





Abstract
There are over 100 known species of cultivated potatoes and their wild relatives. Many of these species, including cultivated potatoes, share the A genome; these species are mainly distributed in South America and are reproductively isolated from Mexican diploid species. The only diploid A-genome species distributed in Mexico is Solanum verrucosum Schlechtendal, which is also a maternal progenitor of Mexican polyploid species. In this study, we constructed a high-quality de novo assembly of the S. verrucosum genome using PacBio long-read sequencing and Hi-C scaffolding technologies. A monohaploid clone (2n = x = 12) of S. verrucosum was used to reduce assembly difficulty due to the heterozygous nature of the species. The final sequence assembly consisted of 780.2 Mb of sequence, 684.0 Mb of which were anchored to the 12 chromosomes, with a scaffold N50 of 55.2 Mb. Putative centromeres were identified using publicly available data obtained via chromatin immunoprecipitation sequencing against a centromere-specific histone 3 protein. Transposable elements accounted for approximately 61.8% (482.1 Mb) of the genome, and 46,904 genes were functionally annotated. High gene synteny and similarity were revealed among the genomes of S. verrucosum, Solanum commersonii, Solanum chacoense, Solanum phureja, Solanum tuberosum, and Solanum lycopersicum. The reference-quality S. verrucosum genome will provide new insights into the evolution of Mexican polyploid species and contribute to potato breeding programs.
Introduction
Potato (Solanum tuberosum L., 2n = 4x = 48) is the most important noncereal food crop in the world. High genetic diversity is observed among primitive cultivated potatoes and the over 100 wild potato species distributed from North and Central America to South America (Hawkes 1990; Spooner et al. 2014). These species are classified into 2 reproductively isolated groups: (1) a group including all Mexican diploid species except for S. verrucosum Schlechtendal and (2) a group including all Mexican polyploid species, S. verrucosum, and all South American species (Hawkes 1958). Based on the meiotic chromosome pairing of interspecific hybrids, the A genome is assigned to the species in the second group (Matsubayashi 1991). Since sexual hybrids between Mexican diploid species and A-genome species are extremely difficult to obtain, their genome affinity has long been debated (Matsubayashi 1991; Pendinen et al. 2008).
S. verrucosum is the only diploid A-genome species from Mexico and is assumed to contribute the A genome of Mexican polyploid species (Hosaka et al. 1984). Most diploid tuber-bearing Solanum species are self-incompatible (Pushkarnath 1942), whereas S. verrucosum is self-compatible (Hawkes 1990). S. verrucosum is cross-compatible with most South American species as the female parent (Eijlander et al. 2000) and with some Mexican diploid species, which provides an opportunity to transfer useful traits from Mexican diploid species to cultivated potatoes as a bridging species (Hermsen and Ramanna 1976; Jansky and Hamernik 2009). The Mexican species, including S. verrucosum, are valuable sources of disease and pest resistance in potato breeding (Hein et al. 2009; Chen et al. 2018).
The first potato genome was sequenced from the DM 1-3 516 R44 clone (hereafter referred to as DM) (Potato Genome Sequencing Consortium 2011). DM resulted from the chromosome doubling of a monoploid derived via anther culture of the cultivated diploid species Solanum phureja Juz. et Buk. (Lightbourn and Veilleux 2007). The homozygous nature of the clone facilitated genome sequencing. Since then, potato whole genomes have been sequenced mainly from cultivated potato species (Kyriakidou, Achakkagari, et al. 2020; Kyriakidou, Anglin, et al. 2020; van Lieshout et al. 2020; Zhou et al. 2020; Yan et al. 2021). Recent advances such as long-read sequencing coupled with high-throughput chromosome conformation capture (Hi-C) scaffolding technologies have resulted in great improvements in the DM genome (DM v6.1; Pham et al. 2020), and chromosome-scale phased assemblies have been obtained from heterozygous diploid and tetraploid potatoes (Zhou et al. 2020; Yan et al. 2021; Hoopes et al. 2022; Sun et al. 2022). However, whole-genome sequencing in wild species has been limited to Solanum commersonii Dun. (Aversano et al. 2015) and Solanum chacoense Bitt. (Leisner et al. 2018), both of which are distributed in the southern marginal distribution area of the South American A-genome species (Hawkes and Hjerting 1969). Only a draft genome sequence has been reported for the Mexican diploid species S. pinnatisectum Dunal (Tiwari et al. 2021).
