, clusters billion of protein sequences within a single day on a single server (Steinegger and Söding 2018); https://github.com/soedinglab/mmseqs2 |
| HH-suite3 | software suite for fast remote homology detection and deep protein annotation (Steinegger et al. 2019); https://github.com/soedinglab/hh-suite |
| Plass | assembles short-read sequencing data on a protein level. Plass is able to extract ten times more proteins from complex metagenomes (Steinegger et al. 2019); https://github.com/soedinglab/plass |
| MetaEuk | enables large-scale eukaryotic metagenomics through reference-based, sensitive taxonomic and functional annotation (Levy Karin, Mirdita and Söding 2020); https://metaeuk.soedinglab.org |
| SpacePHARER | sensitive identification of phages from CRISPR spacers in prokaryotic hosts (Zhang et al. 2021); https://spacepharer.soedinglab.org |
| MMseqs2 software suite | software suite to search and cluster huge protein and nucleotide sequence sets (Steinegger and Söding 2017), sensitive taxonomy assignment (Mirdita et al. 2021), and user-friendly webserver (Mirdita, Steinegger and Söding 2019); https://github.com/soedinglab/mmseqs2 https://search.mmseqs.com |
| EMGB | web-based metagenome annotation browser for large datasets (Henke et al. manuscript in preparation) https://github.com/metagenomics/EMGB2 |
| PHROGs | database of deeply clustered and well-annotated viral protein groups (Terzian et al. 2021 submitted) https://phrogs.lmge.uca.fr |
| Uniclust | database providing functionally homogenous clusters of sequences at clustering depths of 90%, 50%, and 30% sequence identity (Mirdita et al. 2016); https://uniclust.mmseqs.com |
| Soil | Marine Reference Catalogues | catalogues of 2 billion proteins from soil metagenomes (SRC) and 300 million proteins from marine metatranscriptomes assembled (MERC) using Plass http://wwwuser.gwdg.de/∼compbiol/plass |
| BFD | database of over 65 million protein profiles based on combined and clustered 2.5 billion protein sequences from SRC, MERC, Metaclust, and Uniprot https://bfd.mmseqs.com |
| WIsH | prediction of the prokaryotic host of viral contigs as short as3 kb that runs several hundred times faster than standard bioinformatic implementations (Galiez et al. 2017); https://github.com/soedinglab/wish |
| Linclust | first protein clustering algorithm that scales linearly with the input, clusters billion of protein sequences within a single day on a single server (Steinegger and Söding 2018); https://github.com/soedinglab/mmseqs2 |
| HH-suite3 | software suite for fast remote homology detection and deep protein annotation (Steinegger et al. 2019); https://github.com/soedinglab/hh-suite |
| Plass | assembles short-read sequencing data on a protein level. Plass is able to extract ten times more proteins from complex metagenomes (Steinegger et al. 2019); https://github.com/soedinglab/plass |
| MetaEuk | enables large-scale eukaryotic metagenomics through reference-based, sensitive taxonomic and functional annotation (Levy Karin, Mirdita and Söding 2020); https://metaeuk.soedinglab.org |
| SpacePHARER | sensitive identification of phages from CRISPR spacers in prokaryotic hosts (Zhang et al. 2021); https://spacepharer.soedinglab.org |
| MMseqs2 software suite | software suite to search and cluster huge protein and nucleotide sequence sets (Steinegger and Söding 2017), sensitive taxonomy assignment (Mirdita et al. 2021), and user-friendly webserver (Mirdita, Steinegger and Söding 2019); https://github.com/soedinglab/mmseqs2 https://search.mmseqs.com |
| EMGB | web-based metagenome annotation browser for large datasets (Henke et al. manuscript in preparation) https://github.com/metagenomics/EMGB2 |
Virus-X workflow
Platform 1. Metagenome retrieval
A wide range of natural habitats was selected as a source of viral metagenomes. The 50 sites within 12 hydrothermal regions in Iceland explored by scientists from Matis included intertidal geothermal coastal areas and terrestrial hot springs (16–96°C, pH 2.0–9.3) (Table S1, Supporting information). Another team from Institut Pasteur (France) specializing in viruses of Archaea collected 49 samples from hot springs in the western part of Georgia (50–95°C pH 7.0–8.0), Kuju and Beppu regions on Kyushu island in Japan (60–90°C, pH 2.0–7.0), as well as solfataric field in Pozzuoli (87–93°C, pH 1.5–2.0) and hot springs of the Campi Flegrei volcano (81–96°C, pH 1.0–7.0) in Italy (Table S2, Supporting information). Finally, the group from the University of Bergen (Norway) sampled 34 sites that included deep-sea hydrothermal vents located in the Arctic Mid-Ocean Ridge: The Ægir Vent Field—Central Mohn's Ridge and the Loki's Castle Vent Field (Northern Atlantic) and shallow to deep seawaters from the Norwegian sea (Jan Mayen Fracture Zone; −0.8°C — −3.9°C, pH 7.9), the eastern part of the Fram strait (areas around Svalbard; 1.9°C) (Table S3, Supporting information). Sampling of marine sites required access to research cruises equipped with a remote submersible operating robotic vehicle Ægir 6000 with up to 6000 m depth rating (Fig. 2).

