





Abstract
Ganoderma leucocontextum, a newly discovered species of Ganodermataceae in China, has diverse pharmacological activities. Ganoderma leucocontextum was widely cultivated in southwest China, but the systematic genetic study has been impeded by the lack of a reference genome. Herein, we present the first whole-genome assembly of G. leucocontextum based on the Illumina and Nanopore platform from high-quality DNA extracted from a monokaryon strain (DH-8). The generated genome was 50.05 Mb in size with an N50 scaffold size of 3.06 Mb, 78,206 coding sequences, and 13,390 putative genes. Genome completeness was assessed using the Benchmarking Universal Single-Copy Orthologs (BUSCO) tool, which identified 96.55% of the 280 Fungi BUSCO genes. Furthermore, differences in functional genes of secondary metabolites (terpenoids) were analyzed between G. leucocontextum and Ganoderma lucidum. Ganoderma leucocontextum has more genes related to terpenoids synthesis compared to G. lucidum, which may be one of the reasons why they exhibit different biological activities. This is the first genome assembly and annotation for G. leucocontextum, which would enrich the toolbox for biological and genetic studies in G. leucocontextum.
Introduction
Ganoderma leucocontextum (Figure 1) is a newly discovered prize medicinal species of Ganoderma, which was first found in Tibet, China, in 2015. The macroscopic morphological characteristics of the fruiting body of G. leucocontextum are highly similar to Ganoderma lingzhi (Li et al. 2015), which is widely cultivated and used in China (Cao et al. 2012), but there are major differences between these two species in terms of biological characteristics and pharmacological activity. For example, the hyphae of G. leucocontextum displayed an acid-tolerant characteristic; both the mycelium and the fruiting body grew at a lower temperature than that of G. lingzhi (HU et al. 2017; Mo Weipng et al. 2017).

Fruiting bodies of G. leucocontextum. Scale bar: 1.0 cm.
Moreover, some novel bioactive compounds like triterpenes and ganoderols were isolated from G. leucocontextum in recent studies (Wang et al. 2015, 2017; Zhao et al. 2016a, 2016b; Chen et al. 2018a), which have been proved to have pharmacological activities such as anti-tumor (Li et al. 2019; Liu et al. 2018), antioxidant (PAN Jun et al. 2021), antihyperglycemic (Wang et al. 2015, 2017; Chen et al. 2019), hypolipidemic (Wang et al. 2017; Zhang et al. 2018b), neurodegenerative diseases prevention (Chen et al. 2018a; Xiong et al. 2016), anti-aging (Wang et al. 2019), and immune regulation (Gao et al. 2020). Therefore, it is classified as a high-quality category with higher price than the ordinary Ganoderma in Tibet, China (LIU et al. 2020; Shen et al. 2015). These have promoted the research of artificial cultivation of G. leucocontextum and realized the large-scale commercial cultivation since 2016. What is exciting is that it has been confirmed that the content of polysaccharide and triterpenoid of G. leucocontextum is significantly higher than that of G. lingzhi (HU et al. 2017).
Many microorganisms produce natural products like antimicrobials and drugs, while the mining of the genome can quickly screen and obtain new natural products (Gao et al. 2018; Blin et al. 2021). Genome research includes structural genomics, aiming to whole-genome sequencing, and functional genomics, aiming to explore gene function. The fungal genome initiative is the first well-known fungal genome project. It was initiated and launched by the mycology research group in conjunction with the Broad Institute of the United States in 2000, and then issued the white papers of the genome sequencing projects, which were specific to 15 and 44 species of fungi in 2002 and 2003, respectively (Li 2018). Up to May 2021, more than 2000 fungal genome projects, including 595 in basidiomycetes and 1240 in ascomycetes, have been completed and released on the JGI website (https://mycocosm.jgi.doe.gov/mycocosm/home). In terms of the well-known edible and medicinal fungi, such as Ganoderma lucidum (2012), Flammulina velutipes (2015), Lentinula edodes (2016), Cordyceps guangdongensis (2018), Auricularia heimuer (2019), Hericium erinaceus (2020), Morchella sextelata (2019), and Agrocybe cylindracea (2020), the Whole-Genome has been published and some functional genes were predicted and analyzed (Chen et al. 2012; Zeng et al. 2015; Shim et al. 2016; Zhang et al. 2018a; Mei et al. 2019; Yuan et al. 2019; Gong et al. 2020; Liang et al. 2020). Despite progresses have been made toward understanding the cultivation and efficacy, the genome-wide association studies of G. leucocontextum has not been systematically performed. The G. leucocontextum strain in this study was from Nyingchi, Tibet, which was rich in triterpenes compared to G. lucidum (HU et al. 2017). In order to clarify genetic and physiological background of G. leucocontextum, whole-genome sequencing was carried out by Oxford Nanopore technologies; furthermore, functional annotation and gene clusters of secondary metabolites were predicted based on public databases.
Methods and materials
Fungal strains and nucleic acid extraction
In this study, the dikaryon strain (HMGIM-I160015) was isolated from the fruiting body of G. leucocontextum that was collected in Nyingchi by HU Huiping. The monokaryon strain DH-8 was isolated from HMGIM-I160015 using the protoplast-derived method (Li et al. 2021) and preserved in Institute of Microbiology, Guangdong Academy of Sciences. Vegetative mycelium of DH-8 was cultured on Potato Dextrose Agar (PDA) medium (20% potato, 2% glucose, 2% agar, 0.3% KH2PO4, 0.15% MgSO4, trace of vitamin B1) with cellophane at 25°C in darkness for 7 days. Then, the mycelia were frozen in liquid nitrogen and ground to powder for genomic DNA extraction. Genomic DNA was extracted by QIAGEN® Genomic DNA extraction kit (Cat#13323, QIAGEN) according to the manufacturer’s instructions. The extracted DNA was detected by NanoDrop™ One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA) for DNA purity (OD260/280 ranging from 1.8 to 2.0 and OD260/230 is between 2.0 and 2.2) and then Qubit® 3.0 Fluorometer (Invitrogen, USA) was used to quantify DNA accuracy. Total RNA was extracted from the fruiting body tissue by using Plant RNA Purification Reagent, the concentration and purity of the extracted RNA were detected using Nanodrop2000, and the RNA integrity number (RIN) value was determined by Agilent2100.
De novo sequencing and assembly
De novo genome sequencing of DH-8 was performed with a 20-k and 350-bp library size using the Nanopore and Illumina sequel platform at Biomarker Technologies Corporation (Beijing, China), respectively (Wick et al. 2019). The filtered subreads were assembled using NECAT software (https://github.com/xiaochuanle/NECAT). And then the assembled genome was corrected in contrast to the data of the Illumina using Pilon v1.22 (Walker et al. 2014) software, resulting in a more accurate final genome. Burrows-Wheeler Aligner (bwa) (Li and Durbin 2009) and Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.1 (Simão et al. 2015) were used to assess the completeness of genome assembly. RNA sequencing was performed using Illumina HiSeq xten/NovaSeq 6000 sequencer (2 × 150 bp read length), and standard bioinformatics analyses at Shanghai Major Biomedical Technology Co., Ltd. (Shanghai, China). The genome data of DH-8 have been submitted to NCBI, accession number was AHKGY000000000.
Genomic component analysis
Repeat sequence prediction
LTR_FINDER v1.05 (Xu and Wang 2007), MITE-Hunter (Han and Wessler 2010), RepeatScout v1.0.5 (Price et al. 2005), and PILER-DF v2.4 (Edgar and Myers 2005) were applied to construct the repetitive sequence database of the genome of G. leucocontextum based on the structure and ab initio prediction, then the predicted database was categorized by PASTEClassifier (Wicker et al. 2007), and merged as the final repeated sequence database with Repbase (Jurka et al. 2005). The repetitive sequences of G. leucocontextum were predicted by RepeatMasker v4.0.6 (Chen 2004) based on the constructed repeated sequence database.
Protein-coding genes prediction
Gene prediction was conducted through a combination of ab initio prediction, homology-based prediction, and transcriptome-based prediction methods. In detail, ab initio gene predictions were performed using Genscan (Burge and Karlin 1997), Augustus v2.4 (Stanke and Waack 2003), GlimmerHMM v3.0.4 (Majoros et al. 2004), GeneID v1.4 (Blanco et al. 2007), and SNAP (version 2006-07-28) (Korf 2004). Coding gene structures were predicted by GeMoMa v1.3.1 (Keilwagen et al. 2016) based on an alignment of orthologous proteins. Data analysis of RNA-seq Raw reads from Illumina sequencing was trimmed and assessed for quality with Fastp (Chen et al. 2018b), the qualified data were aligned to the genome by Hisat2 (Siren et al. 2014), the aligned reads were assembled into transcripts using Stringtie (Kovaka et al. 2019), and then open reading frames were predicted using PASA (Program to Assemble Spliced Alignments) (Haas et al. 2003). Finally, EVidenceModeler (EVM) (Haas et al. 2008) was used to produce an integrated gene set, the transposable elements (TEs) were removed using TransposonPSI (Urasaki et al. 2017) package (http://transposonpsi.sourceforge.net/), and the miscoded genes were further filtered. All the above software were used with the default parameters.
