





Clostridium difficile causes antibiotic-associated diarrhea and pseudomembranous colitis mainly through two exotoxins TcdA and TcdB that target intestinal epithelial cells. Dendritic cells (DCs) play an important role in regulating intestinal inflammatory responses. In the current study, we explored the interaction of TcdB-intoxicated epithelial cells with mouse bone marrow-derived DCs. TcdB induced cell death and heat shock protein translocation in mouse intestinal epithelial CT26 cells. The intoxicated epithelial cells promoted the phagocytosis and the TNF-α secretion by DCs. Incubation with TcdB-intoxicated CT26 cells stimulated DC maturation. Moreover, TcdB-treated CT26 cells induced DC immigration when they were injected into mice subcutaneously. Taken together, these data demonstrate that TcdB-intoxicated intestinal epithelial cells are able to stimulate DC activation in vitro and attract DCs in vivo, indicating that epithelial cells may be able to regulate DC activation under the exposure of TcdB during C. difficile infection.
INTRODUCTION
Clostridium difficile (C. difficile) is the most common cause of nosocomial antibiotic-associated diarrhea and pseudomembranous colitis (Cloud and Kelly 2007). Clostridium difficile-associated diarrhea and intestinal inflammatory disease (collectively designated CDI) is thought to be mainly mediated by exotoxins TcdA and TcdB (Kelly, Pothoulakis and LaMont 1994; Voth and Ballard 2005; Elliott et al., 2007), which glucosylate low molecular mass GTPases of the Rho family, including Rho, Rac and CDC42 within host cells (Voth and Ballard 2005). TcdB is highly cytotoxic for many cell lines (Voth and Ballard 2005) and also proinflammatory, capable of inducing cytokine or chemokine production in target cells (Flegel et al., 1991; Linevsky et al., 1997). TcdB was initially reported to exhibit no enterotoxic activity in animal models (Lyerly et al., 1982, 1985). However, later studies showed that TcdB is enterotoxic and proinflammatory in human colonic biopsies (Riegler et al., 1995), human intestinal xenografts in immunodeficient (SCID) mice (Savidge et al., 2003) and in hamsters (Lyras et al., 2009). More importantly, TcdA negative TcdB positive (A−B+) strains of C. difficile can cause disease in patients and experimental animal models (Shin et al., 2008; Lyras et al., 2009). Although it has been shown that TcdB can induce inflammatory response in vitro (Flegel et al., 1991) and in vivo (Shin et al., 2008; Lyras et al., 2009), the mechanism regarding TcdB inducing inflammatory disease remains unclear.
Dendritic cells (DCs) play critical roles in innate and adaptive immunity (Steinman 1991). The in vitro maturation process of DCs can be elicited by various inflammatory stimuli (Nakahara et al., 2006). Furthermore, DCs were showed capable of regulating intestinal inflammatory response. The direct activation of DCs through CD40 can result in intestinal inflammation (Uhlig et al., 2006; Coombes and Powrie 2008). It has been reported that TcdA can promote DC maturation and chemokine expression (Lee et al., 2009). Also, TcdB is able to elicit IL-1β production by DCs (Jafari et al., 2013). However, little is known about the regulatory roles of DCs on C. difficile toxin-mediated inflammatory response and their interaction with intoxicated intestinal epithelial cells (IECs). TcdB are capable of inducing cell death of epithelial cells (Matarrese et al., 2007; Chumbler et al., 2012), and the pathway of TcdB induced cell death may dependent on the toxin concentration or the cell types. Additionally, C. difficile toxins are able to induce the expression of proinflammatory cytokines in epithelial cells (Ng et al., 2003). The crosstalk between epithelial cells and DCs is critical for regulating the intestinal immune homeostasis (Rimoldi et al., 2005). Since the intestinal epithelium is the first line exposed to C. difficile toxins, it is therefore important to investigate the interaction of the intoxicated IECs with DCs.