Deep-sea hydrothermal vents located north of Iceland on the Arctic Mid-Ocean Ridge, in the Norwegian-Greenland Sea. White smokers at the Soria Moria vent field (A) and black smokers at the Loki´s Castle vent field (B) host diverse chemosynthetic microbial communities that derive their energy from the chemical disequilibria that form when reduced hot hydrothermal fluids, rich in potential electron donors (e.g. H2, CH4, H2S, mix with cold seawater rich in potential electron acceptors (Dahle et al. 2015; Steen et al. 2016). Photos: ÆGIR team, NORMAR.
In order to identify and characterise new viruses by electron microscopy, enrichment culture techniques based on simulating conditions that favour propagation of both bacterial and archaeal viruses were employed (Liu et al. 2019). It was hence possible to observe diverse virion shapes characteristic of viruses infecting hyperthermophilic members of the phylum Crenarchaota, of the domain Archaea (Fig. 3A). Their morphologies are unique, and such diversity has not been observed among viruses infecting Bacteria and Eukarya. This is matched by the low similarity of thermophilic viruses at the nucleotide sequence level, where the majority of genes show no homology to other viruses or cellular organisms (Wang et al. 2020a; Krupovic et al. 2018). Several new viruses that infect hyperthermophilic archaea and belong to families Globuloviridae, Tristromaviridae, Lipothrixviridae, Rudiviridae and Portogloboviridae were isolated and characterized (Liu et al. 2017; Liu et al. 2019; Baquero et al. 2020). Portogloboviruses were also discovered to carry mini-CRISPR arrays containing spacers targeting each other as well as other viruses, exemplifying a novel mechanism promoting interviral conflicts and superinfection exclusion in extreme environments (Medvedeva et al. 2019). A remarkable novel mode of DNA packaging was observed in the case of the Sulfolobus polyhedral viruses SPV1 and SPV2, the first members of the new family (Portogloboviridae) (Wang et al. 2019). In the icosahedral virions, the double-stranded DNA is wound up sinusoidally into a spherical coil filling the protein capsid (Fig. 3B and C). Electron cryo-microscopy enabled reconstruction at a near-atomic resolution of the virion structure of portoglobovirus SPV1, tristromavirus PFV2, lipothrixvirus SFV1 and rudivirus SSRV1 and revealed that, in all these cases, DNA is packaged in the form of nucleocapsid, where capsid proteins tightly wrap around the DNA and maintain it in A-form (Fig. 3B and C) (Liu et al. 2018; Wang et al. 2019; Wang et al. 2020a; Wang et al. 2020b). This finding is striking as previous reports suggested that, in the case of bacterial viruses, genomic DNA predominantly adopt the B-form (Black and Thomas 2012). The more tightly packed A-form DNA could represent a general mechanism of adaptation to extreme thermal environments.
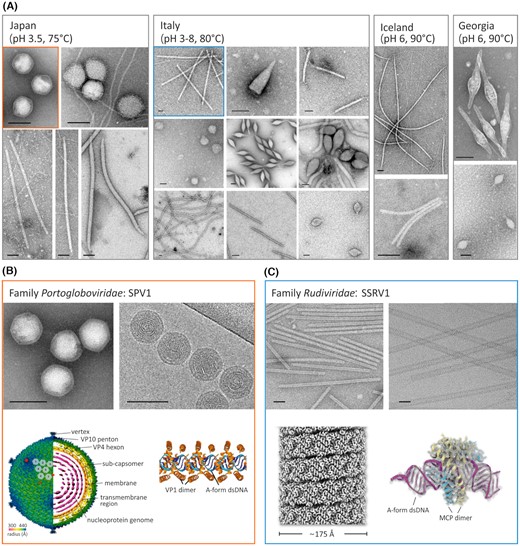
Diversity of virion morphotypes observed by electron microscopy from samples collected from hydrothermal environments at indicated locations. (A) Electron micrographs of virions in established enrichment cultures with denoted pH values and temperatures similar to that of their original environments. Pictures of the samples originated from Japan and Italy are adapted from Liu et al. (Liu et al. 2019) and Baquero et al. (Baquero et al. 2020), respectively. (B) and (C) The cryo-electron microscopy reconstruction of the new viruses isolated from enrichment cultures (framed in the same colors as in (a). SPV1 pictures are adapted from Liu et al. (Liu et al. 2017) and Wang et al. (Wang et al. 2019). Virion components, including structural protein complex, inner membrane, and nucleoprotein genome consisting of A-form dsDNA and virion protein (VP) 1 dimers, are indicated. SSRV1 images are adapted from Wang et al. (Wang et al. 2020b) and Baquero et al. (Baquero et al. 2020). The helical structure of the virion that is composed of A-form dsDNA and the major capsid protein (MCP) homodimers are shown. Bars in electron micrographs, 100 nm.