NcRNAs and pseudogene annotation
To obtain the non-coding RNA (ncRNA) of the genome, tRNAscan-SE (Lowe and Eddy 1997) was used to predict transfer RNAs (tRNAs) with eukaryote parameters. Infernal 1.1 (Nawrocki and Eddy 2013) and RNAmmer (Lagesen et al. 2007) were used to predict ribosomal RNAs (rRNAs) and other RNAs based on Rfam (Griffiths-Jones et al. 2005) databases. Pseudogenes were defined as any gene that had a loss of function mutation anywhere within the coding sequence, though they have similar sequences to functional genes and have lost their original functions due to mutations such as insertions and deletions; however, there are still potential functions of affecting protein (Winglee et al. 2016). The predicted protein sequences were aligned to the protein sequence included in the Swiss-Prot database by using the GenBlastA (She et al. 2009) to find homologous gene sequences (possible genes) in the genome. Then, the immature termination codon and frameshift mutation in the gene sequences were searched to obtain pseudogenes by using the software GeneWise (Birney et al. 2004).
Genome functional annotation
The predicted genes were aligned against the functional databases such as Eukaryotic Orthologous Groups (KOG) (Galperin et al. 2015), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto 2000), Swiss-Prot (Stanke and Waack) (Stanke et al. 2006), TrEMBL (Boeckmann et al. 2003), Non-Redundant Protein Sequence Database (NR) (Deng et al. 2006), transporter classification database (TCDB) (Saier et al. 2006), and the pathogen–host interaction factor database (PHI) (Winnenburg et al. 2006) by BLAST (Altschul et al. 1997) to obtain the results of gene functional annotation. Based on the blast results of NR database, the software Blast2Go (Conesa et al. 2005) was applied to annotate the function of Gene Ontology (GO) (Ashburner et al. 2000) database. In addition, gene function annotation analysis was performed on Clusters of Orthologous Groups (COG), KEGG metabolic pathway enrichment analysis, and GO function enrichment analysis. Hmmer (Eddy 1998) was used for functional annotation of carbohydrate-related enzymes based on the database of carbohydrate-active enzymes (CAZymes) (Cantarel et al. 2009). Additionally, the online software antiSMASH (https://antismash.secondarymetabolites.org/#!/start) (Medema et al. 2011) was employed to predict gene clusters of secondary metabolites. KEGG Mapper (Reconstruct Pathway) (Minoru Kanehisa and Sato 2019) was also used to comparative analysis of pathway in metabolism of terpenoids and polyketides between G. leucocontextum and G. lucidum.
Results and discussion
Genome assembly and evaluation
A total of 47.5 μg DNA were obtained, in which A260/280 was 1.88 and OD260/230 was 2.26 according to Nanodrop detection, indicating that the extracted DNA was pollution-free and there was no protein or other contamination. A total of 8.58 Gb and 15.45 Gb raw data were obtained using the Illumina and Nanopore’s sequel platform, respectively. A total of 14.42 Gb clean data from Nanopore were obtained by filtering ploy-N, adapters, and low-quality reads from raw data. The quality statistics on the raw data and clean data was shown in Supplementary Table S1. The assembled genome was 50.05 Mb with a GC content of 55.85%; the sequencing depth was 288.16× and consisted of 58 scaffolds with an N50 of 3.06 Mb (Supplementary Table S2). Clean data from Illumina were aligned to the assembled genome by bwa (Li and Durbin 2009) to assess the genome assembly quality, and the coverage of assembled genome was 96.13%. The result of assessment for genome integrity by BUSCO (Simão et al. 2015) analyses was 96.55% (Table 1), indicating that vast majority of the conserved core genes of fungi were predicted, which revealed the high reliability of the prediction. Compared with similar species of the gene of Ganoderma, the genome size of G. leucocontextum was larger than G. lucidum (43.3 Mb), G. tsugae (45.5 Mb), and G. sinense (48.96 Mb) (Table 2). The number of scaffolds and Contig N50 revealed that we got the better genome assembly of G. leucocontextum in this study.
The quality assessment for genome assembly of G. leucocontextum
| Quality assessment | Values |
|---|---|
| Librarya | 350 bp |
| Mapped (%)b | 99.39 |
| Properly mapped (%)c | 97.66 |
| Coverage (%)d | 96.13 |
| Depth (X)e | 74.36 |
| Complete BUSCOs (C)f | 280 (96.55%) |
| Complete and single-copy BUSCOs (S)g | 276 (95.17%) |
| Complete and duplicated BUSCOs (D)h | 4 (1.38%) |
| Fragmented BUSCOs (F)i | 1 (0.34%) |
| Missing BUSCOs (M)j | 9 (3.10%) |
| Total lineage BUSCOsk | 290 |
| Quality assessment | Values |
|---|---|
| Librarya | 350 bp |
| Mapped (%)b | 99.39 |
| Properly mapped (%)c | 97.66 |
| Coverage (%)d | 96.13 |
| Depth (X)e | 74.36 |
| Complete BUSCOs (C)f | 280 (96.55%) |
| Complete and single-copy BUSCOs (S)g | 276 (95.17%) |
| Complete and duplicated BUSCOs (D)h | 4 (1.38%) |
| Fragmented BUSCOs (F)i | 1 (0.34%) |
| Missing BUSCOs (M)j | 9 (3.10%) |
| Total lineage BUSCOsk | 290 |
Represents Illumina sequencing library size.
Represents the percentage of clean reads mapped to the genome assembly of G. leucocontextum to all clean reads.
Represents the paired-end sequencing; sequences were all located on the genome assembly of G. leucocontextum and the distance was consistent with the length distribution of the sequenced fragments.
Represents genome coverage of data from Illumina sequence.
Represents genome coverage depth of data from Illumina sequence.
Represents the number and percentage of complete genes found in the database (contains 290 conserved core genes of fungi).
Represents the number and percentage of complete single-copy genes.
Represents the number and percentage of complete duplicated genes.
Represents the number of predictions for incomplete genes.
Represents the unpredicted number of genes.
Represents the number of conserved gene sets in fungi from the database of fungi_odb9.
The quality assessment for genome assembly of G. leucocontextum
| Quality assessment | Values |
|---|---|
| Librarya | 350 bp |
| Mapped (%)b | 99.39 |
| Properly mapped (%)c | 97.66 |
| Coverage (%)d | 96.13 |
| Depth (X)e | 74.36 |
| Complete BUSCOs (C)f | 280 (96.55%) |
| Complete and single-copy BUSCOs (S)g | 276 (95.17%) |
| Complete and duplicated BUSCOs (D)h | 4 (1.38%) |
| Fragmented BUSCOs (F)i | 1 (0.34%) |
| Missing BUSCOs (M)j | 9 (3.10%) |
| Total lineage BUSCOsk | 290 |
| Quality assessment | Values |
|---|---|
| Librarya | 350 bp |
| Mapped (%)b | 99.39 |
| Properly mapped (%)c | 97.66 |
| Coverage (%)d | 96.13 |
| Depth (X)e | 74.36 |
| Complete BUSCOs (C)f | 280 (96.55%) |
| Complete and single-copy BUSCOs (S)g | 276 (95.17%) |
| Complete and duplicated BUSCOs (D)h | 4 (1.38%) |
| Fragmented BUSCOs (F)i | 1 (0.34%) |
| Missing BUSCOs (M)j | 9 (3.10%) |
| Total lineage BUSCOsk | 290 |
Represents Illumina sequencing library size.
Represents the percentage of clean reads mapped to the genome assembly of G. leucocontextum to all clean reads.
Represents the paired-end sequencing; sequences were all located on the genome assembly of G. leucocontextum and the distance was consistent with the length distribution of the sequenced fragments.
Represents genome coverage of data from Illumina sequence.
Represents genome coverage depth of data from Illumina sequence.
Represents the number and percentage of complete genes found in the database (contains 290 conserved core genes of fungi).
Represents the number and percentage of complete single-copy genes.
Represents the number and percentage of complete duplicated genes.
Represents the number of predictions for incomplete genes.
Represents the unpredicted number of genes.
Represents the number of conserved gene sets in fungi from the database of fungi_odb9.