In this study, we generate a chromosome-scale assembly of the genome of the Mexican diploid species S. verrucosum using PacBio long-read sequencing and Hi-C scaffolding technologies. A monohaploid S. verrucosum clone (2n = x = 12) was used to reduce complexity caused by the heterozygous nature of the species. The constructed reference-quality genome will provide new insights into the evolutionary process in Mexican polyploid species and contribute to potato breeding programs.
Materials and methods
Plant material
A monohaploid clone of S. verrucosum (11H23, available as PI 666968 from the U.S. Potato Genebank) that was derived from anther culture (Irikura and Sakaguchi 1972) and maintained in vitro in our laboratory (Sanetomo and Hosaka 2021) was used for sequencing.
DNA extraction
A plant grown in vitro was transferred to a pot filled with soil and further grown for DNA extraction. Fresh leaves were collected, frozen in liquid nitrogen, and ground into powder with a mortar and pestle. The powder was suspended in 7 ml of 2× CTAB buffer (100 mM Tris-Cl buffer pH 8.0, 20 mM EDTA pH 8.0, 1.4 M NaCl, 2% CTAB, 1% PVP-40, and 0.2% 2-mercaptoethanol) and incubated at 60°C for 30 min. The suspension was gently mixed with 5 ml of chloroform: isoamyl alcohol (24:1) and centrifuged at 10,000 rpm for 5 min at 20°C. Using a wide-bore pipet tip, the supernatant was transferred to a 50-ml tube containing 5 ml of isopropanol and mixed gently by inverting the tube. Aggregated DNA strands were hooked and drawn up using a Pasteur pipet modified by flaming the tip and bending it into a U shape, after which they were transferred to a tube containing 10 ml of 75% ethanol washed for 30 min. Then, the aggregated DNA was dissolved in 2 ml of TE buffer (10 mM Tris-Cl buffer pH 8.0 and 1 mM EDTA pH 8.0) and incubated with 5 μl of RNase (10 mg/ml) for 3 h at room temperature. After complete dissolution, 100 μl of 5 M NaCl and 700 μl of 99.9% ethanol were added, followed by mixing and incubation at 4°C overnight to precipitate polysaccharides. After centrifugation at 10,000 rpm for 5 min at 4 °C, the supernatant was collected and mixed gently with 9 ml of 75% ethanol with 10 mM ammonium acetate. The aggregated DNA strands were hooked and drawn up using a U-shaped Pasteur pipet, transferred to a tube containing 10 ml of 75% ethanol, and washed for 30 min. Then, the DNA was dried completely while hanging on the U-shaped Pasteur pipet and dissolved in 100 μl of sterile water.
Genome sequencing and assembly
The quality of the extracted DNA was measured with a Genomic DNA ScreenTape System (Agilent) and a Qubit Fluorometer (Thermo Fisher Scientific). A long-read DNA library was prepared with the SMRTbell Express Template Prep Kit 2.0 (PacBio) and sequenced using the PacBio Sequel lle system in CCS mode (PacBio). The resulting raw data were converted to FASTQ format using BAM2fastx 1.3.1 (PacBio). Reads longer than 5 kb were extracted with SeqKit 0.15.0 (Shen et al. 2016) and used for genome assembly with the Hifiasm 0.15.5-r350 assembler (Cheng et al. 2021).The -l 0 option was specified to disable the purge haplotigs function since the plant was monohaploid.