Despite an increase in general knowledge on viral diversity, biogeography data is still scarce or almost missing from some specific geographic areas such as deep-sea vent systems (Le Moine Bauer et al. 2018) or arctic waters (Sandaa et al. 2018). Preliminary analysis of viromes from extreme marine habitats in Norwegian territories confirmed the distribution of dominant taxa in the ocean (order Caudovirales and family Phycodnaviridae) (Brum et al. 2015; Mihara et al. 2018; Endo et al. 2020), but due to our novel sampling and preparation protocol, we were also able to capture viruses with different genome types and large differences in capsid size in the same sample (Blanc-Mathieu et al. 2021).
Platform 2. High‐throughput sequencing, assembly and annotation
Metagenomic samples collected in the first phase of the project were processed to isolate genetic material for sequencing. One of our approaches in the Virus-X team has been to capture the total viral diversity within marine samples from arctic and deep-sea vent systems, including viruses with all genome types and within all size ranges. The protocol developed was based on less rigorous filtration steps (only 0.45 µm) to capture even the largest virions, and the extraction of total nucleic acids (TNA) rather than double-stranded DNA only. The RNA fractions were enzymatically converted to cDNA prior to metagenomic library preparation for high-throughput sequencing. For the remaining samples, we used consecutive steps of microfiltration to remove cellular organisms, followed by concentrating the samples and DNA extraction using standard procedures. Viral genomic DNA can be different from cellular DNA in many aspects. It often contains a high fraction of modified bases and complex genomic structures such as extremely long direct or inverted repeats and terminal redundancies (Li et al. 2014; Davison 2015; Weigele and Raleigh 2016). This leads to particular challenges both in the sequencing procedure and the downstream assembly with short sequence reads and uneven sequence depth (Klumpp, Fouts and Sozhamannan 2012; Beaulaurier et al. 2020). Sequencing efforts were guided by various parameters such as overall diversity of reads, estimated coverage based on the size distribution of contigs, rarefaction analysis, using several selected indicator genes, and the proportion of single reads. In the project, various NGS platforms (Illumina MiSeq, HiSeq; Oxford Nanopore) were used for metagenomic total DNA analysis as well as for assembly of individual viral genomes out of the complex viral metagenome sequence data.
In one exemplary study, metagenomic sequencing of polyhedral and filamentous viruses that infect archaea belonging to the order Sulfolobales yielded seven complete or near-complete genomes (Liu et al. 2019). They were assigned at the nucleotide sequence level to viruses belonging to the family of polyhedral Portogloboviridae (SPV1, SPV2), filamentous Rudiviridae (SBRV1), and Lipothrixviridae (SBFV3). In addition, two genomes of filamentous viruses (SBFV1 and SBFV2) could not be assigned to any family and hence form a new group of filamentous archaeal viruses. The same applies to the seventh viral genome (SBV1), which is likely to represent a new virus family. A striking feature was the observation that ca. 75% of genes contained in analysed genomes show no homology to genes in other viruses or cellular organisms, making archaeal viruses a valuable source of unknown genes. A similar conclusion was reached from the analysis of genome content of bacterial viruses from Iceland metagenomic samples. Out of 2015 genes from 22 phages that infect bacteria of genus Thermus, 129 genes (6%) were assigned as type-A (known function), 157 (8%) were type-B (putative function), and 1727 genes (86%) were classified as type-C (no known function).