The current data in this study show that TcdB-intoxicated IECs are capable of activating DCs in vitro and have the ability to attract DCs in vivo, indicating that the epithelial cells exposed to TcdB may play roles in regulating DC function and subsequent intestinal inflammation during C. difficile infection.
MATERIALS AND METHODS
Mice, cell lines and toxins
Six- to ten-week-old male BALB/c mice were purchased from the Medical Experimental Animal Center (Guangdong, China) or Harlan Laboratories (Maryland USA). All animals were handled and cared for according to China Animal Care and Use Committee guidelines or Institutional Animal Care and Use Committee guidelines and in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Murine IECs CT26 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (Invitrogen), 100 U mL−1 penicillin, 100 μg mL−1 streptomycin (Invitrogen), 2 mM L-glutamine (Invitrogen) and 1 mM pyruvate acid (Invitrogen). Full-length recombinant TcdB were purified from total crude extract of Bacillus megaterium as described previously (Yang et al., 2008). The highly purified recombinant TcdB that appeared as a single band on SDS-PAGE, and was absent of detectable Toll-like receptor 2 (TLR2) and 4 (TLR4) ligand activity as determined by bioassays (Yang et al., 2008; Sun et al., 2009) was used in this study.
Cytotoxicity assays
Subconfluent CT26 cells were exposed to different doses of TcdB. The cytopathic change (cell rounding) was assessed using scanning electron microscopy after 4 h of toxin treatment (10 ng mL−1). Coverslips with adherent CT26 cells were fixed in 2.5% glutaraldehyde in PBS, post-fixed in 1% osmium tetroxide (OsO4) in phosphate buffered saline (PBS) and then dehydrated in a graded series of ethanol using a Pelco Biowave Microwave Processor. Samples destained for scanning electron microscopy (SEM) were then critical-point dried, mounted on stubs with carbon tabs and coated with 3 nm platinum for high-resolution imaging. SEM images were acquired using a Zeiss 1555VP field emission electron microscope. MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) viability assay was used to determine the cytotoxicity of the toxins as described previously (He et al., 2009a).
Fluorescence detection of cell surface calreticulin
Cell surface expression of calreticulin (CRT) was examined as described previously (Obeid et al., 2007). Briefly, CT26 cells were incubated with 500 ng mL−1 TcdB or 10 ug mL−1 of Mitomycin C for 24 h before harvesting and then stained with rabbit anti-mouse CRT antibody (dilution 1:1000; Abcam). The secondary antibody used was Alexa Fluor 488 conjugated goat anti-rabbit IgG (H+L) (dilution 1:100; Invitrogen). After staining, the cells were analyzed for surface CRT of propidium iodide (PI)-negative cells by flow cytometry. In some experiments, the cells were fixed in 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) in 1 × PBS, counterstained with Hoechst 33342 (Invitrogen), and examined for CRT fluorescence using a Nikon TE300 microscope (Tokyo, Japan) and Bio-Rad 1024 MRC confocal imaging system (Bio-Rad, Hercules, CA, USA).
Western blotting for heat shock protein upregulation or secretion
CT26 cells were incubated with 500 ng mL−1 of TcdB for different periods of time. Cells were harvested and lysed and total heat shock proteins (HSPs) were examined by immunoblotting assays. To measure the secretion of proteins by epithelial cells, cells were exposed to TcdB in serum-free medium for different periods of time before harvesting the supernatant for immunoblotting analysis. The primary antibodies used in this assay were rabbit anti-HSP70 (1 μg mL−1; Abcam, Cambridge, MA, USA), or anti-HSP90 (1 μg mL−1; Abcam). The secondary antibody used was goat anti-rabbit IgG conjugated with horseradish peroxidase (HRP) (dilution 1:5,000; Calbiochem, San Diego, CA, USA). Anti-actin antibody (dilution 1:1000; Abcam) was used as a control for equal loading in total cell lysate or necrotic death of cells in cell culture supernatants.