Genomic comparison of important species of Ganoderma
| Ganoderma sp. | Strain | GenBank assembly accession | Genome size (Mb) | Number of scaffolds | GC% | Genome coverage | Assembly level | Contig N50/bp | Sequencing technology |
|---|---|---|---|---|---|---|---|---|---|
| G. leucocontextum | HMGIM-I160015 | AHKGY000000000 | 50.05 | 58 | 55.85 | 288.16x | Scaffold | 3,064,430 | Illumina, Nanopore |
| G. lucidum | BCRC 36111 | GCA_012655175.1 | 48.91 | 173 | 55.1 | 97.73x | Contig | 1,281,108 | PacBio |
| G. lucidum | BCRC 37177 | GCA_000338035.1 | 44.08 | 3,275 | 55.5 | 824x | Contig | 63,041 | Illumina |
| G. lucidum | G.260125-1 | GCA_000271565.1 | 43.29 | 82 | 56.1 | 440x | Scaffold | 649,708 | Illumina |
| G. lucidum | Xiangnong No.1 | GCA_000262775.1 | 39.95 | 634 | 55.3 | 70x | Scaffold | 80,796 | Illumina |
| G. tsugae | s90 | GCA_003057275.1 | 45.50 | 6,638 | — | 101.0x | Scaffold | 11,659 | Illumina HiSeq |
| G. sinense | ZZ0214-1 | GCA_002760635.1 | 48.96 | 69 | 55.6 | 500.0x | Scaffold | 753,893 | 454; Illumina HiSeq |
| G. multipileum | BCRC 37180 | GCA_000338015.1 | 46.38 | 6,173 | 55.3 | 824x | Contig | 50,471 | Roche/454; Illumina/ABI |
| G. sp. | BRIUMSc | GCA_008694245.1 | 52.28 | 12,158 | 55.6 | 99.35x | Contig | 6,197 | Illumina |
| G. boninense | NJ3 | GCA_001855635.1 | 60.33 | 18,903 | 55.9 | 20.0x | Contig | 6,116 | Illumina HiSeq; 454 |
| G. boninense | G3 | GCA_002900995.2 | 79.19 | 495 | 55.9 | 50.0x | Contig | 272,644 | PacBio |
| Ganoderma sp. | Strain | GenBank assembly accession | Genome size (Mb) | Number of scaffolds | GC% | Genome coverage | Assembly level | Contig N50/bp | Sequencing technology |
|---|---|---|---|---|---|---|---|---|---|
| G. leucocontextum | HMGIM-I160015 | AHKGY000000000 | 50.05 | 58 | 55.85 | 288.16x | Scaffold | 3,064,430 | Illumina, Nanopore |
| G. lucidum | BCRC 36111 | GCA_012655175.1 | 48.91 | 173 | 55.1 | 97.73x | Contig | 1,281,108 | PacBio |
| G. lucidum | BCRC 37177 | GCA_000338035.1 | 44.08 | 3,275 | 55.5 | 824x | Contig | 63,041 | Illumina |
| G. lucidum | G.260125-1 | GCA_000271565.1 | 43.29 | 82 | 56.1 | 440x | Scaffold | 649,708 | Illumina |
| G. lucidum | Xiangnong No.1 | GCA_000262775.1 | 39.95 | 634 | 55.3 | 70x | Scaffold | 80,796 | Illumina |
| G. tsugae | s90 | GCA_003057275.1 | 45.50 | 6,638 | — | 101.0x | Scaffold | 11,659 | Illumina HiSeq |
| G. sinense | ZZ0214-1 | GCA_002760635.1 | 48.96 | 69 | 55.6 | 500.0x | Scaffold | 753,893 | 454; Illumina HiSeq |
| G. multipileum | BCRC 37180 | GCA_000338015.1 | 46.38 | 6,173 | 55.3 | 824x | Contig | 50,471 | Roche/454; Illumina/ABI |
| G. sp. | BRIUMSc | GCA_008694245.1 | 52.28 | 12,158 | 55.6 | 99.35x | Contig | 6,197 | Illumina |
| G. boninense | NJ3 | GCA_001855635.1 | 60.33 | 18,903 | 55.9 | 20.0x | Contig | 6,116 | Illumina HiSeq; 454 |
| G. boninense | G3 | GCA_002900995.2 | 79.19 | 495 | 55.9 | 50.0x | Contig | 272,644 | PacBio |
Data of G. leucocontextum were from this study; other data were from NCBI.
Genomic comparison of important species of Ganoderma
| Ganoderma sp. | Strain | GenBank assembly accession | Genome size (Mb) | Number of scaffolds | GC% | Genome coverage | Assembly level | Contig N50/bp | Sequencing technology |
|---|---|---|---|---|---|---|---|---|---|
| G. leucocontextum | HMGIM-I160015 | AHKGY000000000 | 50.05 | 58 | 55.85 | 288.16x | Scaffold | 3,064,430 | Illumina, Nanopore |
| G. lucidum | BCRC 36111 | GCA_012655175.1 | 48.91 | 173 | 55.1 | 97.73x | Contig | 1,281,108 | PacBio |
| G. lucidum | BCRC 37177 | GCA_000338035.1 | 44.08 | 3,275 | 55.5 | 824x | Contig | 63,041 | Illumina |
| G. lucidum | G.260125-1 | GCA_000271565.1 | 43.29 | 82 | 56.1 | 440x | Scaffold | 649,708 | Illumina |
| G. lucidum | Xiangnong No.1 | GCA_000262775.1 | 39.95 | 634 | 55.3 | 70x | Scaffold | 80,796 | Illumina |
| G. tsugae | s90 | GCA_003057275.1 | 45.50 | 6,638 | — | 101.0x | Scaffold | 11,659 | Illumina HiSeq |
| G. sinense | ZZ0214-1 | GCA_002760635.1 | 48.96 | 69 | 55.6 | 500.0x | Scaffold | 753,893 | 454; Illumina HiSeq |
| G. multipileum | BCRC 37180 | GCA_000338015.1 | 46.38 | 6,173 | 55.3 | 824x | Contig | 50,471 | Roche/454; Illumina/ABI |
| G. sp. | BRIUMSc | GCA_008694245.1 | 52.28 | 12,158 | 55.6 | 99.35x | Contig | 6,197 | Illumina |
| G. boninense | NJ3 | GCA_001855635.1 | 60.33 | 18,903 | 55.9 | 20.0x | Contig | 6,116 | Illumina HiSeq; 454 |
| G. boninense | G3 | GCA_002900995.2 | 79.19 | 495 | 55.9 | 50.0x | Contig | 272,644 | PacBio |
| Ganoderma sp. | Strain | GenBank assembly accession | Genome size (Mb) | Number of scaffolds | GC% | Genome coverage | Assembly level | Contig N50/bp | Sequencing technology |
|---|---|---|---|---|---|---|---|---|---|
| G. leucocontextum | HMGIM-I160015 | AHKGY000000000 | 50.05 | 58 | 55.85 | 288.16x | Scaffold | 3,064,430 | Illumina, Nanopore |
| G. lucidum | BCRC 36111 | GCA_012655175.1 | 48.91 | 173 | 55.1 | 97.73x | Contig | 1,281,108 | PacBio |
| G. lucidum | BCRC 37177 | GCA_000338035.1 | 44.08 | 3,275 | 55.5 | 824x | Contig | 63,041 | Illumina |
| G. lucidum | G.260125-1 | GCA_000271565.1 | 43.29 | 82 | 56.1 | 440x | Scaffold | 649,708 | Illumina |
| G. lucidum | Xiangnong No.1 | GCA_000262775.1 | 39.95 | 634 | 55.3 | 70x | Scaffold | 80,796 | Illumina |
| G. tsugae | s90 | GCA_003057275.1 | 45.50 | 6,638 | — | 101.0x | Scaffold | 11,659 | Illumina HiSeq |
| G. sinense | ZZ0214-1 | GCA_002760635.1 | 48.96 | 69 | 55.6 | 500.0x | Scaffold | 753,893 | 454; Illumina HiSeq |
| G. multipileum | BCRC 37180 | GCA_000338015.1 | 46.38 | 6,173 | 55.3 | 824x | Contig | 50,471 | Roche/454; Illumina/ABI |
| G. sp. | BRIUMSc | GCA_008694245.1 | 52.28 | 12,158 | 55.6 | 99.35x | Contig | 6,197 | Illumina |
| G. boninense | NJ3 | GCA_001855635.1 | 60.33 | 18,903 | 55.9 | 20.0x | Contig | 6,116 | Illumina HiSeq; 454 |
| G. boninense | G3 | GCA_002900995.2 | 79.19 | 495 | 55.9 | 50.0x | Contig | 272,644 | PacBio |
Data of G. leucocontextum were from this study; other data were from NCBI.
Genome structure analysis
Repeat sequence annotation
The total number of repeat sequences was 4231, covering 12.64% of the genome. In detail, TEs of DNA and RNA account for 1.02% and 8.11%, respectively. Whereas the proportion of long terminal repeats was 5.06%, the proportion of unknown repeat sequences was 3.26%.
Coding protein genes prediction
The annotations of 13,390 protein-coding genes were supported by the public databases. The total sequences length for all the protein-coding genes were 29,573,839 bp, average of gene length was 2,208.65 bp, number of exon and intron were 82,221 and 68,831, respectively. Detailed gene information statistics were shown in Table 3. The number of predicted genes by homology and transcriptome prediction was 12,837, accounting for 95.87% of the annotated genes, which reveals high quality of the gene prediction. The annotations with NR, COG, KEGG, etc. were shown in Supplementary Table S3.