In total, metagenomic sequencing within the Virus-X project reached a final output of 290 Gbases. The computational analysis started with assembly and binning, which allowed the preliminary assessment for extensive downstream analysis (Fig. 4). We implemented a modern workflow using the Common Workflow Language (CWL) to keep it maintainable and portable across computing environments (Amstutz et al. 2016). Briefly, the workflow consists of the following steps: (i) quality control to trim or even discard the raw reads based on the quality score of their individual bases; (ii) assembly of input reads into longer contigs. The MEGAHIT assembler (Li et al. 2015) was chosen for this task based on its evaluation within the Critical Assessment of Metagenome Interpretation challenge (Sczyrba et al. 2017); (iii) mapping of reads to assembled contigs to determine sequencing coverage of each contig; (iv) gene prediction stage to identify potential genes within the assembled contigs with the use of Prodigal (Hyatt et al. 2010); (v) taxonomic and functional annotation of predicted genes using DIAMOND (Buchfink, Xie and Huson 2015) and MEGAN (Huson et al. 2016) tools. The functional annotation also included a prediction of functional domains using the PFAM database (El-Gebali et al. 2019), pathways using KEGG (Kanehisa et al. 2017), as well as the metagenomic tools developed within the project (vi; see below). In step (vii), binning of assembled contigs into metagenome-assembled genomes (MAGs) was performed using MetaBAT 2 (Kang et al. 2019), followed (viii) by taxonomic classification of MAGs utilizing GTDB-Tk (Chaumeil et al. 2020). Freely accessible bioinformatic tools developed within the Virus-X project are summarized in Table 1.

Core elements of the bioinformatics workflow (A). The web-based project data browser EMGB (B). It integrates all workflow results and features tailored filtering and inspection tools for screening large metagenomic datasets. One particular important inspection tool is the contig viewer (C), which merges gene annotations and taxonomic assignments with reading coverage graphs, thus enabling a quick inspection of neighbouring genes and a comparison of coverage distributions across samples of a combined assembly.
Applying the workflow to all Virus-X datasets resulted in a total assembly size of 23 Gbases, 38417734 contigs, and 54106508 predicted genes. In addition, 3591 viral metagenome datasets publicly available from NCBI's Sequence Read Archive (SRA) were also processed and predicted genes included in the subsequent clustering step (S1 file, Supporting information).
From this extensive database, it was possible to extract 157428937 open reading frames (ORFs, genetic sequences that are potentially translated to proteins) with a minimum length of 60 amino acids. To deal with the high degree of redundancy in this data set, amino-acid sequences were clustered using MMseqs2 (Steinegger and Söding 2017) down to 30% of pairwise sequence identity and using a conservative 90% minimum coverage threshold, resulting in 56790072 clusters. To substantially increase the protein sequence recovery, the open-source de-novo protein-level assembler (Plass) was developed (Steinegger, Mirdita and Söding 2019). Of vital importance to the project was gene annotation, as viral genes display extensive sequence divergence making homology detection exceedingly difficult. At the same time, virus genes are ideal targets for homology‐based function prediction because the functions and structures of viral enzymes are usually very well conserved despite the high sequence divergence. As a consequence, enzymes from bacteria or viruses detected as homologous to a viral protein usually still have a similar molecular function. Therefore, heavy emphasis in Virus‐X was placed on gathering extensive expertise in gene annotation to develop a new methodology for assigning functions to gene products, both by in-depth bioinformatics and by an experimental pipeline for selected genes. The deep annotation of ORFs was performed using the Uniboost database and HHboost procedure based on reverse profile-sequence search to detect proteins that show remote homology at the amino-acid sequence level (Mirdita et al. 2017; Steinegger et al. 2019). To further increase sensitivity, a consensus sequence was computed among each of the clusters eliminating sequencing errors and microdiversity. This database of ORFs, the Uniboost matches, and the annotations of each Uniboost entry, as well as the outputs of the workflow described above, were integrated into the main Virus-X EMGB data browser at CeBiTec at Bielefeld University, Germany (Fig. 4). This data is browsable to select gene targets of potential interest for cloning and expression. To further help in their annotation, ORFs were compared to PHROGs (Prokaryotic Virus Remote Homologous Groups), a database of deeply clustered and well-annotated viral protein groups (https://phrogs.lmge.uca.fr/).
In parallel to the discovery and analysis of single genes, metagenomic data were used to increase our understanding of the composition and dynamics of viral communities in these under-explored ecosystems such as deep-sea vents or arctic waters. Using a metabarcoding approach on 42 arctic seawater samples, covering the water column from 0 to 1000 m, and both the polar day and night, it was possible to gain a first glimpse into the viral ecology in these arctic environments (Fig. 5). Unlike with cellular organisms, there is no universal marker gene that targets all viruses. Using two genes targeting common groups of marine viruses, namely g23, capturing T4-like bacteriophages (Filee et al. 2005) and the mcp gene of large dsDNA phytoplankton viruses (Larsen et al. 2008), we demonstrated that seasonality is a key factor shaping arctic viral communities. Viral diversity and virus-to-host ratios dropped substantially at the beginning of the spring, then increased during the season, with the highest rates observed during the winter (Sandaa et al. 2018). In order to extend this analysis to the whole viral community, 20 arctic metagenomes were generated. Tailed bacteriophages belonging to the Caudovirales were the most dominant group, followed by giant dsDNA viruses belonging to the nucleocytoplasmic large DNA viruses (NCLDV). Viruses with other genome forms, such as ssDNA, dsRNA and ssRNA, were also detected. Viral operational taxonomic units (vOTU) were generated and compared to marine virus populations from Tara expeditions (Gregory et al. 2019) and to the RefSeq database. We confirmed the influence of seasonality over arctic viral communities, even if sampling depths and sampling location also seemed to have a substantial impact on the differentiation of these viruses.