Phagocytosis assay
BALB/c mouse bone marrow DCs were generated as described previously (Feng et al., 2002, 2003). More than 90% of these cells were CD11c+ DCs (Feng et al., 2005). CT26 cells were stained with carboxyfluorescein diacetate succinimidyl esters (CFSE; Invitrogen) following the method described previously (Feng et al., 2005). The CFSE-stained CT26 cells were exposed to TcdB (100 ng mL−1, 4 h) before extensive washing and then co-cultured with DCs for 24 h before harvesting. The mixed cells were stained with R-phycoerythrin (PE) -conjugated anti-CD11c antibody (clone HL3; BD PharMingen, San Diego, CA, USA) before analysis by flow cytometry using FACSCalibur and CellQuest software (BD Biosciences, Mountain View, CA, USA).
TNF-α production
Intracellular cytokine staining was used to determine the TNF-α production in DCs as described previously (Sun et al., 2009). CT26 cells were treated with PBS or TcdB (100 ng mL−1) for 4 h before extensive washing to remove free toxins. In some groups, cells were treated with goat antitoxin polyclonal antibodies as a negative control (Techlab Inc., Blacksburg, VA, USA) for 5 min on ice after washing. Cells were then pulsed to DCs for 36 h. In the last 12 h of co-culture, cells were treated with 20 μM brefeldin A. Cells were then harvested and TNF-α production was determined by intracellular staining using an Alexa-647-conjugated anti-mouse-TNF-α antibody, followed by flow cytometry analysis.
FACS analysis of DC maturation
Immature DCs were co-incubated with live CT26 cells or CT26 dying cells induced by TcdB at a 1:1 ratio for 24 h. In some experiments, DCs were treated with lipopolysaccharide (LPS; Sigma-Aldrich, St. Louis, MO, USA) for 6 h as a positive control. Mixed cells or LPS-matured DCs were harvested and stained with PE-conjugated anti-CD11c (clone HL3; BD PharMingen) and FITC-conjugated anti-CD86 (clone 1C10; Beckman Coulter, Fullerton, CA, USA), anti-CD80 (clone 1G10; Beckman Coulter) or anti-CD40 (clone 2D10; Beckman Coulter). After 30 min of incubation at 4°C, the cells were washed three times in PBS containing 1% bovine serum albumin and analyzed by flow cytometry using FACS Calibur (BD Biosciences) and CellQuest software (BD Biosciences).
Immunohistochemistry assay for DC immigration
Balb/c mice were injected with PBS or TcdB-intoxicated CT26 cells (106 cells/mouse) with or without anti-toxin pretreatment subcutaneously into the neck-notum with 20-gauge needles. Mice were sacrificed 12, 24 or 48 h later by cervical dislocation with standard performance, and the tissues at the injection site were removed and fixed in 4% formaldehyde/0.1 M PBS buffer overnight at 4°C. A tissue block was embedded with paraffin, and cut and sectioned at 5 μm with a slicing machine (KD-2508; Zhejiang Kedi, Zhejiang, China). The deparaffinized slices were treated with 0.3% hydrogen peroxide in methanol for 10 min at room temperature to block endogenous peroxidase activity, and blocked in normal sheep serum. The sections were incubated with rabbit anti-CD11c antibodies (clone HL3; BD PharMingen) followed by HRP-labeled secondary antibody staining and developed with DAB solution from EnvisionTM Detection Kit (GK500705; DAKO, Glostrup, Denmark) according to the manufacturer's instructions. Following three washes in ddH2O, the sections were processed using hematoxylin to visualize the nucleus. Tissue sections were examined using a Zeiss bright-field microscope, and imaged with a Nikon digital camera attached to the microscope.
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM) unless otherwise indicated. Statistical analysis was performed by unpaired two-tailed t-test or one-way ANOVA using the Prism statistical software program (GraphPad Software, Inc., San Diego, CA, USA).
