





Abstract
Muscadinia rotundifolia cv. Trayshed is a valuable source of resistance to grape powdery mildew. It carries 2 powdery mildew resistance-associated genetic loci, Run1.2 on chromosome 12 and Run2.2 on chromosome 18. The purpose of this study was to identify candidate resistance genes associated with each haplotype of the 2 loci. Both haplotypes of each resistance-associated locus were identified, phased, and reconstructed. Haplotype phasing allowed the identification of several structural variation events between haplotypes of both loci. Combined with a manual refinement of the gene models, we found that the heterozygous structural variants affected the gene content, with some resulting in duplicated or hemizygous nucleotide-binding leucine-rich repeat genes. Heterozygous structural variations were also found to impact the domain composition of some nucleotide-binding leucine-rich repeat proteins. By comparing the nucleotide-binding leucine-rich repeat proteins at Run1.2 and Run2.2 loci, we discovered that the 2 loci include different numbers and classes of nucleotide-binding leucine-rich repeat genes. To identify powdery mildew resistance-associated genes, we performed a gene expression profiling of the nucleotide-binding leucine-rich repeat genes at Run1.2b and Run2.2 loci with or without powdery mildew present. Several nucleotide-binding leucine-rich repeat genes were constitutively expressed, suggesting a role in powdery mildew resistance. These first complete, haplotype-resolved resistance-associated loci and the candidate nucleotide-binding leucine-rich repeat genes identified by this study are new resources that can aid the development of powdery mildew-resistant grape cultivars.
Introduction
Grapevine powdery mildew (PM) is a devastating fungal disease caused by Erysiphe necator Schwein. (syn. Uncinula necator), an obligate biotrophic ascomycete that can infect all green organs of a grapevine (Gadoury et al. 2012). Cultivated grapevines that belong to Vitis vinifera (ssp. vinifera) are highly susceptible to PM. Fungicide sprays are applied prophylactically to control the disease but are costly (Sambucci et al. 2019). Natural resistance to PM exists in several wild grapes. Thirteen PM resistance-associated loci were identified in the last 2 decades (Dry et al. 2019; Karn et al. 2021). Vitis includes several PM-resistant species, including Vitis romanetii (Ramming et al. 2011; Riaz et al. 2011) and Vitis piasezkii (Pap et al. 2016), which are native to China, V. vinifera ssp. sylvestris from Central Asia (Riaz et al. 2020), the North American Vitis cinerea (Dalbó et al. 2001), and the muscadine grape, Muscadinia rotundifolia (Pauquet et al. 2001; Riaz et al. 2011; Feechan et al. 2013).
Muscadinia rotundifolia is closely related to Vitis (Small 1913). Muscadine grapes are native to the southeastern United States where they are cultivated for fruit, juice, and wine production (Olien 1990). Muscadinia rotundifolia is resistant to several diseases in addition to PM (Olmo 1971, 1986), including downy mildew, Pierce’s disease, and phylloxera. Two major genetic loci associated with PM resistance were found in M. rotundifolia. Resistance to U. necator 1 (Run1), located on chromosome 12, and its alternative form, Run1.2, were identified in M. rotundifolia G52 and Trayshed, respectively (Pauquet et al. 2001; Riaz et al. 2011). Bacterial artificial chromosome sequencing of the Run1 haplotype from M. rotundifolia G52 resulted in the partial reconstruction of the locus, a ∼1.2 Mb region composed of 7 TIR-NBS-LRR genes (Feechan et al. 2013). Run2.1 and Run2.2 were identified on chromosome 18 of M. rotundifolia Magnolia and Trayshed, respectively (Riaz et al. 2011). Both haplotypes of Trayshed’s Run1.2 were associated with PM resistance and designated Run1.2a and Run1.2b (Feechan et al. 2015).