Hi-C sequencing and scaffolding
The Hi-C library was prepared using the Dovetail Omni-C Kit (Dovetail Genomics) following the Proximity Ligation Assay Nonmammalian Samples Protocol version 1.0. The prepared library was sequenced on the NovaSeq 6000 (Illumina) platform. The read quality was assessed using FastQC 0.11.8 (Andrews 2010) and MultiQC v1.8 (Ewels et al. 2016) and then filtered using Trimmomatic 0.39 (Bolger et al. 2014) with the “ILLUMINACLIP: TruSeq3-PE.fa : 2:30:10 TRAILING : 20 SLIDINGWINDOW : 4:15 HEADCROP : 10 MINLEN : 50” options. The trimmed reads were aligned to the contigs using Juicer 1.6 (Durand et al. 2016). Since DNase I was used to digest fixed nucleosomes, the “-s none -y none” options were specified. The generated contact maps were then used for scaffolding with a 3D-DNA pipeline (Dudchenko et al. 2017) with the default parameters. The scaffolds were manually corrected using JuiceBox 1.11.08 (https://github.com/aidenlab/Juicebox). The corrected scaffolds were aligned to the DM v6.1 reference genome using D-GENIES (Cabanettes and Klopp 2018). The identities and directions of the scaffolds were determined based on the alignment.
Identification of organelle sequences
To identify regions or contigs derived from organelle genomes, sequences of chloroplast genome of S. verrucosum (MH021593.1; Huang et al. 2019) and mitochondrial genome of S. tuberosum cultivar Désirée (MN104801, MN104802, and MN104803; Varré et al. 2019) were obtained from the National Center for Biotechnology Information (NCBI), and a nucleotide homology search was performed against S. verrucosum contigs using BLASTN 2.12.0 (Altschul et al. 1990) with the “-outfmt 6 -evalue 0.0001” options. Regions with more than 10 kb of alignment length and with more than 90% homology were selected and reformatted to BED files. Overlapped regions were merged using BEDTools 2.30.0 (Quinlan and Hall 2010).
Identification of putative centromeres
To identify centromeres, sequence reads generated via chromatin immunoprecipitation sequencing (ChIP-seq) against a centromere-specific histone 3 (CENH3) protein that were publicly available from NCBI were obtained for S. verrucosum (SRR18548893; Zhang et al. 2014) and S. phureja (SRR18548894; Gong et al. 2012) and aligned to their genomes using Bowtie2 (Langmead and Salzberg 2012) in single-end mode. The resulting BAM files were converted to BigWig files using DeepTools 3.5.1 (Ramírez et al. 2016) for visualization on the IGV 2.11.0 genome browser (Robinson et al. 2011). Centromeric regions of chromosomes were manually identified with IGV.
Annotation
Transposable elements (TEs) were identified using EDTA 1.9.6 (Ou et al. 2019), and the defined TE regions were hard masked. To evaluate assembly completeness, the long terminal repeat (LTR) assembly index (LAI) score (Ou et al. 2018) was calculated using the EDTA output files. Tandem repeats were defined using Tandem Repeats Finder v4.09 (Benson 1999) with the default parameters, and the defined repeats were soft masked using BEDTools. The masked scaffolds were subjected to gene prediction using the MAKER 3.01.03 (Cantarel et al. 2008) annotation pipeline by providing mRNA and protein sequences of DM v6.1 (Pham et al. 2020) and pretrained AUGUSTUS (Stanke et al. 2004) gene models of tomato. The functional annotation of the predicted proteins was performed using Hayai-Annotation Plants v.2 (Ghelfi et al. 2019). The density of the annotated TE families, Miniature Inverted-repeat Transposable Element (MITE) derivatives, genes, and CENH3 ChIP-seq reads within every 1 Mb segment was calculated using BEDTools and visualized in a circular heatmap generated by Circos (Krzywinski et al. 2009).