RESULTS
Cytotoxicity of TcdB on CT26 cells
The action of C. difficile toxins on cultured cells leads to reorganization of the cytoskeleton and cell rounding (Voth and Ballard 2005). We examined the susceptibility of CT26 cells to TcdB. Exposure of CT26 cells to 10 ng mL−1 TcdB resulted in a rapid cell rounding, as determined by SEM (Fig. 1a), while the CT26 cells treated with medium maintain a normal shape. After 3 days of exposure to the toxin, TcdB induced death of most CT26 cells in the pg mL−1 range, as determined by the MTT viability assay (Fig. 1b). These data indicate that mouse IECs CT26 are highly sensitive to the cytotoxic activity of TcdB.
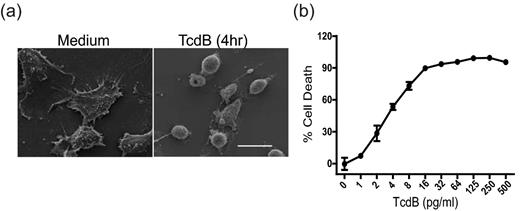
Cytopathic and cytotoxic effects of TcdB on CT26 cells. (a) SEM shows the rapid CT26 cell rounding triggered by TcdB exposure. CT26 cells were untreated (left panel) or exposed to 10 ng mL−1 of TcdB (right panel) for 4 h before harvesting for imaging. Scale bar: 20 μm. (b) CT26 cells are highly sensitive to TcdB in a MTT viability assay. CT26 cells were exposed to the indicated concentrations of TcdB for 72 h, and cell viability was measured by the MTT assay. These data represent the mean of three independent determinations ±SEM.
Translocation of CRT onto the cell surface of dying CT26 cells
The translocation of a calcium-binding protein CRT, which is normally located in the endoplasmic reticulum or Golgi apparatus, onto the plasma membrane is associated with increasing phagocytosis of dying cells by DCs (Clarke and Smyth 2007; Obeid et al., 2007). We therefore examined whether TcdB exposure of CT26 cells results in the plasma membrane translocation of CRT. Fig. 2a shows that CRT translocated to the surface of CT26 cells after 24 h of TcdB exposure, as determined by confocal microscopy. Mitomycin C treatment failed to induce translocation of the protein (Fig. 2a) although it induced cell death of CT26 cells (data not shown). Flow cytometry analysis of CT26 cells, using fluorochrome-conjugated antibodies against CRT, showed a similar result as observed by confocal microscopy (Fig. 2b). Quantitative analysis of three independent flow cytometry experiments confirmed that TcdB, but not mitomycin C, treatment of CT26 cells, significantly enhanced cell surface translocation of CRT (Fig. 2c).
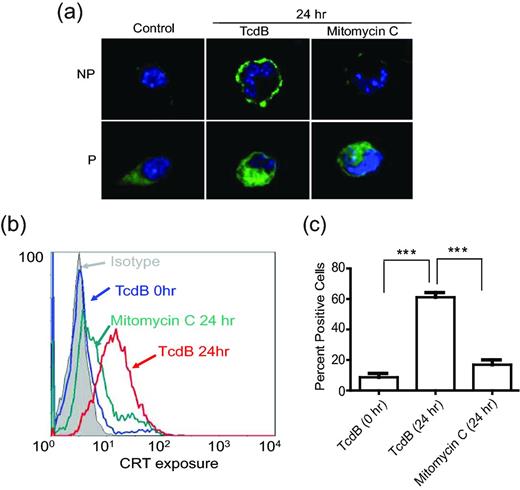
Cell surface translocation of CRT in TcdB-treated CT26 cells. (a) Immunofluorescence detection of CRT on the cell surface (in non-permeabilized live cells; NP) or within the cell (with fixation and permeabilization; P) was performed using confocal microscopy (100×). The nuclei were visualized with Hoechst33342 (blue). (b and c) Flow cytometry analysis of PI-negative cells for surface expression of CRT. Quantitative analysis of three independent results from flow cytometry is shown in c (Data were analyzed by unpaired two-tailed t-test; ** represents P < 0.01, *** represents P < 0.001; error bars, SEM).