Muscadinia rotundifolia is an ideal partner for breeding PM-resistant grapevines that are durably resistant and require few fungicidal applications. This can be done by introgressing functionally diverse PM resistance-associated genes into V. vinifera (Michelmore et al. 2013). In wild grapes, PM resistance is associated with a programmed cell death-mediated response in infected epidermal cells. This suggests that PM resistance is based on an intracellular recognition of E. necator’s effectors by disease resistance (R) proteins that activate effector-triggered immunity (Qiu et al. 2015; Dry et al. 2019).
Most R genes encode nucleotide-binding leucine-rich repeat (NLR) proteins (Dubey and Singh 2018). NLRs are intracellular receptors that recognize and interact directly with pathogen-derived effectors, detect modifications in host cellular targets, or detect molecular decoys triggered by effectors (Dangl et al. 2013). NLR activation leads to the induction of immune responses that can restrict pathogen spread (Jones and Dangl 2006). These include calcium oscillations, a rapid burst of reactive oxygen species, extensive transcriptional reprogramming that leads to cell wall modifications, and the synthesis of pathogenesis-related proteins and antimicrobial compounds (Jones and Dangl 2006; Dangl et al. 2013; Kretschmer et al. 2019). Effector-triggered immunity is often associated with a hypersensitive response and programmed death of infected plant cells that restricts further pathogen development (Jones and Dangl 2006). NLR intracellular receptors are typically composed of 3 domains: a C-terminal leucine-rich repeat (LRR) domain, a central nucleotide-binding site domain (NBS), and a variable N-terminal domain (Meyers et al. 1999; McHale et al. 2006). The variable N-terminal domain distinguishes NLR classes. The 3 main NLR classes are the TIR-NBS-LRRs, CC-NBS-LRRs, and RPW8-NBS-LRRs; these possess N-terminal toll/interleukin-1 receptor-like (TIR), Coiled-coil (CC), and resistance to PM 8 (RPW8) domains, respectively (Meyers et al. 1999; Xiao et al. 2001; McHale et al. 2006; Michelmore et al. 2013). Only 2 TIR-NBS-LRR genes, MrRPV1 and MrRUN1, have been functionally characterized in grapes (Feechan et al. 2013). MrRPV1 and MrRUN1 are at the Run1/Rpv1 locus of M. rotundifolia G52 and confer resistance to downy mildew and PM, respectively.
The first diploid chromosome-scale genome assembly of a muscadine grape was recently published and is a valuable resource for identifying candidate PM resistance-associated NLR genes from other genetic loci in M. rotundifolia (Cochetel et al. 2021). A first analysis of the Run1.2 locus suggested an expansion of TIR-NBS-LRR genes in M. rotundifolia Trayshed relative to Cabernet Sauvignon (Cochetel et al. 2021). Which of these TIR-NBS-LRR are involved in Trayshed’s PM resistance and to which haplotype they belong, Run1.2a or Run1.2b, is unknown. The goal of this study was to identify candidate resistance genes in each haplotype of M. rotundifolia Trayshed Run1.2 and Run2.2. Haplotypes of Trayshed’s R loci were differentiated and reconstructed with deep sequencing data from 2 backcrossed V. vinifera lines, e6-23 (Run1.2b+) and 08391-029 (Run2.2+). Gene models in both loci were manually curated to identify the genes encoding NLRs. The 2 haplotypes of each R locus were compared to determine the effect of heterozygous structural variations on NLR gene content. To determine NLR genes associated with PM resistance, NLR genes’ expression in Run1.2b and Run2.2 were profiled with and without PM present using RNA-sequencing (RNA-seq).
Materials and methods
Plant material
We used 2 V. vinifera backcrossed lines in this study, e6-23 carrying Run1.2b (Feechan et al. 2015) and 08391-029 possessing Run2.2 (Riaz et al. 2011). Both e6-23 (Run1.2b+) and 08391-029 (Run2.2+) are V. vinifera backcrosses derived from the T6 population series developed by Dr. Harold P. Olmo at University of California Davis (Riaz et al. 2011). Information about the lineage of each genotype is provided in Fig. 1. For each grape accession, 3 plants were inoculated with E. necator C-strain and 3 plants were mock-inoculated as described in Amrine et al. (2015). Two leaves from each plant were collected 1 and 5 days postinoculation (dpi) and immediately frozen in liquid nitrogen. Leaves from an individual plant were pooled together and constitute a biological replicate. Three biological replications were obtained for each treatment.