Genome synteny and orthologs
The genome of S. verrucosum was compared with those of S. phureja (DM v6.1; Pham et al. 2020), diploid S. tuberosum (Solyntus v1.1; van Lieshout et al. 2020), S. chacoense (M6; Leisner et al. 2018), S. commersonii (Aversano et al. 2015), and Solanum lycopersicum L. (Hosmani et al. 2019). Syntenic gene pairs were searched using MCScan (python version) (Tang et al. 2008) with the default parameters, and syntenic blocks containing more than 30 genes were visualized. The orthologous relationships of S. verrucosum genes were assessed using OrthoFinder (Emms and Kelly 2015, 2019). All protein-coding genes except for those encoding isoforms or sequences shorter than 10 amino acids were compared. Intersections of orthogroups were visualized with UpSetR 1.4.0 (Lex et al. 2014; Conway et al. 2017).
Results and discussion
Genome assembly
We obtained 46.5 Gb of HiFi reads using a PacBio Sequel IIe system with an N50 read size of 15.6 kb and an average read size of 14.9 kb. Reads longer than 5 kb were used for assembly with Hifiasm. The resulting assembly consisted of 1,437 contigs with an N50 contig size of 21.0 Mb (Table 1). The contigs were error corrected and scaffolded with 101 million Omni-C read pairs using Juicer and a 3D-DNA pipeline (Supplementary Fig. 1). The final sequence assembly consisted of 780.2 Mb, among which 684.0 Mb were anchored to the 12 chromosomes, with a scaffold N50 of 55.2 Mb (Table 1). Of the remaining 1,535 unanchored contigs (a total of 96.3 Mb in size), 688 contigs (33.3 Mb) and 102 contigs (3.6 Mb) showed high homology to the chloroplast and mitochondrial genomes, respectively (Supplementary File 1). The dot plot analysis using these contigs against the organelle genomes indicated that these contigs were fragments of the organelle genomes (Supplementary Fig. 2). This is in accordance with previous studies that most of smaller contigs from Hifiasm assembly corresponded to small portions of the chloroplast and mitochondrial genomes (Sharma et al. 2022; Sun et al. 2022). The other unanchored contigs (59.4 Mb) consisted mostly of TEs (88.6%) and showed homology to localized regions in the chromosomes (Supplementary File 1 and Fig. 1).
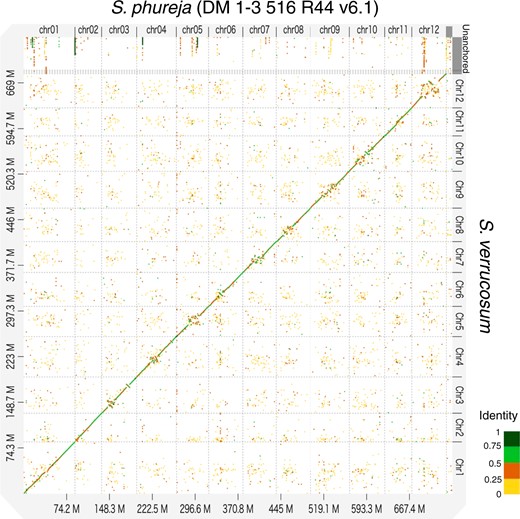
Dot plot analysis between S. verrucosum and S. phureja using D-GENIES with the “hide noise” option.
Assembly statistics.
| Primary contigs with PacBio reads | Final scaffolded contigs after Hi-C sequencing | |
|---|---|---|
| Number of contigs | 1,437 | 1,547 |
| Total size, bp | 779,910,189 | 780,238,689 |
| Longest size, bp | 55,137,318 | 84,109,000 |
| Mean size, bp | 542,735 | 504,356 |
| N50 size, bp | 20,992,750 | 55,157,000 |
| Primary contigs with PacBio reads | Final scaffolded contigs after Hi-C sequencing | |
|---|---|---|
| Number of contigs | 1,437 | 1,547 |
| Total size, bp | 779,910,189 | 780,238,689 |
| Longest size, bp | 55,137,318 | 84,109,000 |
| Mean size, bp | 542,735 | 504,356 |
| N50 size, bp | 20,992,750 | 55,157,000 |
Assembly statistics.