Gene information statistics of G. leucocontextum
| Gene statistics | Values |
|---|---|
| Gene number | 13,390 |
| CDs number | 78,206 |
| Exon number | 82,221 |
| Intron number | 68,831 |
| Gene length | 29,573,839 |
| CDs length | 19,331,970 |
| Exon length | 23,157,769 |
| Intron length | 6,416,070 |
| Average gene length | 2,208.65 |
| Average CDs length | 247.19 |
| Average exon length | 281.65 |
| Average intron length | 93.21 |
| Average CDs number | 5.84 |
| Average exon number | 6.14 |
| Average intron number | 5.14 |
| Gene statistics | Values |
|---|---|
| Gene number | 13,390 |
| CDs number | 78,206 |
| Exon number | 82,221 |
| Intron number | 68,831 |
| Gene length | 29,573,839 |
| CDs length | 19,331,970 |
| Exon length | 23,157,769 |
| Intron length | 6,416,070 |
| Average gene length | 2,208.65 |
| Average CDs length | 247.19 |
| Average exon length | 281.65 |
| Average intron length | 93.21 |
| Average CDs number | 5.84 |
| Average exon number | 6.14 |
| Average intron number | 5.14 |
Gene information statistics of G. leucocontextum
| Gene statistics | Values |
|---|---|
| Gene number | 13,390 |
| CDs number | 78,206 |
| Exon number | 82,221 |
| Intron number | 68,831 |
| Gene length | 29,573,839 |
| CDs length | 19,331,970 |
| Exon length | 23,157,769 |
| Intron length | 6,416,070 |
| Average gene length | 2,208.65 |
| Average CDs length | 247.19 |
| Average exon length | 281.65 |
| Average intron length | 93.21 |
| Average CDs number | 5.84 |
| Average exon number | 6.14 |
| Average intron number | 5.14 |
| Gene statistics | Values |
|---|---|
| Gene number | 13,390 |
| CDs number | 78,206 |
| Exon number | 82,221 |
| Intron number | 68,831 |
| Gene length | 29,573,839 |
| CDs length | 19,331,970 |
| Exon length | 23,157,769 |
| Intron length | 6,416,070 |
| Average gene length | 2,208.65 |
| Average CDs length | 247.19 |
| Average exon length | 281.65 |
| Average intron length | 93.21 |
| Average CDs number | 5.84 |
| Average exon number | 6.14 |
| Average intron number | 5.14 |
NcRNAs and pseudogene annotation
NcRNAs have no or limited protein-coding capacity but as potent and multifunctional regulators (Lekka and Hall 2018), including tRNAs, rRNAs, and long ncRNAs (lncRNAs). According to the structural characteristics of ncRNAs, different strategies are used. For ncRNA, 350 tRNAs, 78 rRNAs, and 72 other ncRNAs were predicted. The number of tRNA family based on the different anticodon was 51. Among the tRNAs, 15 were pseudo anticodons, 1 was undetermined anticodon, and the remaining anticodon tRNAs correspond to the 20 common amino acid codons. A total of 180 pseudogenes were predicted, the total size of Pseudogenes was 443,628 bp, and the average length was 2,464.6 bp.
Genome functional annotation
A total of 12,724 non-redundant genes were annotated from the public databases. Among the annotated genes, 12,703 genes were annotated in the NR database, followed by 12,566 (TrEMBL), 7,894 (Pfam), 6,345 (Swiss-Prot), 5,658 (KOG), 3,401 (KEEG), and 3,110 (GO) (Table 4). These homologous protein genes represent 95.03% of the predicted genes of assembled genome.
Functional annotation of G. leucocontextum genes from public databases
| Public database | Number of genes | Percentage |
|---|---|---|
| GO | 3,110 | 24.4 |
| KEGG | 3,401 | 26.7 |
| KOG | 5,658 | 44.5 |
| Pfam | 7,894 | 62.0 |
| Swiss-Prot | 6,345 | 49.9 |
| TrEMBL | 12,566 | 98.8 |
| Nr | 12,703 | 99.8 |
| All annotated | 12,724 | 100.0 |
| Public database | Number of genes | Percentage |
|---|---|---|
| GO | 3,110 | 24.4 |
| KEGG | 3,401 | 26.7 |
| KOG | 5,658 | 44.5 |
| Pfam | 7,894 | 62.0 |
| Swiss-Prot | 6,345 | 49.9 |
| TrEMBL | 12,566 | 98.8 |
| Nr | 12,703 | 99.8 |
| All annotated | 12,724 | 100.0 |
Functional annotation of G. leucocontextum genes from public databases
| Public database | Number of genes | Percentage |
|---|---|---|
| GO | 3,110 | 24.4 |
| KEGG | 3,401 | 26.7 |
| KOG | 5,658 | 44.5 |
| Pfam | 7,894 | 62.0 |
| Swiss-Prot | 6,345 | 49.9 |
| TrEMBL | 12,566 | 98.8 |
| Nr | 12,703 | 99.8 |
| All annotated | 12,724 | 100.0 |
| Public database | Number of genes | Percentage |
|---|---|---|
| GO | 3,110 | 24.4 |
| KEGG | 3,401 | 26.7 |
| KOG | 5,658 | 44.5 |
| Pfam | 7,894 | 62.0 |
| Swiss-Prot | 6,345 | 49.9 |
| TrEMBL | 12,566 | 98.8 |
| Nr | 12,703 | 99.8 |
| All annotated | 12,724 | 100.0 |
Genomics analysis of KOG annotations
KOG is a gene orthology database for eukaryotes. In this study, 5658 genes were assigned to the KOG categories while the majority of genes was classified into the “General function prediction only,” followed by “Posttranslational modification, protein turnover, chaperones,” “Signal transduction mechanisms,” “Secondary metabolites biosynthesis, transport, and catabolism.” There were fewer genes in “Nuclear structure” and “Defense mechanisms,” and much fewer genes in “Cell motility” and “Extracellular structures” (Figure 2). A total of 1842 (32.55% of the total) predicted genes were involved in metabolic processes, and 374 (6.61% of the total) predicted genes were related to “secondary metabolites biosynthesis, transport, and catabolism.” The closely related species G. lucidum has the similar number of functional genes by KOG annotation (Chen et al. 2012). In addition, the number of genes related to “Secondary metabolites biosynthesis, transport, and catabolism” in G. leucocontextum was much more than that of mycorrhizal and straw-rotting fungus, such as Laccaria bicolor (Martin et al. 2008) and Agaricus bisporus (Kerrigan et al. 2013), and also it was much more than that of G. lucidum (Liu et al. 2012). Although the number of functional genes cannot determine the number of active ingredients, the result reveals the possible genetic basis of G. leucocontextum being rich in secondary metabolites. According to the results of previous research (Xia et al. 2014; Kladar et al. 2016), the species which belongs to the genus of Ganoderma was rich in triterpenes and other metabolites, and this seemed to be consistent with the results of the KOG annotation.
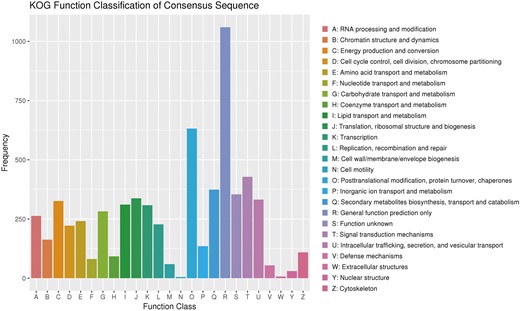
Annotated result classification chart by KOG database.
Genomics analysis of GO annotations
To shed light on the potential roles of the predicted genes, GO enrichment analysis was performed. A total of 3110 GO annotations were matched and distributed in three functional categories: “biological process,” “cellular components,” and “molecular function.” The top four categories of GO were “catalytic activity,” “metabolic process,” “binding,” and “cellular process” (Figure 3), similar to Hericium erinaceus (Gong et al. 2020) and Auricularia heimuer (Yuan et al. 2019) but different to Agaricus bisporus (Kerrigan et al. 2013). On the other hand, the number of genes associated with the categories of “Nutrient reservoir activity,” “Reproduction,” “Reproductive process,” and “developmental process” was fewer, which may reflect from one side why G. leucocontextum can only distributes in a narrow area.
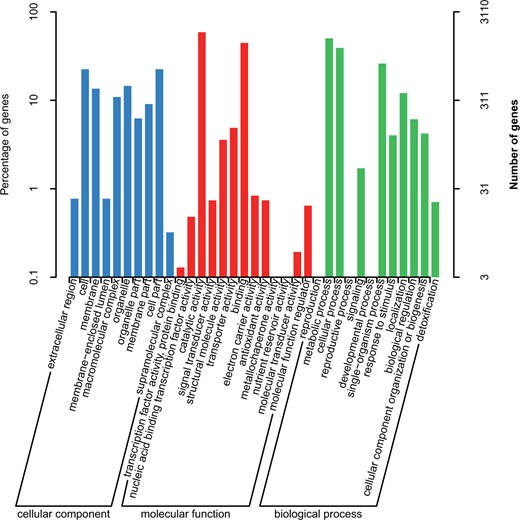
Annotated result classification chart by GO database.
Genomics analysis of KEEG annotations
KEGG is a comprehensive database that collects information on genomes, pathways, and compounds of organisms, which can help to further understand the gene functions in G. leucocontextum. According to the result of KEGG function annotation, 3401 genes were annotated to four physiological processes including “Metabolism,” “Cellular processes,” “Genetic information processing,” and “Environmental information processing” (Figure 4). In the second layer of KEGG pathway terms, we found that G. leucocontextum had much more genes associated to “RNA transport” (116 genes), “Biosynthesis of amino acids” (111 genes), “Ribosome” (109 genes), “Carbon metabolism” (102 genes), and “Protein processing in endoplasmic reticulum” (101 genes). These results may reveal the genetic basis of G. leucocontextum being rich in the secondary metabolites.
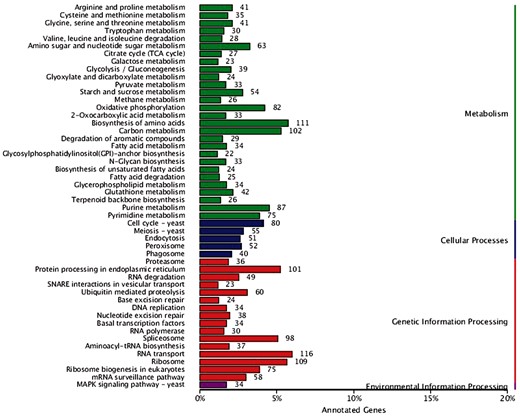
Annotated result classification chart by KEEG database.