Upregulation and secretion of HSPs in TcdB-treated CT26 cells
HSPs play important roles in modulating both innate and adoptive immunity, including the regulation of DC activation and the secretion of proinflammatory cytokines or chemokines (Feng et al., 2002; McNulty et al., 2013). Inducible HSP70 (HSP72) and HSP90 are two main members of the HSP family that are associated with DC activation (Lehner et al., 2004; Spisek et al., 2007; Tesniere et al., 2008). After exposure of TcdB, the HSP70 was upregulated in CT26 in a time-dependent manner (Fig. 3a). Total levels of HSP70 in CT26 cells were significantly elevated after 24 h of toxin exposure. In contrast to HSP70, the total expression of HSP90 did not change in response to TcdB exposure (data not shown). Since the secretion of these HSPs is correlated with the activation of DCs (Mycko et al., 2004; Martin et al., 2009) or microphages (Vega et al., 2008), we examined the release of HSP70 and HSP90 in the culture medium of the intoxicated CT26 cells. Both HSP70 and HSP90 were released in the culture media of CT26 cells after 16 or 24 h of toxin treatments (Fig. 3b). The release of these HSPs was not mainly due to necrotic death of the cells, but rather to secretion since comparatively little actin was released in the culture media (Fig. 3b lower panel). No significant differences in the cell surface expression of HSP70 or HSP90 were observed between TcdB-exposed and untreated CT26 cells (data not shown).
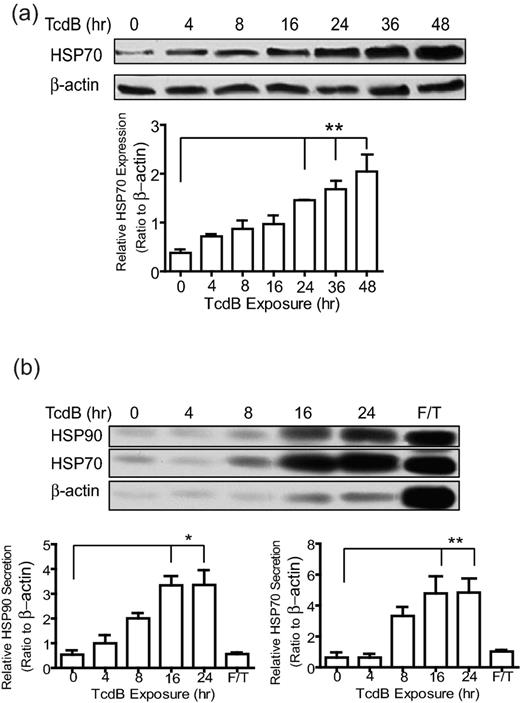
Upregulation and secretion of HSP in CT26 cells induced by TcdB intoxication. CT26 cells were incubated with 500 ng mL−1 of TcdB for the indicated times. (a) Cells were lysed and the expression of HSP70 was measured using Western blotting. Quantitative data (lower panel) from three independent experiments were analyzed by one-way ANOVA (** represents P < 0.01). Error bars show the standard error of mean (SEM). (b) The supernatant was collected and concentrated for measuring the secretion of HSP70 or HSP90 by intoxicated CT26 cells by Western blotting. Quantitative data (lower panel) from three independent were analyzed by one-way ANOVA (* represents P < 0.05; ** represents P < 0.01). Error bars, SEM. F/T means repeatedly frozen-thawed cell lysate.
The phagocytosis of TcdB-treated CT26 cells by DCs
Since C. difficile toxins are proinflammatory and able to induce IECs to translocate CRT to the cell surface (Fig. 2) and release cytokines/chemokines (Ng et al., 2003; Savidge et al., 2003) and HSPs (Fig. 3b), we hypothesized that TcdB-treated CT26 cells are capable of activating DCs. We first examined whether TcdB-intoxicated cells were susceptible of being phagocytosed by mouse bone marrow-derived DCs (BMDCs). As shown in Fig. 4a, CD11c-positive BMDCs phagocytosed TcdB-exposed CSFE-labeled CT26 cells, as indicated by the double positive population in the upper-right quadrant of the dotplots (Fig. 4a lower panel), whereas the untreated CT26 cells remained a separate population from the DCs (Fig. 4a upper panel). These data suggested that after toxin intoxication, the epithelial cells become ready to be phagocytosed by DCs.