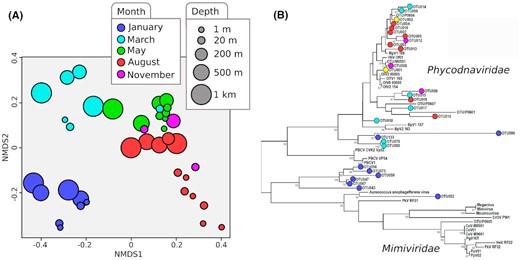
Nonmetric multidimensional scaling (NMDS) analysis of OTU diversity for major capsid protein (gp23) of T4-like myoviruses (A). Sampling depths are represented by circle sizes and sampling month by colors. Bray–Curtis dissimilarity was used to compare OTU composition between samples. Maximum likelihood tree constructed from the 29 most abundant major capsid protein OTUs of giant algal dsDNA viruses (B). Each OTU is identified by a circle, colored by the sampling month in which the OTU was found as dominant. The two yellow circles represent cosmopolitan OTU, significantly present in samples from more than three different months.
Platform 3. Protein selection, production and characterisation
Access to an extensive genetic database necessitates a careful selection of gene products for further analysis. All putative genes in the Virus-X database were divided into three categories with respect to assigned function: (i) type-A: assigned function through significant sequence identity at the amino-acid sequence level; (ii) type-B: putative function based on weak sequence similarity, and (iii) type-C: genes with no assigned function. Genes from all three categories were selected for cloning and expression for subsequent functional and/or structure determination. Genes that fall into categories A or B were evaluated and ranked according to the interest of the consortium members. In this process, particular emphasis was given to the SME partners to select targets with biotechnological potential. The selection of type-C target genes was also based on the following considerations. Genes located in a cluster of structural proteins were excluded, and genes showing extended conservation among different viral genomes but still with unknown function were prioritized. The particular focus was on gene targets coding for proteins with biotech potential that meet the following criteria: (i) heat or salt tolerance, (ii) cold-active/heat lability, (iii) high substrate specificity, (iv) high affinity, (v) strand displacement activity, (vi) DNA/RNA modifying/synthesizing/interacting, (vii) high fidelity, (viii) processivity and robustness in complex reaction settings. Most features outlined above are impossible to determine solely from nucleotide sequences alone. However, our approach involved experimental characterisation of target proteins as well as structural analysis by X-ray crystallography (Fig. 6) to shed light on protein function and allow for further optimization of annotation protocols.
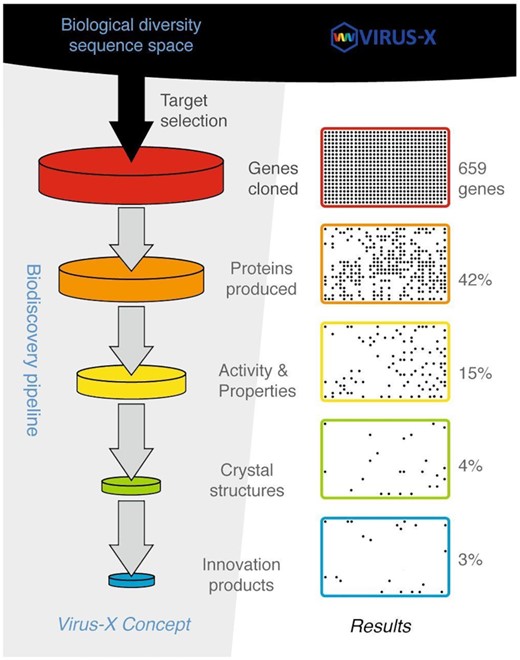
The Virus-X project's conceptual framework is the biodiscovery pipeline with consecutive steps from biological diversity sequence space to innovations as seen on the left. The available exploration of sequences is provided by the metagenomic approach as well as isolated genomes of viruses. The concept is basically a funnel-shaped pipeline based on expected fall-off in numbers of targets as we move from one step to the next using, as a guideline, previously reported cases of comparable implementations such as seen in structural genomics projects (Christendat et al. 2000). The progress of the project and results, as seen on the right, reflects this expected outcome, and having large enough input into the pipeline ensures control of the final output and provides certain flexibility in terms of difficulties in moving a particular target from one step to another. An example was a persistent problem in getting soluble proteins in expression experiments, apparently reflecting some inherent difficulties in expression of our viral protein targets. However, the funnel-shaped approach provided optimized use of efforts as challenging targets could be omitted in the process unless additional measures were justified for targets of particular value, such as a strong potential for becoming an innovation product, e.g. commercial enzyme.