Carbohydrate-active enzymes annotation
The CAZymes database includes glycoside hydrolases (GHs), glycosyltransferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), and auxiliary activities (AAs). Genes were annotated against the CAZy database to further understand the carbohydrate degradation capacity of G. leucocontextum. A total of 614 genes were assigned to CAZymes families as defined in CAZy database (Table 5 and Figure 5). Like other species’ CAZymes, the GHs were the most abundant enzymes of G. leucocontextum, in which 273 genes were predicted (Table 6). Furthermore, G. leucocontextum had more CAZymes genes than other fungi, including wood-rotting fungi [G. lucidum (Liu et al. 2012), Auricularia heimuer (Yuan et al. 2019), and H. erinaceus (Gong et al. 2020)], straw-rotting fungi (M. sextelata) (Mei et al. 2019), Mycorrhizal fungi (L. bicolor), and entomogenous fungi (Cordyceps militaris) (Zheng et al. 2011). It was interesting to note that 42 genes in G. leucocontextum were annotated to GH16, which was associated with the growth and development of fungi, and plays an important role in drought and other stresses (Sun et al. 2019), while 30 genes were assigned to GH18, which mainly contains the function of catalyzing the decomposition of chitin, but the number was less than G. lucidum, which has the highest genes number (40) annotated to GH18 among the known basidiomycetes (Liu et al. 2012). In particular, a number of CEs of G. leucocontextum were more than other fungus; 50 genes were annotated to CE10, which is related to the activities in aryl esterase, carboxyl esterase, acetylcholinesterase, cholinesterase, sterol esterase, and brefeldin A esterase, this also maybe the genetic basis of G. leucocontextum for the abundant sterols and other secondary metabolites.
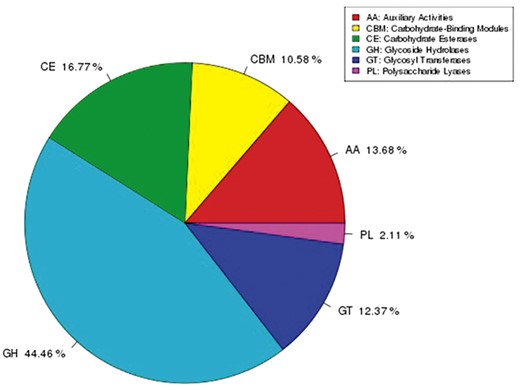
The CAZymes annotation of G. leucocontextum.
Annotation results of carbohydrate-active enzymes
| Type | Number | Percentage |
|---|---|---|
| AAs | 84 | 13.68 |
| CBMs | 65 | 10.58 |
| CEs | 103 | 16.77 |
| GHs | 273 | 44.46 |
| GTs | 76 | 12.37 |
| PLs | 13 | 2.11 |
| Type | Number | Percentage |
|---|---|---|
| AAs | 84 | 13.68 |
| CBMs | 65 | 10.58 |
| CEs | 103 | 16.77 |
| GHs | 273 | 44.46 |
| GTs | 76 | 12.37 |
| PLs | 13 | 2.11 |
Annotation results of carbohydrate-active enzymes
| Type | Number | Percentage |
|---|---|---|
| AAs | 84 | 13.68 |
| CBMs | 65 | 10.58 |
| CEs | 103 | 16.77 |
| GHs | 273 | 44.46 |
| GTs | 76 | 12.37 |
| PLs | 13 | 2.11 |
| Type | Number | Percentage |
|---|---|---|
| AAs | 84 | 13.68 |
| CBMs | 65 | 10.58 |
| CEs | 103 | 16.77 |
| GHs | 273 | 44.46 |
| GTs | 76 | 12.37 |
| PLs | 13 | 2.11 |
The gene distribution of CAZymes in six different fungi
| Nutritional ecological type | Species | The proportion of match number and percentage | ||||||
|---|---|---|---|---|---|---|---|---|
| GHs | GTs | PLs | CEs | CBMs | AAs | Totals | ||
| Wood-rotting fungi | G. leucocontextum | 273 (44.46%) | 76 (12.37%) | 13 (2.11%) | 103 (16.77%) | 65 (10.58%) | 84 (13.68%) | 614 |
| Wood-rotting fungi | G. lucidum | 288 (58.90) | 70 (14.31%) | 10 (2.04%) | 30 (6.13) | 53 (10.84) | 38 (7.77%) | 489 |
| Wood-rotting fungi | A. heimuer | 106 (31.55%) | 29 (8.63%) | 14 (4.17%) | 22 (6.55%) | 103 (30.65%) | 66 (18.45%) | 340 |
| Wood-rotting fungi | H. erinaceus | 161 (47.21%) | 59 (17.30%) | 7 (2.05%) | 26 (7.62%) | 4 (1.17%) | 84 (24.63%) | 341 |
| Straw-rotting fungi | M. sextelata | 159 (47.60%) | 41 (12.28%) | 20 (5.99%) | 13 (3.89%) | 57 (17.07%) | 44 (13.17%) | 334 |
| Mycorrhizal fungi | L. bicolor | 112 (51.85%) | 41 (18.98) | 5 (2.31) | 5 (2.31%) | 21 (9.72%) | 32 (14.81%) | 216 |
| Entomogenous fungi | C. militaris | 159 (50.15%) | 84 (26.49%) | 4 (1.26%) | 13 (4.10%) | 2 (0.63%) | 55 (17.35%) | 317 |
| Nutritional ecological type | Species | The proportion of match number and percentage | ||||||
|---|---|---|---|---|---|---|---|---|
| GHs | GTs | PLs | CEs | CBMs | AAs | Totals | ||
| Wood-rotting fungi | G. leucocontextum | 273 (44.46%) | 76 (12.37%) | 13 (2.11%) | 103 (16.77%) | 65 (10.58%) | 84 (13.68%) | 614 |
| Wood-rotting fungi | G. lucidum | 288 (58.90) | 70 (14.31%) | 10 (2.04%) | 30 (6.13) | 53 (10.84) | 38 (7.77%) | 489 |
| Wood-rotting fungi | A. heimuer | 106 (31.55%) | 29 (8.63%) | 14 (4.17%) | 22 (6.55%) | 103 (30.65%) | 66 (18.45%) | 340 |
| Wood-rotting fungi | H. erinaceus | 161 (47.21%) | 59 (17.30%) | 7 (2.05%) | 26 (7.62%) | 4 (1.17%) | 84 (24.63%) | 341 |
| Straw-rotting fungi | M. sextelata | 159 (47.60%) | 41 (12.28%) | 20 (5.99%) | 13 (3.89%) | 57 (17.07%) | 44 (13.17%) | 334 |
| Mycorrhizal fungi | L. bicolor | 112 (51.85%) | 41 (18.98) | 5 (2.31) | 5 (2.31%) | 21 (9.72%) | 32 (14.81%) | 216 |
| Entomogenous fungi | C. militaris | 159 (50.15%) | 84 (26.49%) | 4 (1.26%) | 13 (4.10%) | 2 (0.63%) | 55 (17.35%) | 317 |
The gene distribution of CAZymes in six different fungi
| Nutritional ecological type | Species | The proportion of match number and percentage | ||||||
|---|---|---|---|---|---|---|---|---|
| GHs | GTs | PLs | CEs | CBMs | AAs | Totals | ||
| Wood-rotting fungi | G. leucocontextum | 273 (44.46%) | 76 (12.37%) | 13 (2.11%) | 103 (16.77%) | 65 (10.58%) | 84 (13.68%) | 614 |
| Wood-rotting fungi | G. lucidum | 288 (58.90) | 70 (14.31%) | 10 (2.04%) | 30 (6.13) | 53 (10.84) | 38 (7.77%) | 489 |
| Wood-rotting fungi | A. heimuer | 106 (31.55%) | 29 (8.63%) | 14 (4.17%) | 22 (6.55%) | 103 (30.65%) | 66 (18.45%) | 340 |
| Wood-rotting fungi | H. erinaceus | 161 (47.21%) | 59 (17.30%) | 7 (2.05%) | 26 (7.62%) | 4 (1.17%) | 84 (24.63%) | 341 |