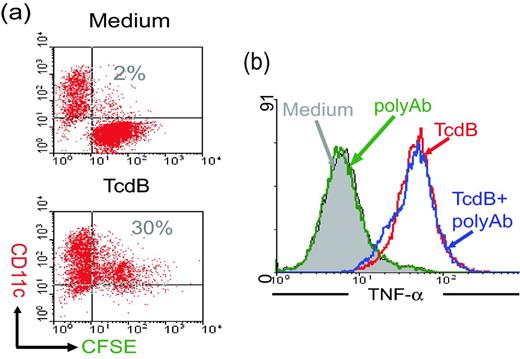
Phagocytosis of intoxicated CT26 cells and TNF-α production by DCs. (a) DC phagocytosis of intoxicated CT26 cells. After co-culturing TcdB-intoxicated and CFSE-labeled CT26 cells with CD11c-positive mouse BMDCs, the cell mixture was harvested and analyzed by FACS. (b) The production of TNF-α by DCs was detected by flow cytometry. CT26 cells alone or TcdB-intoxicated epithelial cells with or without anti-TcdB polysera (polyAb) treatment were used to stimulated DCs. Brefeldin A was added in the last 12 h of co-culture, and the cells were harvested to measure intracellular TNF-α expression. These data show the gated CD11c-positive cells under FACS analysis.
Maturation of DCs and TNF-α production after stimulation by TcdB-treated CT26 cells
We next determined DC maturation induced by the intoxicated epithelial cells through measuring the expression of surface co-stimulatory molecules CD86, CD80 and CD40. Similar to LPS, TcdB-treated CT26 cells induced the upregulation of the surface markers CD86, CD80 and CD40 as compared to the untreated epithelial cells (Fig. 5), suggesting that the intoxicated CT26 cells have the potent capacity to induce DC maturation. Furthermore, co-culturing BMDCs with TcdB-exposed, but not with untreated CT26 cells, stimulated their production of TNF-α (Fig. 4b). The cytokine-driven effect was not a result of possible toxins bound to the surface of CT26 cells, because the exposure of the intoxicated cells to neutralizing antibodies did not block the induction of TNF-α by DCs (Fig. 4b). Neutralizing antibodies alone with untreated CT26 cells did not induce TNF-α production (Fig. 4b). These in vitro data showing DC maturation and their production of TNF-α induced by TcdB exposed-CT26 cells indicated the activity of TcdB-intoxicated epithelial cells to trigger DC activation.
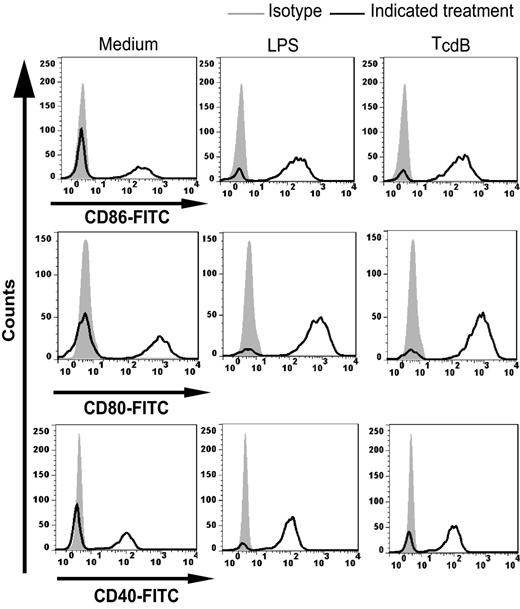
Maturation of DCs incubated with TcdB-intoxicated CT26 cells. DCs were incubated with CT26 cells alone (medium), TcdB-treated CT26 cells, or LPS. The cell mixtures were harvested and the cell surface expression of CD40, CD80 or CD86 for the CD11c-positive gated population was analyzed by FACS.