| Primary contigs with PacBio reads | Final scaffolded contigs after Hi-C sequencing | |
|---|---|---|
| Number of contigs | 1,437 | 1,547 |
| Total size, bp | 779,910,189 | 780,238,689 |
| Longest size, bp | 55,137,318 | 84,109,000 |
| Mean size, bp | 542,735 | 504,356 |
| N50 size, bp | 20,992,750 | 55,157,000 |
| Primary contigs with PacBio reads | Final scaffolded contigs after Hi-C sequencing | |
|---|---|---|
| Number of contigs | 1,437 | 1,547 |
| Total size, bp | 779,910,189 | 780,238,689 |
| Longest size, bp | 55,137,318 | 84,109,000 |
| Mean size, bp | 542,735 | 504,356 |
| N50 size, bp | 20,992,750 | 55,157,000 |
Putative centromeres
The dot plot analysis performed between the genome of S. verrucosum and that of S. phureja DM v6.1 using D-GENIES showed significant consistency in the distal regions of each chromosome, while the central regions diverged considerably (Fig. 1). This is because centromere sequences evolve rapidly (Henikoff et al. 2001) and might be distinct between S. verrucosum and S. phureja (Gong et al. 2012; Zhang et al. 2014). To precisely determine the centromeres and compare these structures between the 2 species, we used publicly available ChIP-seq data generated against the CENH3 protein. The sequence reads from S. verrucosum (SRR18548893) and S. phureja (SRR18548894) were aligned to their genomes. Strong signals were observed on each chromosome, and the mapping rates were high (>97%) and comparable between the genomes of the 2 species, indicating that the centromere sequences were properly assembled in both genomes (Supplementary Table 1, Supplementary Fig. 3 and Fig. 2a). It was noted that 17.2% of the mapped reads were aligned to unanchored contigs, suggesting that some centromere sequences could not be assembled into chromosomes, possibly due to their highly repetitive nature.
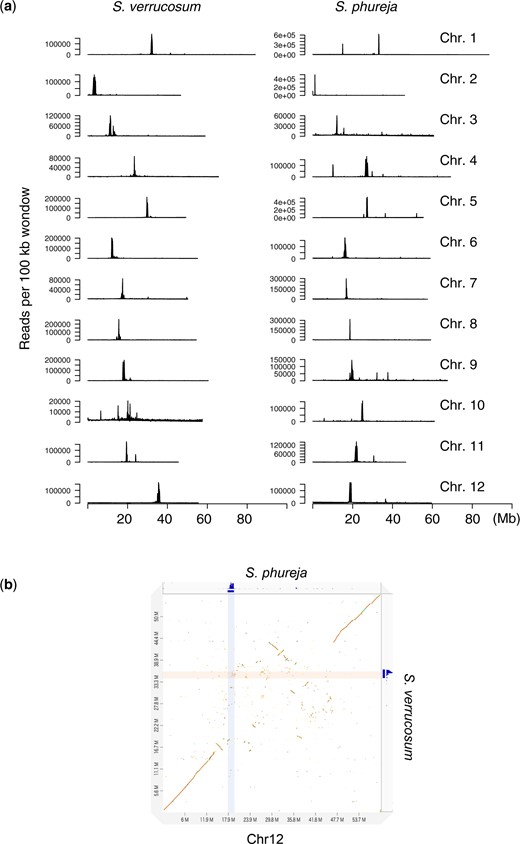
Putative centromeres. a) Distribution of CENH3 ChIP-seq signals in every 100 kb window in S. verrucosum and S. phureja. b) Dot plot between S. verrucosum and S. phureja for chromosome 12. ChIP-seq signals against the CENH3 proteins of S. verrucosum and S. phureja are shown on the right and at the top of the plot, respectively, and are highlighted on the plot.
In accordance with a previous report (Zhang et al. 2014), centromere regions showed little conservation between the sequences of S. verrucosum and S. phureja (Supplementary Fig. 4). In particular, the centromere position on chromosome 12 differed between S. verrucosum and S. phureja indicating that a massive rearrangement occurred during their speciation (Fig. 2b).