| Straw-rotting fungi | M. sextelata | 159 (47.60%) | 41 (12.28%) | 20 (5.99%) | 13 (3.89%) | 57 (17.07%) | 44 (13.17%) | 334 |
| Mycorrhizal fungi | L. bicolor | 112 (51.85%) | 41 (18.98) | 5 (2.31) | 5 (2.31%) | 21 (9.72%) | 32 (14.81%) | 216 |
| Entomogenous fungi | C. militaris | 159 (50.15%) | 84 (26.49%) | 4 (1.26%) | 13 (4.10%) | 2 (0.63%) | 55 (17.35%) | 317 |
| Nutritional ecological type | Species | The proportion of match number and percentage | ||||||
|---|---|---|---|---|---|---|---|---|
| GHs | GTs | PLs | CEs | CBMs | AAs | Totals | ||
| Wood-rotting fungi | G. leucocontextum | 273 (44.46%) | 76 (12.37%) | 13 (2.11%) | 103 (16.77%) | 65 (10.58%) | 84 (13.68%) | 614 |
| Wood-rotting fungi | G. lucidum | 288 (58.90) | 70 (14.31%) | 10 (2.04%) | 30 (6.13) | 53 (10.84) | 38 (7.77%) | 489 |
| Wood-rotting fungi | A. heimuer | 106 (31.55%) | 29 (8.63%) | 14 (4.17%) | 22 (6.55%) | 103 (30.65%) | 66 (18.45%) | 340 |
| Wood-rotting fungi | H. erinaceus | 161 (47.21%) | 59 (17.30%) | 7 (2.05%) | 26 (7.62%) | 4 (1.17%) | 84 (24.63%) | 341 |
| Straw-rotting fungi | M. sextelata | 159 (47.60%) | 41 (12.28%) | 20 (5.99%) | 13 (3.89%) | 57 (17.07%) | 44 (13.17%) | 334 |
| Mycorrhizal fungi | L. bicolor | 112 (51.85%) | 41 (18.98) | 5 (2.31) | 5 (2.31%) | 21 (9.72%) | 32 (14.81%) | 216 |
| Entomogenous fungi | C. militaris | 159 (50.15%) | 84 (26.49%) | 4 (1.26%) | 13 (4.10%) | 2 (0.63%) | 55 (17.35%) | 317 |
Secondary metabolism analysis
Mushroom have been widely used as food and medicine in different part of the world for centuries. The main reason was that mushroom can not only be used as the nutritional source, such as dietary fiber, proteins, fats, amino acids, minerals, and vitamins, but also be used as potential pharmaceutical applications owing to the bioactive metabolites, including polysaccharides, terpenoids, fungal immunomodulatory proteins, and many other low-molecular-weight substances, which were widespread in the fruiting body (Elsayed et al. 2014; Zhao et al. 2020). The encoding genes for the biosynthesis of these active compounds were often organized as biosynthetic gene clusters (Blin et al. 2019). Previous studies on functional gene clusters, such as terpene synthases (Chen et al. 2011; Quin et al. 2014), non-ribosomal peptide synthetases (NRPS) (Finking and Marahiel 2004) (Schwarzer et al. 2003), and polyketide synthases (PKS) (Sims and Schmidt 2008; Lackner et al. 2012), have provided the references of gene clusters for genome mining. By using antiSMASH, one NRPS, one beta-lactone, three T1PKS, six NRPS-like, and ten terpene gene clusters were identified in the genome of G. leucocontextum; the details of secondary metabolite gene clusters are listed in Supplementary Table S4. This revealed that abundant terpenoid gene synthesis clusters may be the genetic basis for fruiting bodies of G. leucocontextum to produce rich terpenoids. Triterpenoids are a highly diverse group of natural products that are widely distributed in eukaryotes, and many triterpenoids have beneficial properties for human health. Ganoderma lucidum has the most diverse and abundant triterpenoid content of all examined fungi (Bishop et al. 2015). Ergosterol compounds are one of the major groups of therapeutic compounds in Ganoderma species. Previous studies in G. lucidum have identified 24 key genes involved in the biosynthesis of ergosterol compounds (Chen et al. 2012). In this study, Terpenoid backbone biosynthesis (26 genes) and Sesquiterpenoid and triterpenoid biosynthesis (3 genes) were identified in the genome of G. leucocontextum. Compared to G. lucidum (Chen et al. 2012), G. leucocontextum almost have all the genes required in the whole synthesis pathway of ganoderic acids and ergosterol, which were the two important secondary metabolites except for the genes ERG11-2, but it can be substituted by ERG11-1 (Table 7). Based on previous research (Cai et al. 2021), the triterpenoid and ergosterol biosynthesis pathways of G. leucocontextum were deduced (Figure 6). Considering that terpenoids are bioactive natural products widespread in fungi, especially in the fruiting body of G. lucidum (Su et al. 2020), we compared and analyzed the genes and enzymes involved in the pathways of metabolism of terpenoids and polyketides between G. leucocontextum and G. lucidum (Table 8). The core genes for C10–C20 isoprenoid biosynthesis (non-plant eukaryotes) were all present in the two species, and the pathway of “Sesquiterpenoid and triterpenoid biosynthesis” was intact in G. leucocontextum but incomplete in G. lucidum (Figure 7). The results were not quite similar to previous research results (Gu et al. 2017), that is, genes of isopentenyl diphosphate isomerase (IDI) and mevalonate kinase were present in G. lucidum. We suspect that there may be two reasons: first, poor-quality assembly of the G. lucidum genome hamper downstream analyses; second, there may be other metabolic pathways being used for the synthesis of terpenoids and polyketides in G. lucidum. Irrespectively, we have demonstrated that there is a difference in the active ingredients between the two species as they show different effects in cell experiments (the results have not published).
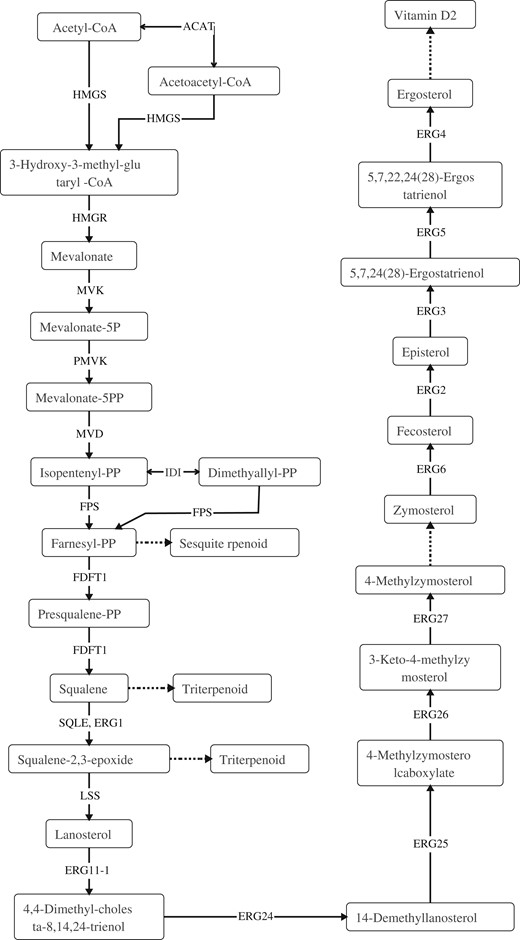
Triterpenoid and ergosterol biosynthesis pathways of G. leucocontextum.
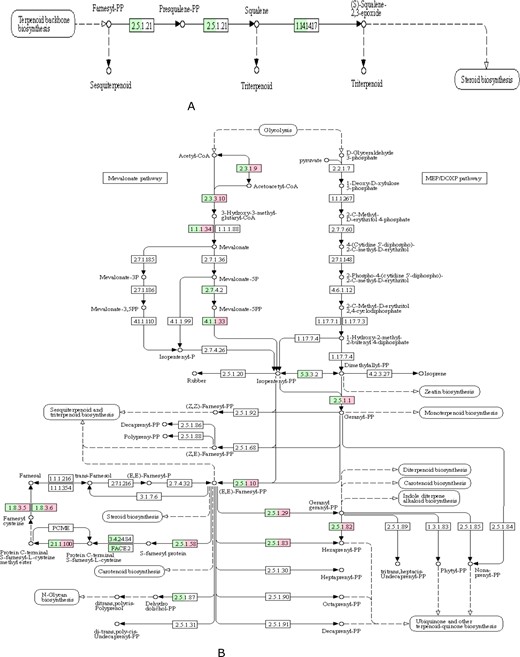
Enzyme in G. leucocontextum and G. lucidum involved in the pathway of Sesquiterpenoid and triterpenoid biosynthesis (map00909, A) and Terpenoid backbone biosynthesis (map00900, B). The colored box indicated existing homologous genes of the enzyme; green represent G. leucocontextum, red represent G. lucidum, white box means not, the same below.