DCs were attracted by TcdB-treated CT26 cells in vivo
Finally, we investigated whether TcdB-intoxicated epithelial cells can induce DC migration locally in vivo, a key step for DC activation developed in vivo. TcdB-treated CT26 cells significantly enhanced DC recruitment 24 h after subcutaneous injection of toxin-intoxicated epithelial cells (Fig. 6, P < 0.05). DC recruitment was not due to the presence of carcinogenic epithelial cells since injection of CT26 cells treated with antitoxin polyserum-neutralized TcdB failed to recruit DCs to the injection site (Fig. 6). The local DC accumulation was reduced after 48 h post-injection (Fig. 6a). This may be because some recruited DCs migrate to draining lymph nodes after their activation and engulfment of toxin-exposed epithelial cells. Thus, TcdB-intoxicated CT26 cells are able to attract DC migration in vivo.
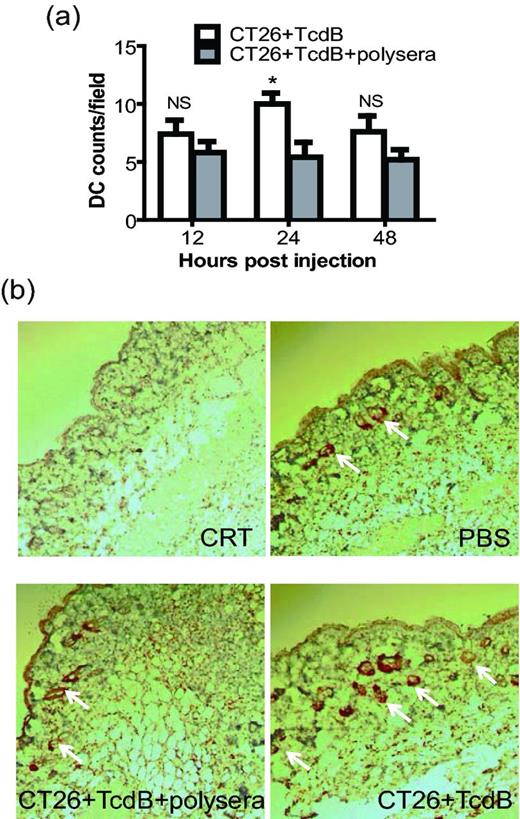
DC immigration induced by TcdB-exposed CT26 cells in vivo. Mouse tissues were collected 12, 24 or 48 h after injection of TcdB-treated CT26 cells. Tissue DCs were detected by immunohistochemistry performed on sections from paraffin-embedded tissue blocks using CD11c antibodies and DAB visualization; the nuclei were stained with hematoxylin. (a) Quantitative analysis of the number of DCs from five equivalent fields in each tissue section. These data represent the mean of five-field determinations ± SEM and were analyzed by unpaired two-tailed t-test. * represents P < 0.05 for TcdB group (CT26 + TcdB) vs antitoxin pretreatment group (CT26 + TcdB + polysera). (b) The images from representative fields, for different groups at 24 h, were taken with a Nikon digital camera attached to a microscope (200×) and arrows point CD11c-positive staining.
DISCUSSION
Clostridium difficile can proliferate and colonize the gut when the intestinal microbiota equilibrium is disrupted (Lawley et al., 2009; Reeves et al., 2012). The toxins released by the bacteria (TcdA and TcdB) are the main factors to induce intestinal damage and inflammatory disease (Voth and Ballard 2005). The toxins are proinflammatory and able to induce cytokines/chemokines by IECs (Ng et al., 2003). They also disrupt epithelial barriers and disseminate into the lamina propria allowing their direct interaction with immune cells. Clostridium difficile toxins can induce the production of cytokines or chemokines by immune cells, including DCs (Flegel et al., 1991; Linevsky et al., 1997; Jafari et al., 2013). Immune response caused by C. difficile toxins plays important roles in regulating the disease progression of CDI. While the adaptive immune response against toxins provides hosts the protection against C. difficile infection (Kelly and Kyne 2011), the innate immune response with the production of proinflammatory cytokines is critical to the pathophysiology of CDI (Kelly and Kyne 2011; Czepiel et al., 2014). Besides interacting with immune cells directly, C. difficile toxins may stimulate immune response through IECs by inducing their productions of inflammatory mediators. In this study, we investigated the interaction of TcdB with IECs and subsequent interaction with DCs. Our results show that TcdB-intoxicated IECs are capable of activating DCs in vitro and have an ability to attract DCs in vivo.