Gene prediction
Since TEs are one of the major forces driving genome evolution (Hosaka and Kakutani 2018), the quantity and diversity of TEs were analyzed using the EDTA transposon annotation pipeline. TEs accounted for approximately 61.8% (482.1 Mb) of the S. verrucosum genome, among which LTR-type retrotransposons accounted for 39.9%, and terminal inverted repeat (TIR)-type transposons accounted for 9.7% (Supplementary Table 2). MITEs, which are nonautonomous derivatives of TIR-type transposons, were identified in 20.5% of the TIR-type transposons. The most abundant TEs were Gypsy elements (24.2%), as reported previously in other Solanum species (Aversano et al. 2015; Gaiero et al. 2019; Hosmani et al. 2019). Putative protein-coding genes were searched in the genome using the MAKER pipeline, and their functions were predicted using the Hayai-Annotation Plants v2 pipeline. As a result, 64,294 genes were predicted, and 46,904 genes were functionally annotated. Their chromosomal distributions and the correlations of their locations are shown in Fig. 3. The genes were densely distributed in telomeric and subtelomeric regions. Some class II transposons, such as Tc1_Mariner, hAT, helitron, and MITEs, were distributed in a pattern similar to that of genes. In contrast, Gypsy and unknown LTR retrotransposons were densely distributed in pericentromeric and centromeric regions. Similar distribution patterns have been reported in S. phureja DM v4.03 (Zavallo et al. 2020).
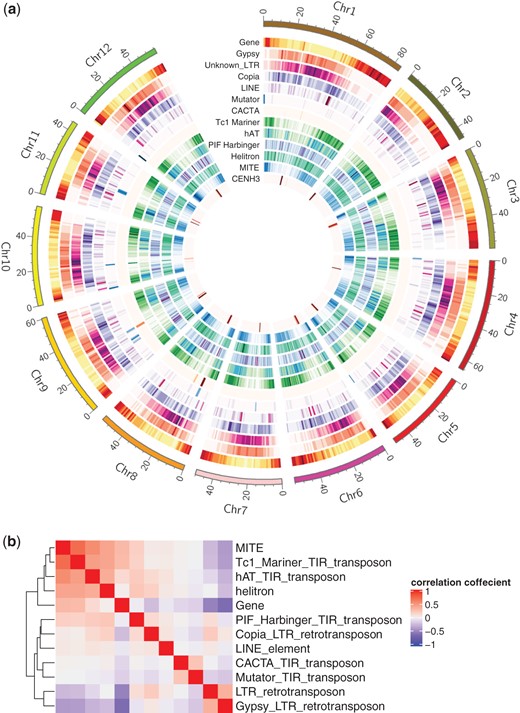
Chromosomal distribution of genes and transposons (a) and the correlations of their locations (b).
Assembly completeness and quality assessment
The LAI score used to measure assembly completeness was 11.97, which was slightly lower than that of DM v6.1 but much higher than that of DM v4.04 (LAI scores of 13.56 and 7.87, respectively; Pham et al. 2020). Higher LAI scores correspond to more complete genome assemblies, and genome LAI scores between 10 and 20 are considered to indicate reference genome quality (Ou et al. 2018). Thus, the S. verrucosum genome was highly contiguous and is categorized as showing reference genome quality.
The quality of the gene predictions was assessed using the Benchmarking Universal Single-Copy Orthologs (BUSCO) database. Among 5,950 BUSCOs that are conserved in Solanales species, the number of complete BUSCOs identified was 5,759 (96.8%) in genome mode and 5,490 (92.3%) in protein mode. The number of missing BUSCOs identified in protein mode was 272 (4.6%), which was almost equivalent to the numbers found in the genomes of the other 5 species, ranging from 2.6% in S. phureja to 15.4% in S. tuberosum (Supplementary Table 3). Thus, most of the known BUSCOs were identified in the S. verrucosum genome, demonstrating robust representation of protein-coding genes.