Genes of ganoderic acids and ergosterol biosynthesis in G. leucocontextum and G. lucidum
| Secondary metabolite | Gene name | Gene number | |
|---|---|---|---|
| G. lucidum | G. leucocontextum | ||
| Ganoderic acids | AACT-1 | GL23502 | EVM0010239.1 |
| AACT-2 | GL26574 | EVM0000725.1 | |
| FPS-1 | GL22068 | EVM0004420.2 | |
| FPS-2 | GL25499 | EVM0009008.1 | |
| HMGR | GL24088 | EVM0008852.1 | |
| HMGS | GL24922 | EVM0003163.1 | |
| IDI | GL29704 | EVM0010983.1 | |
| LSS | GL18675 | EVM0005702.1 | |
| MVD | GL25304 | EVM0004601.1 | |
| MVK | GL17879 | EVM0008302.1 | |
| PMVK | GL17808 | EVM0009209.1 | |
| SE | GL23376 | EVM0008824.1 | |
| SQS | GL21690 | EVM0010547.1 | |
| Ergosterol | ERG11-1 | GL26139 | EVM0001186.1 |
| ERG11-2 | GL22375 | / | |
| ERG2 | GL22516 | EVM0010304.1 | |
| ERG24 | GL23832 | EVM0011570.1 | |
| ERG25 | GL23074 | EVM0000338.1 | |
| ERG26 | GL16838 | EVM0012405.1 | |
| ERG27 | GL22371 | EVM0011171.1 | |
| ERG3 | GL26052 | EVM0000544.3 | |
| ERG4 | GL21870 | EVM0000917.1 | |
| ERG5 | GL30444 | EVM0010271.1 | |
| ERG6 | GL18323 | EVM0002811.1 | |
| Secondary metabolite | Gene name | Gene number | |
|---|---|---|---|
| G. lucidum | G. leucocontextum | ||
| Ganoderic acids | AACT-1 | GL23502 | EVM0010239.1 |
| AACT-2 | GL26574 | EVM0000725.1 | |
| FPS-1 | GL22068 | EVM0004420.2 | |
| FPS-2 | GL25499 | EVM0009008.1 | |
| HMGR | GL24088 | EVM0008852.1 | |
| HMGS | GL24922 | EVM0003163.1 | |
| IDI | GL29704 | EVM0010983.1 | |
| LSS | GL18675 | EVM0005702.1 | |
| MVD | GL25304 | EVM0004601.1 | |
| MVK | GL17879 | EVM0008302.1 | |
| PMVK | GL17808 | EVM0009209.1 | |
| SE | GL23376 | EVM0008824.1 | |
| SQS | GL21690 | EVM0010547.1 | |
| Ergosterol | ERG11-1 | GL26139 | EVM0001186.1 |
| ERG11-2 | GL22375 | / | |
| ERG2 | GL22516 | EVM0010304.1 | |
| ERG24 | GL23832 | EVM0011570.1 | |
| ERG25 | GL23074 | EVM0000338.1 | |
| ERG26 | GL16838 | EVM0012405.1 | |
| ERG27 | GL22371 | EVM0011171.1 | |
| ERG3 | GL26052 | EVM0000544.3 | |
| ERG4 | GL21870 | EVM0000917.1 | |
| ERG5 | GL30444 | EVM0010271.1 | |
| ERG6 | GL18323 | EVM0002811.1 | |
Genes of ganoderic acids and ergosterol biosynthesis in G. leucocontextum and G. lucidum
| Secondary metabolite | Gene name | Gene number | |
|---|---|---|---|
| G. lucidum | G. leucocontextum | ||
| Ganoderic acids | AACT-1 | GL23502 | EVM0010239.1 |
| AACT-2 | GL26574 | EVM0000725.1 | |
| FPS-1 | GL22068 | EVM0004420.2 | |
| FPS-2 | GL25499 | EVM0009008.1 | |
| HMGR | GL24088 | EVM0008852.1 | |
| HMGS | GL24922 | EVM0003163.1 | |
| IDI | GL29704 | EVM0010983.1 | |
| LSS | GL18675 | EVM0005702.1 | |
| MVD | GL25304 | EVM0004601.1 | |
| MVK | GL17879 | EVM0008302.1 | |
| PMVK | GL17808 | EVM0009209.1 | |
| SE | GL23376 | EVM0008824.1 | |
| SQS | GL21690 | EVM0010547.1 | |
| Ergosterol | ERG11-1 | GL26139 | EVM0001186.1 |
| ERG11-2 | GL22375 | / | |
| ERG2 | GL22516 | EVM0010304.1 | |
| ERG24 | GL23832 | EVM0011570.1 | |
| ERG25 | GL23074 | EVM0000338.1 | |
| ERG26 | GL16838 | EVM0012405.1 | |
| ERG27 | GL22371 | EVM0011171.1 | |
| ERG3 | GL26052 | EVM0000544.3 | |
| ERG4 | GL21870 | EVM0000917.1 | |
| ERG5 | GL30444 | EVM0010271.1 | |
| ERG6 | GL18323 | EVM0002811.1 | |
| Secondary metabolite | Gene name | Gene number | |
|---|---|---|---|
| G. lucidum | G. leucocontextum | ||
| Ganoderic acids | AACT-1 | GL23502 | EVM0010239.1 |
| AACT-2 | GL26574 | EVM0000725.1 | |
| FPS-1 | GL22068 | EVM0004420.2 | |
| FPS-2 | GL25499 | EVM0009008.1 | |
| HMGR | GL24088 | EVM0008852.1 | |
| HMGS | GL24922 | EVM0003163.1 | |
| IDI | GL29704 | EVM0010983.1 | |
| LSS | GL18675 | EVM0005702.1 | |
| MVD | GL25304 | EVM0004601.1 | |
| MVK | GL17879 | EVM0008302.1 | |
| PMVK | GL17808 | EVM0009209.1 | |
| SE | GL23376 | EVM0008824.1 | |
| SQS | GL21690 | EVM0010547.1 | |
| Ergosterol | ERG11-1 | GL26139 | EVM0001186.1 |
| ERG11-2 | GL22375 | / | |
| ERG2 | GL22516 | EVM0010304.1 | |
| ERG24 | GL23832 | EVM0011570.1 | |
| ERG25 | GL23074 | EVM0000338.1 | |
| ERG26 | GL16838 | EVM0012405.1 | |
| ERG27 | GL22371 | EVM0011171.1 | |
| ERG3 | GL26052 | EVM0000544.3 | |
| ERG4 | GL21870 | EVM0000917.1 | |
| ERG5 | GL30444 | EVM0010271.1 | |
| ERG6 | GL18323 | EVM0002811.1 | |
Pathway of metabolism of terpenoids and polyketides in G. leucocontextum and G. lucidum
| Pathway of metabolism of terpenoids and polyketides | Gene name | Definition | KO term | EC number | G. leucocontextum | G. lucidum |
|---|---|---|---|---|---|---|
| Terpenoid backbone biosynthesis/map00900 | ACAT, atoB | Acetyl-CoA C-acetyltransferase | K00626 | EC:2.3.1.9 | Present | Present |
| E2.3.3.10 | Hydroxymethylglutaryl-CoA synthase | K01641 | EC:2.3.3.10 | Present | Present | |
| HMGCR | Hydroxymethylglutaryl-CoA reductase (NADPH) | K00021 | EC:1.1.1.34 | Present | Present | |
| E2.7.4.2, mvaK2 | Phosphomevalonate kinase | K00938 | EC:2.7.4.2 | Present | Absent | |
| MVD, mvaD | Diphosphomevalonate decarboxylase | K01597 | EC:4.1.1.33 | Present | Present | |
| idi, IDI | Isopentenyl-diphosphate Delta-isomerase | K01823 | EC:5.3.3.2 | Present | Absent | |
| FDPS | Farnesyl-diphosphate synthase | K00787 | EC:2.5.1.1, EC:2.5.1.10 | Present | Present | |
| PCYOX1, FCLY | Prenylcysteine oxidase/farnesylcysteine lyase | K05906 | EC:1.8.3.5, EC:1.8.3.6 | Present | Present | |
| ICMT, STE14 | Protein-S-isoprenylcysteine O-methyltransferase | K00587 | EC:2.1.1.100 | Present | Present | |
| STE24 | STE24 endopeptidase | K06013 | EC:3.4.24.84 | Present | Absent | |
| RCE1, FACE2 | Prenyl protein peptidase | K08658 | EC:3.4.22.- | Present | Absent | |
| FNTB | Protein farnesyltransferase subunit beta | K05954 | EC:2.5.1.58 | Present | Present | |
| DHDDS, RER2, SRT1 | Ditrans, polycis-polyprenyl diphosphate synthase | K11778 | EC:2.5.1.87 | Present | Absent | |
| GGPS1 | Geranylgeranyl diphosphate synthase, type III | K00804 | EC:2.5.1.29 | Present | Present | |
| hexPS, COQ1 | Hexaprenyl-diphosphate synthase | K05355 | EC:2.5.1.82, EC:2.5.1.83 | Present | Present | |
| Sesquiterpenoid and triterpenoid biosynthesis/map00909 | FDFT1 | Farnesyl-diphosphate farnesyltransferase | K00801 | EC:2.5.1.21 | Present | Absent |
| SQLE, ERG1 | Squalene monooxygenase | K00511 | EC:1.14.14.17 | Present | Absent |
| Pathway of metabolism of terpenoids and polyketides | Gene name | Definition | KO term | EC number | G. leucocontextum | G. lucidum |
|---|---|---|---|---|---|---|
| Terpenoid backbone biosynthesis/map00900 | ACAT, atoB | Acetyl-CoA C-acetyltransferase | K00626 | EC:2.3.1.9 | Present | Present |
| E2.3.3.10 | Hydroxymethylglutaryl-CoA synthase | K01641 | EC:2.3.3.10 | Present | Present | |
| HMGCR | Hydroxymethylglutaryl-CoA reductase (NADPH) | K00021 | EC:1.1.1.34 | Present | Present | |
| E2.7.4.2, mvaK2 | Phosphomevalonate kinase | K00938 | EC:2.7.4.2 | Present | Absent | |
| MVD, mvaD | Diphosphomevalonate decarboxylase | K01597 | EC:4.1.1.33 | Present | Present | |
| idi, IDI | Isopentenyl-diphosphate Delta-isomerase | K01823 | EC:5.3.3.2 | Present | Absent | |
| FDPS | Farnesyl-diphosphate synthase | K00787 | EC:2.5.1.1, EC:2.5.1.10 | Present | Present | |
| PCYOX1, FCLY | Prenylcysteine oxidase/farnesylcysteine lyase | K05906 | EC:1.8.3.5, EC:1.8.3.6 | Present | Present | |
| ICMT, STE14 | Protein-S-isoprenylcysteine O-methyltransferase | K00587 | EC:2.1.1.100 | Present | Present | |
| STE24 | STE24 endopeptidase | K06013 | EC:3.4.24.84 | Present | Absent | |
| RCE1, FACE2 | Prenyl protein peptidase | K08658 | EC:3.4.22.- | Present | Absent | |
| FNTB | Protein farnesyltransferase subunit beta | K05954 | EC:2.5.1.58 | Present | Present | |
| DHDDS, RER2, SRT1 | Ditrans, polycis-polyprenyl diphosphate synthase | K11778 | EC:2.5.1.87 | Present | Absent | |
| GGPS1 | Geranylgeranyl diphosphate synthase, type III | K00804 | EC:2.5.1.29 | Present | Present | |