DCs play critical roles in regulating intestinal immune homeostasis, and inappropriate DC function may cause the pathogenesis of intestinal inflammatory disease (Coombes and Powrie 2008). Aberrant properties of DCs may be elicited by their local tissue environment, including pathogens and miscommunication between epithelial cells and DCs (Coombes and Powrie 2008). The IECs can sense the environment conditions, such as the presence of pathogenic bacterial strains and their products, and subsequently interact with DCs. TcdB can induce cytokine/chemokine production in epithelial cells (Savidge et al., 2003). Interestingly, we found that TcdB-induced HSP secretion and the cell-surface translocation of CRT in IECs CT26. These TcdB-induced signals by IECs may subsequently regulate DC function. The translocation of CRT and HSPs (HSP70 and HSP90) has been reported to play roles in promoting the antigen-phagocytosis and maturation of DCs (Clarke and Smyth 2007; Tesniere et al., 2008). As the current study, incubation of DCs with TcdB-exposed, but not untreated, CT26 cells stimulated DCs phagocytosis of these intoxicated epithelial cells. Furthermore, TcdB-treated CT26 cells induced the upregulation of the surface markers CD86, CD80 and CD40 as compared to the untreated epithelial cells. Thus, TcdB interacts with host IECs in turn activating DCs via production of cytokines/chemokines and regulation of HSP translocation.
In this study, we found that TcdB-exposed CT26 cells stimulated DCs to produce TNF-α. We previously found that both TcdA and TcdB possess potent ability to directly stimulate murine macrophages to produce TNF-α (Sun et al. 2009). TNF-α is a potent proinflammatory cytokine playing roles in C. difficile-mediated colitis (Darkoh et al., 2014) and other intestinal inflammation such as inflammatory bowel disease (Uhlig et al., 2006). TNF-α inhibitors were capable of decreasing TcdB-induced neutrophil migration significantly (Carneiro-Filho et al., 2001), suggesting that TNF-α may be important for TcdB-induced inflammation. Moreover, TNF-α has an ability to induce DC maturation (Trevejo et al., 2001; Baldwin et al., 2010), thus the TNF-α production of DCs may further augment DCs’ role in the intestinal inflammatory process.
To determine the possibility of crosstalk between toxin-exposed epithelial cells and DCs in vivo, we injected mice with intoxicated CT26 cells subcutaneously and assess their ability to induce DC migration. These intoxicated CT26 cells are able to attract DCs to the site of injection within 24 h post-injection. These data suggested that epithelial cells may be able to regulate DC function after TcdB intoxication and subsequently contribute to the intestinal inflammatory disease. Further work will be needed to confirm this hypothesis in CDI model and investigate the mechanism by which the TcdB-intoxicated epithelial cells regulate DC function and contribute to the inflammation during bacterial infection. The crosstalk between pathogens, epithelial cells and DCs may be complicated and it may be a challenge to distinguish the contribution of toxins and that of toxin-intoxicated epithelial cells to DC activation in CDI model. Furthermore, except for the toxins released by C. difficile, bacteria or other bacterial products may also interact with IECs and/or DCs, and subsequently play roles in host defense and pathogenesis in CDI (Ryan et al., 2011; Jafari et al., 2013). Nevertheless, understanding the crosstalk of major virulence factors with host IECs and immune cells may be helpful to design new treatment strategies against CDI.
FUNDING
This work was supported by awards R01AI088748, R01DK084509, R56AI99458, and U19 AI109776, funded from the National Institute of Allergy and Infectious Diseases and National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (NIH).
Conflict of interest statement. None declared.
REFERENCE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}