As structure-based multiple sequence alignments are more accurate than sequence-based alignments (Carpentier and Chomilier 2019), we employed the 3DM information systems developed by our partner Bio-Prodict that collectively cover the entire structural space for a given enzyme family (Kuipers et al. 2010). This repository was designed to allow the integration of target proteins in these 3DM systems, which allows the detailed analysis of new protein sequences in the context of all the information of complete protein families. This approach facilitated our target selection process and was proven to be essential to gain insights into the specific adaptations of the viral proteins in comparison to known cellular proteins. The second internal database developed within the project was the protein Tiki Wiki that holds all experimental information from cloning, characterisation to structure determination of over 800 target proteins.
To facilitate speed in recombinant protein production, Escherichia coli was selected as a priority production system, based on its fast growth on cheap media, available cloning vectors, various promoters, and alternative solubility and purification tags, as well as co-plasmids with chaperonins to facilitate folding. Plasmid vectors used to express cloned target genes selected from the Virus-X database were mainly chosen to include tightly controlled inducible promoters like positively controlled L-rhamnose rhaPBAD (Wegerer, Sun and Altenbuchner 2008) or T7 ϕ10 promoter (Studier and Moffatt 1986; Studier et al. 1990). In cases where the protein overproduction yielded mainly insoluble protein, the creation of fusion proteins by adding genes coding for the maltose-binding protein (MBP) improved the synthesis of soluble targets, as shown previously (Motejadded and Altenbuchner 2009; Wang et al. 2013). Another successfully applied strategy was lowering the temperature at the induction step to18°C–25°C. In other cases, a number of different E. coli expression strains, including those supplemented with a plasmid pRARE (Novagen) coding for rare-codon tRNAs, were tested, and the use of strains containing additional chaperone genes to aid protein folding (Nishihara et al. 1998; de Marco and de Marco 2004; de Marco et al. 2007) proved very beneficial for targets from deep-sea metagenomes.
In total, 659 genes were cloned into expression vectors and 478 tested in production trials. The majority of them, 65%, resulted in the production of recombinant proteins; however, only approximately 42% were produced in high yields in a soluble form (Fig. 6). These soluble target proteins were expressed in standard shaking incubators, usually at temperatures between 18°C and 25°C and purified by a combination of affinity chromatography (immobilized metal affinity chromatography (IMAC) for His-tagged proteins or amylose affinity chromatography for MBP-fusion proteins) with size exclusion chromatography or ion exchange chromatography. After purification, activity testing and characterisation of recombinant MBP-fused proteins was, in the majority of cases, performed without MBP removal, while solubility tags were removed before recombinant protein was forwarded to crystallisation. Protein production was successfully upscaled for a majority of targeted constructs. In some cases, target genes were cloned, generating two or more variants that were subjected for protein production upscaling in parallel. For those type B and type C targets where structural solutions with ab initio methods were unsuccessful, selenomethionine substituted protein was produced as described before (Turner et al. 2007; Russo et al. 2009) to enable structure determination by multiple anomalous diffraction (MAD) methods (Hendrickson 1991).
Protein characterisation was divided into biophysical techniques that included Electron Spray Ionisation mass spectrometry (ESI-MS) to investigate protein integrity and covalent modifications, circular dichroism (CD) to confirm proper folding, and Thermal Shift Analysis (TSA) to study protein stability (Groftenhauge et al. 2015). TSA is beneficial to identify optimal storage and transport conditions as well as to guide subsequent crystallisation trials (Bruce et al. 2019) (Fig. 7A).