Synteny and phylogenetic analyses
Structural variation and gene similarity were compared between the S. verrucosum genome and the other 5 genomes. The chromosome lengths varied by species (Supplementary Table 4 and Fig. 4a). Each of the S. chacoense chromosomes except for the chromosome 7 was shorter than the corresponding chromosomes of the 5 other species, likely because S. chacoense showed the largest number of unanchored sequences (Leisner et al. 2018). The highest size similarity was observed between S. verrucosum and S. phureja, with a Pearson’s correlation coefficient of 0.978.
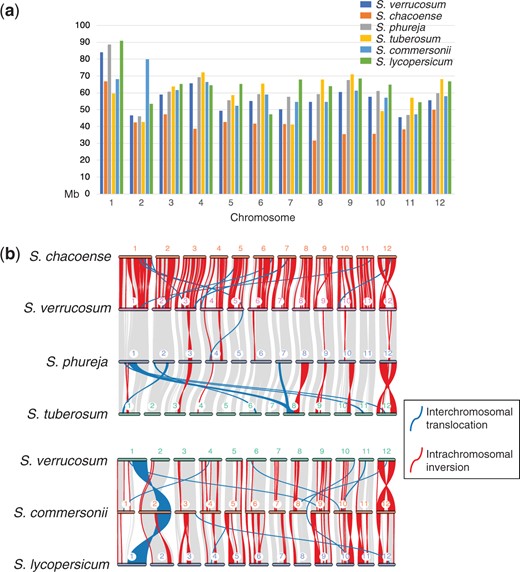
Size and structural differences among 6 species genomes. a) Chromosome size in Mb. b) Synteny plot between S. verrucosum and the other species.
The gene collinearity analyses between S. verrucosum and the other 5 species showed putative inversions and interchromosomal translocations, indicating that genome rearrangements have occurred (Fig. 4b). The S. verrucosum genome was most syntenic to the S. phureja genome. Furthermore, all genomes except for that of S. commersonii showed similar gene synteny. The S. commersonii genome showed a large translocated segment on chromosome 2, which might indicate that a unique genome rearrangement occurred in this species, or this could be a result of simple misassembly.
The analysis of orthologous relationships using OrthoFinder showed that 288,020 genes (90.1%) among the 319,562 genes identified in the 6 species were assigned to 38,937 orthogroups (Supplementary Table 5). Among the 38,937 orthogroups, 16,964 (43.6%) were present in all the species analyzed, while only 1,027 (2.6%) were present in S. verrucosum (Fig. 5a). These S. verrucosum-specific orthogroups included 7,103 genes, of which 77.4% lacked functional annotations and 8.9% had similarities to the genes encoded in TEs based on the Hayai-Annotation Plants v2 pipeline. S. verrucosum presented the second largest number of shared orthogoups (28,568) after S. commersonii.
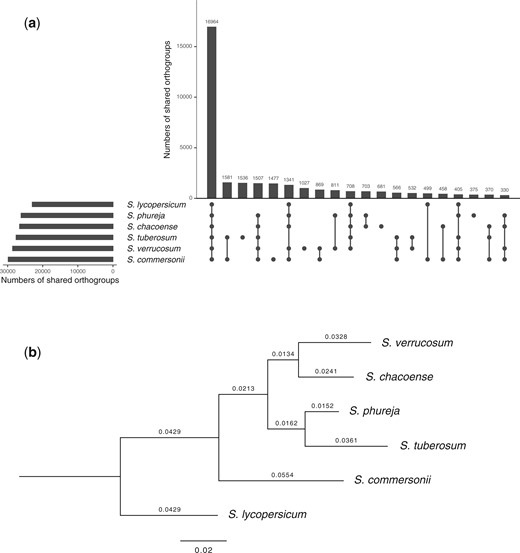
Orthologous relationships among 6 species genomes. a) UpSet plot of the shared orthogroups among 38,937 orthogroups assigned from a total of 288,020 genes identified in the 6 species genomes. The top 20 orthogroup intersections are shown. b) OrthoFinder-generated phylogenetic tree constructed using 16,964 orthogroups present in all 6 species. S. lycopersicum was used as the outgroup species.