| hexPS, COQ1 | Hexaprenyl-diphosphate synthase | K05355 | EC:2.5.1.82, EC:2.5.1.83 | Present | Present | |
| Sesquiterpenoid and triterpenoid biosynthesis/map00909 | FDFT1 | Farnesyl-diphosphate farnesyltransferase | K00801 | EC:2.5.1.21 | Present | Absent |
| SQLE, ERG1 | Squalene monooxygenase | K00511 | EC:1.14.14.17 | Present | Absent |
Pathway of metabolism of terpenoids and polyketides in G. leucocontextum and G. lucidum
| Pathway of metabolism of terpenoids and polyketides | Gene name | Definition | KO term | EC number | G. leucocontextum | G. lucidum |
|---|---|---|---|---|---|---|
| Terpenoid backbone biosynthesis/map00900 | ACAT, atoB | Acetyl-CoA C-acetyltransferase | K00626 | EC:2.3.1.9 | Present | Present |
| E2.3.3.10 | Hydroxymethylglutaryl-CoA synthase | K01641 | EC:2.3.3.10 | Present | Present | |
| HMGCR | Hydroxymethylglutaryl-CoA reductase (NADPH) | K00021 | EC:1.1.1.34 | Present | Present | |
| E2.7.4.2, mvaK2 | Phosphomevalonate kinase | K00938 | EC:2.7.4.2 | Present | Absent | |
| MVD, mvaD | Diphosphomevalonate decarboxylase | K01597 | EC:4.1.1.33 | Present | Present | |
| idi, IDI | Isopentenyl-diphosphate Delta-isomerase | K01823 | EC:5.3.3.2 | Present | Absent | |
| FDPS | Farnesyl-diphosphate synthase | K00787 | EC:2.5.1.1, EC:2.5.1.10 | Present | Present | |
| PCYOX1, FCLY | Prenylcysteine oxidase/farnesylcysteine lyase | K05906 | EC:1.8.3.5, EC:1.8.3.6 | Present | Present | |
| ICMT, STE14 | Protein-S-isoprenylcysteine O-methyltransferase | K00587 | EC:2.1.1.100 | Present | Present | |
| STE24 | STE24 endopeptidase | K06013 | EC:3.4.24.84 | Present | Absent | |
| RCE1, FACE2 | Prenyl protein peptidase | K08658 | EC:3.4.22.- | Present | Absent | |
| FNTB | Protein farnesyltransferase subunit beta | K05954 | EC:2.5.1.58 | Present | Present | |
| DHDDS, RER2, SRT1 | Ditrans, polycis-polyprenyl diphosphate synthase | K11778 | EC:2.5.1.87 | Present | Absent | |
| GGPS1 | Geranylgeranyl diphosphate synthase, type III | K00804 | EC:2.5.1.29 | Present | Present | |
| hexPS, COQ1 | Hexaprenyl-diphosphate synthase | K05355 | EC:2.5.1.82, EC:2.5.1.83 | Present | Present | |
| Sesquiterpenoid and triterpenoid biosynthesis/map00909 | FDFT1 | Farnesyl-diphosphate farnesyltransferase | K00801 | EC:2.5.1.21 | Present | Absent |
| SQLE, ERG1 | Squalene monooxygenase | K00511 | EC:1.14.14.17 | Present | Absent |
| Pathway of metabolism of terpenoids and polyketides | Gene name | Definition | KO term | EC number | G. leucocontextum | G. lucidum |
|---|---|---|---|---|---|---|
| Terpenoid backbone biosynthesis/map00900 | ACAT, atoB | Acetyl-CoA C-acetyltransferase | K00626 | EC:2.3.1.9 | Present | Present |
| E2.3.3.10 | Hydroxymethylglutaryl-CoA synthase | K01641 | EC:2.3.3.10 | Present | Present | |
| HMGCR | Hydroxymethylglutaryl-CoA reductase (NADPH) | K00021 | EC:1.1.1.34 | Present | Present | |
| E2.7.4.2, mvaK2 | Phosphomevalonate kinase | K00938 | EC:2.7.4.2 | Present | Absent | |
| MVD, mvaD | Diphosphomevalonate decarboxylase | K01597 | EC:4.1.1.33 | Present | Present | |
| idi, IDI | Isopentenyl-diphosphate Delta-isomerase | K01823 | EC:5.3.3.2 | Present | Absent | |
| FDPS | Farnesyl-diphosphate synthase | K00787 | EC:2.5.1.1, EC:2.5.1.10 | Present | Present | |
| PCYOX1, FCLY | Prenylcysteine oxidase/farnesylcysteine lyase | K05906 | EC:1.8.3.5, EC:1.8.3.6 | Present | Present | |
| ICMT, STE14 | Protein-S-isoprenylcysteine O-methyltransferase | K00587 | EC:2.1.1.100 | Present | Present | |
| STE24 | STE24 endopeptidase | K06013 | EC:3.4.24.84 | Present | Absent | |
| RCE1, FACE2 | Prenyl protein peptidase | K08658 | EC:3.4.22.- | Present | Absent | |
| FNTB | Protein farnesyltransferase subunit beta | K05954 | EC:2.5.1.58 | Present | Present | |
| DHDDS, RER2, SRT1 | Ditrans, polycis-polyprenyl diphosphate synthase | K11778 | EC:2.5.1.87 | Present | Absent | |
| GGPS1 | Geranylgeranyl diphosphate synthase, type III | K00804 | EC:2.5.1.29 | Present | Present | |
| hexPS, COQ1 | Hexaprenyl-diphosphate synthase | K05355 | EC:2.5.1.82, EC:2.5.1.83 | Present | Present | |
| Sesquiterpenoid and triterpenoid biosynthesis/map00909 | FDFT1 | Farnesyl-diphosphate farnesyltransferase | K00801 | EC:2.5.1.21 | Present | Absent |
| SQLE, ERG1 | Squalene monooxygenase | K00511 | EC:1.14.14.17 | Present | Absent |
Conclusion
As a newly discovered prize medicinal mushroom, the pharmacological activity and cultivation characteristics of G. leucocontextum have been studied. However, the study of functional and biological properties at the genome level remains unknown. The genome of G. leucocontextum in this study provided the gene information and laid the foundation for further understanding of the reasons behind its activity and function. Based on the advancing technologies of sequencing and analyses, genes related to secondary metabolites biosynthesis, transport, and catabolism of G. leucocontextum were obtained, and the number of some specific genes in G. leucocontextum was much more than other edible and medical fungus; the relationship between these genes and the biological characteristics and pharmacological activities remains to be further studied. Like the rich gene of CEs in G. leucocontextum, we speculate that this characteristic was to better adapt to the special climate of the plateau. As a newly identified member of Ganoderma, there will be abundant of active ingredients and metabolic genes to be excavated and utilized. Although incomplete, the results of G. leucocontextum genome in this study provide a preliminary insight to the biosynthesis of active secondary metabolites and can be used as a theoretical reference for the development and application of Ganoderma industry.
Author statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. All the authors have seen the manuscript and approved to submit to your journal. Neither the entire paper nor any part of its content has been published or has been accepted elsewhere. It is not being submitted to any other journal as well.
Data availability
The assembled genome sequence of G. leucocontextum has been provided to NCBI with the BioProject ID PRJNA729903 and accession number JAHKGY000000000. Supplementary material is available at figshare: https://doi.org/10.25387/g3.16545636.
Acknowledgments
We thank Guan-shen Liu (Biomarker Technologies, Beijing, China) for providing us with valuable technical and analytical assistance. We also thank Feng Chen (Yanke Biotechnology Co., Ltd. Guangzhou, China) for recommendations for data analysis. The author thanks Tamdrin Tsering for his assistance during the process of specimen collection.
Conceptualization: Yuanchao Liu, Huiping Hu, and Qingping Wu; Methodology: Longhua Huang and Xiaowei Liang; Validation: Qingping Wu and Yizhen Xie; Formal Analysis: Manjun Cai; Investigation: Xiangmin Li, Chun Xiao, Shaodan Chen, Honghui Pan, and Xiong Gao; Resources: Yizhen Xie and Zhi Zhang; Data Curation: Diling Chen and Tianqiao Yong; Writing—Original Draft Preparation: Yuanchao Liu, Longhua Huang, and Huiping Hu; Writing—Review and Editing: Qingping Wu; Visualization: Yuanchao Liu and Huiping Hu; Supervision: Qingping Wu; Project Administration: Qingping Wu; Funding Acquisition: Qingping Wu and Yizhen Xie.
Funding
This study was supported by Science and Technology Planning Project of Guangdong Province, China (2019B121202005), Key-Area Research and Development Program of Guangdong Province (2018B020205001), and the Program for Guangdong YangFan Introducing Innovative and Enterpreneurial Teams (2017YT05S115).
Conflicts of interest
The authors declare no conflicts of interest.
Literature cited
Author notes
These authors are co-first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}