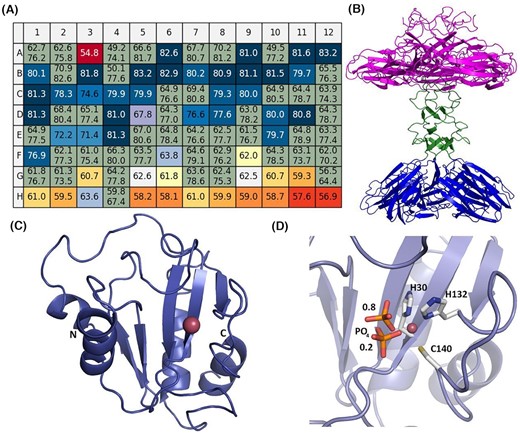
Protein characterisation and crystal structure analysis. (A) Thermal shift analysis using the Durham pH screen to identify the optimal pH range and buffer type for protein storage of an exemplary single-stranded DNA binding protein (SSB). The melting temperature in the water of 76°C (A1 and A2) is increased at low pH to over 81°C (colour-coded by a change to darker shades to blue) and decreased to over 58°C at high pH over 9 (colour-coded from yellow to red). Data analysis was performed with NAMI (Groftehauge et al. 2015). (B) Ribbon diagram of the crystal structure of XepA determined at 2.1 Å resolution (Freitag-Pohl et al. 2019). The five N-terminal domains shown in magenta are presumably responsible for the antibacterial activity and are connected via a linker region (shown in green) to the C-terminal domains that have been suggested to connect to the corners of the viral capsids to aid its escape. (C) The crystal structure of Ph2119 endolysin of Thermus scotoductus MAT2119 bacteriophage Ph2119 determined to 1.2 Å resolution (PDB entry: 6SU5) with Zn2+ (pink sphere) in a conserved peptidoglycan binding site (Plotka et al. 2020). This enzyme represents the first thermostable endolysin with amidase activity and shows a similar architecture to eukaryotic peptidoglycan recognition proteins (PGRPs) and T7 lysozyme. (D) Close view of the Zn2+ binding site with coordinating side chains (H30, H132 and C140) and a phosphate group (in sticks representation) shown in two positions with occupancies of 0.8 and 0.2, as indicated by the numbers in the figure.
Protein structure determination through X‐ray crystallography complemented bioinformatics for selected targets in all categories: i) to provide structural insight in atomic detail into gene products of established function but of particular interest to the consortium members; ii) to verify putative functional assignment, and iii) to provide important clues to the possible function of hypothetical genes that lack significant similarity to known sequences and have no assigned putative function. As a result, 24 crystal structures of target proteins have been determined, 7 in category A, 11 in B and 6 in C, respectively. In addition, 16 targets are at the final stage of analysis, where we have already obtained crystals and collected diffraction data sets. Perfect examples of the collaborative effort are the functional and structural analysis of two enzymes with an antimicrobial activity where several Virus-X consortium members contributed: Bacillus subtilis prophage PBSX lytic cassette protein XepA (Fig. 7B) (Freitag-Pohl et al. 2019) and highly thermostable Ph2119 endolysin from extremophilic Ph2119 bacteriophage of Thermus scotoductus (Plotka et al. 2020) (Fig. 7C and D). The latter is N-acetylmuramoyl-L-alanine amidase (EC 3.5.1.28), with unique structural features among thermophilic phages lytic enzymes. It contains characteristic Zn2+ binding site and shows similarity to T3 and T7 phage lysozymes and also to eukaryotic peptidoglycan recognition proteins (PGRPs), which are engaged in innate immunity and are highly conserved from invertebrates to mammals (Dziarski and Gupta 2006a; Dziarski and Gupta 2006b; Wang et al. 2007). In the case of Ph2119 endolysin, the conserved motif is formed by three amino acid residues: His30-His132-Cys140 that coordinate Zn2+ (Fig 7D).
The Virus-X strategy to identify proteins with novel functions within the type B and C targets was based on two main elements. First, optimized sequence analysis followed by structure-based studies to unveil sequence-function data. Second, experimental characterisation based on the combination of biochemical and biophysical tests. This kind of approach allowed to thoroughly characterize 97 proteins from all categories, including novel thermostable DNA polymerases with strand displacement activity, robust lytic enzymes with high thermal stability that can be considered as perspective antibacterial agents (Plotka et al. 2019a, Plotka et al. 2019b; Freitag-Pohl et al. 2019; Plotka et al. 2020), and single-strand DNA binding proteins that increase specificity either in PCR-based or isothermal amplification of DNA (Werbowy et al. 2020). Some of these proteins, including a set of novel thermostable heat-shock proteins that are capable of stabilizing proteins and supporting their folding process, have led to the patent application filed in 2020.
Platform 4. Invention to innovation
Research projects are often invention‐rich; however, the apparent gap between scientific research and commercialization is often difficult to bridge and has been called ‘The valley of Death’ (Ehlers 1998). Virus-X partners have therefore developed an exploitation plan with clear commercialization goals recognizing the importance of turning inventions into innovations and exploitable intellectual property (IP). The project addressed real opportunity markets for the SMEs. The current molecular diagnostic methods face constraints such as DNA/RNA extraction from samples, low DNA content in a sample, inhibition of the polymerase chain reaction (PCR), mutation of amplified PCR products, and DNA contamination (Yang and Rothman 2004; Afzal 2020). Novel enzymes dedicated to molecular biology, such as robust thermostable strand‐displacement DNA polymerases and DNA/RNA replication proteins, are in constant demand. New products, technologies, and knowledge that can lead to better diagnostics and research tools will, in the long run, save time and money and improve standards of living.