The species phylogeny was inferred from the similarity of the 16,964 orthogroups present in all the 6 species using OrthoFinder with the Species Tree inference from All Genes (STAG) algorithm (Emms and Kelly 2018). S. commersonii was distantly related among tuber-bearing species (Fig. 5b). In tuber-bearing Solanum species, the interspecific crossing barrier is explained by the Endosperm Balance Number (EBN) hypothesis (Johnston et al. 1980; Ehlenfeldt and Ortiz 1995). According to this hypothesis, a 2:1 ratio of maternal to paternal EBN in the endosperm is necessary for normal endosperm development (Johnston et al. 1980). S. commersonii shows an EBN of 1, whereas most of the other A-genome diploid species show an EBN of 2 (Ehlenfeldt and Hanneman 1988; Hanneman 1994). An imbalanced EBN causes endosperm abortion following interspecific hybridization, which is one of the major reproductive barriers among potato species (Johnston et al. 1980; Hawkes and Jackson 1992; Hanneman 1999). S. verrucosum was observed to be most closely related to S. chacoense, indicating that the 2 species have relatively similar gene sequences while frequent rearrangements by intrachromosomal inversions were observed (Fig. 4b). Interestingly, geographical distributions of the 2 species are most distant among the A-genome species (Hawkes 1990).
Conclusions
We constructed a high-quality de novo assembly of the geographically isolated A-genome species S. verrucosum with a scaffold N50 of 55.2 Mb. The evaluation of variability within the A genome, including that of S. verrucosum, encompassed the geographic range of these species from the north (S. verrucosum in Mexico) to the south (S. chacoense in Argentina), which will be useful for understanding genomic differentiation among A-genome species. Since S. verrucosum has been considered a maternal progenitor of Mexican polyploid species (Hosaka et al. 1984; Spooner et al. 1991; Rodríguez and Spooner 2009; Sanetomo and Hosaka 2013), this whole-genome sequence will be a valuable resource for understanding polyploid evolution. Furthermore, the whole-genome sequence of S. verrucosum will facilitate the exploration of its unique crossing behaviors, such as self-compatibility and unilateral cross-compatibility (Hawkes 1990; Eijlander et al. 2000), which will help us understand its function as a bridging species (Hermsen and Ramanna 1976; Hamernik et al. 2001; Dinu et al. 2005; Jansky and Hamernik 2009; Bamberg et al. 2021) and will promote the introgression of useful traits from reproductively isolated Mexican diploid species into cultivated potatoes.
Data availability
The raw DNA sequencing reads, genome assembly, and annotation have been deposited into the National Center for Biotechnology Information under BioProject Number PRJNA820895.
Supplemental material is available at G3 online.
The Hawkes (1990) classification system is tentatively adopted throughout the text.
Acknowledgments
KH and RS designed the research. KH prepared the DNA sample for sequencing. AH performed genome sequencing, assembly, and annotation and comparative genome analysis. AH and KH wrote the manuscript with input from all authors. Supercomputing resources were provided by the Human Genome Center at the University of Tokyo. We thank Dr Jiming Jiang, Michigan State University, for releasing the raw data from CENH3 ChIP-seq. All the authors have read and approved the final manuscript.
Funding
This research was supported in part by Calbee, Inc., Hokkaido Potato Growers Association, Kewpie Corp., KENKO Mayonnaise Co., Ltd., and the Japan Snack Cereal Foods Association.
Conflicts of interest
None declared.
Literature cited
Huang B, Ruess H, Liang Q, Colleoni C, Spooner DM. Analyses of 202 plastid genomes elucidate the phylogeny of Solanum section Petota. Sci Pep. 2019;9:4454.
Pushkarnath.
Author notes
Present address for Awie J. Hosaka: Nihon BioData Corporation, Takatsu, Kawasaki, Kanagawa 213-0012, Japan.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}