In addition, Virus-X put a strong emphasis on demonstration and dissemination activities for increasing visibility of commercially exploitable results to end-users, building on experience, established channels, and proven strategies in previous projects. Highlights of this work include the ‘Going to Extremes’ session at the multidisciplinary Euroscience Open Forum 2018 in Toulouse, France, participation in the Europe-wide Researcher's Night in October 2019 in Reykjavik (http://virus-x.eu/uncategorized/virus-x-on-researchers-night/), a feature on the Futuris program of the international TV channel, Euronews (https://www.euronews.com/2020/09/14/virus-hunters-explore-iceland-s-geothermal-hot-springs-for-solutions) as well as a popular science publication (Ævarsson 2018).
COVID-19 task force
Since January 2020 the World has been struggling with the Coronavirus pandemic (COVID-19) caused by severe acute respiratory syndrome coronavirus SARS-Cov-2 (Zhu et al. 2020). In order to join the global efforts to combat the disease, the Virus-X COVID-19 task force was established. It aims to leverage our knowledge and expertise to support COVID-19 research by (i) participating in large-scale sequencing of virus isolates in different countries; (ii) tracking SARS-CoV-2 evolution; (iii) the development of quick, high-throughput diagnostic methods, and (iv) supporting structure-based drug discovery for specific antiviral treatments. The Virus-X research group at the University of Bielefeld has taken an early lead in complete genome SARS-CoV-2 sequencing and sequence analysis in Germany as part of the Deutsche Covid-19 Omics consortium (Schulte-Schrepping et al. 2020). The group at the University of Clermont, Auvergne, France, uses their bioinformatics programs to analyse hundreds of SARS-CoV-2 genomes published in open databases; phylogenetic analysis of local clusters provides essential information on the epidemiology of the disease. SARomics (Lund, Sweden) cofounded the LundaGUARD, a private-public consortium in Sweden developing a platform for the rapid scientific response to the COVID-19 crisis and future pandemic threats. Bio-Prodict (Netherlands) has released in open access their customized expert system 3DM for several proteins encoded by SARS-CoV-2, including viral enzymes, potential key targets for drugs, and the surface proteins fundamental for the development of vaccines. The Virus-X SME partner A&A Biotechnology (Poland) stepped up to the challenge and quickly increased the capacity of their RNA extraction kits used as the first step in the testing regime (Caruana et al. 2020). The company is also taking part in the development and implementation of coronavirus test kits. While the current gold standard tests are based mainly on RT-PCR, the consortium has also focused on new technologies, namely reverse transcriptase Loop-mediated isothermal amplification (RT-LAMP). As shown recently by other groups, this method offers a fast and reliable test for viral RNA in a single step at one temperature (Dao Thi et al. 2020; Ganguli et al. 2020; Huang et al.2020). The Virus-X SME, ArcticZymes in Norway in close collaboration with the grous in Bielefeld (Germany) and Durham (UK) uses the unique project resources to develop new tools for molecular diagnostics.
CONCLUSIONS
The Horizon 2020 research programme was set up by the European Union to support the goal to become the world's most competitive knowledge-based economy by coupling excellent research and innovation. The results obtained within the frame of the Virus-X project have expanded our knowledge and understanding in many areas: an exploration of new genetic territory and sequence diversity, development of new approaches and tools for viral metagenomics, understanding of microbial communities, identification of commercially valuable genes, and the corresponding impact on European biotech industry and companies involved in the project towards new marketable products and services. The project combined basic research questions with real market opportunities for the participating SME partners. The development of novel enzymes for molecular biology allows for the design and development of new tools for genetic engineering. Our investigations of thermophilic enzymes such as strand‐displacement DNA polymerases and DNA/RNA replication proteins could significantly improve their current use as diagnostic tools that are of vital importance, particularly in times of a new pandemic. Current molecular diagnostic methods for COVID-19 face significant challenges, including DNA/RNA extraction from a range of sources, low DNA content in a sample, inhibition of the polymerase chain reaction, mutation of amplified PCR products, and DNA contamination. New products, technologies, and knowledge that can lead to better diagnostics and research tools will save time and money and improve standards of living. It is evident that the legacy of the Virus-X project in terms of vast sequences, cloned genes, and produced target proteins, as well as the bioinformatics software that was developed, will continue to be explored and exploited for years to come.
ACKNOWLEDGEMENTS
We would like to thank the many undergraduate students involved at several Universities for their hard work and dedication. We are also very grateful to our technical staff for their great support during the project.
FUNDING
Funding was provided by the Europan Union's Horizon 2020 Research and Innovation Programme Virus-X project: Viral Metagenomics for Innovation Value (grant no. 685778). This work was supported by the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) (031A537B, 031A533A, 031A538A, 031A533B, 031A535A, 031A537C, 031A534A, 031A532B).
Conflicts of Interest
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}