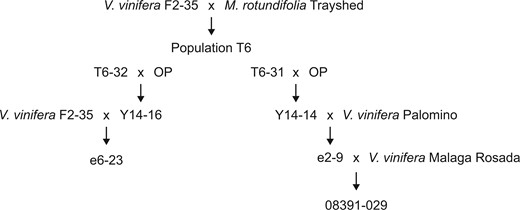
Detailed parentage of e6-23 (Run1.2b+) and 08391-029 (Run2.2+). F2-35 was produced by crossing V. vinifera Cabernet Sauvignon with V. vinifera Carignane. OP, open pollinator which is assumed V. vinifera.
DNA and RNA extraction, library preparation, and sequencing
Deep sequencing of e6-23 (Run1.2b+) and 08391-029 (Run2.2+) was done to distinguish the 2 haplotypes of each Trayshed’s locus. Genomic DNA was extracted from mock-inoculated leaves of e6-23 and 08391-029 and libraries were prepared as in Massonnet et al. (2020). Final libraries were sequenced on the Illumina HiSeqX Ten system in paired-end 150-bp reads (IDseq, Davis, CA, USA; Supplementary Table 1).
Gene expression in mock- and PM-inoculated e6-23 (Run1.2b+) and 08391-029 (Run2.2+) leaves was assessed by RNA-seq. RNA extraction and library preparation were performed as in Amrine et al. (2015). cDNA libraries were sequenced using an Illumina HiSeq4000 sequencer (DNA Technologies Core, University of California, Davis, CA, USA) in 50-bp single-end reads (Supplementary Table 2).
Locus reconstruction
The Run1.2 and Run2.2 haplotypes were located by aligning the primers for Run1.2-associated markers, VMC4f3.1 and VMC8g9, and Run2.2-associated markers, VMC7f2 and UDV108, onto the diploid, chromosome-scale genome of M. rotundifolia Trayshed (Riaz et al. 2011; Cochetel et al. 2021). Whole-genome DNA sequencing reads from e6-23 (Run1.2b+) and 08391-029 (Run2.2+) were used to identify Run1.2b and Run2.2 sequences. Low-quality DNA sequencing reads were removed and adapter sequences were trimmed using Trimmomatic v.0.36 (Bolger et al. 2014) with the following settings: LEADING:3 TRAILING:3 SLIDINGWINDOW:10:20 MINLEN:36 CROP:150. High-quality, paired-end reads were aligned onto the diploid genome of M. rotundifolia Trayshed (Cochetel et al. 2021) using BWA v.01.17 (Li and Durbin 2009) and default parameters. Reads aligning onto the reference genome with no edit distance (0 mismatch) were selected using bamtools filter v.2.5.1 (Barnett et al. 2011) and the tag “NM : 0.” These alignments were used as input for evaluating base coverage with genomecov (BEDTools v2.29.1; Quinlan 2014). Coverage from bases located in repetitive elements was removed using BEDTools intersect v2.29.1 (Quinlan 2014). Median coverage per 10-kb window was calculated using BEDTools map v2.29.1 (Quinlan 2014) and normalized by dividing by the sequencing coverage (Supplementary Table 1). Sequences were removed from the locus and labeled “unplaced” if DNA sequencing reads did not cover a primary contig or its alternative haplotigs. Each haplotype was fragmented into 1 kb sequences using seqkit sliding v.0.16.1 (Shen et al. 2016) and aligned to itself using Minimap2 v.2.12-r847-dirty (Li 2018). Sequence overlaps between contigs were removed from the locus. DNA sequencing coverage along the 4 haplotypes was manually inspected by visualizing alignments using Integrative Genomics Viewer (IGV) v.2.4.14 (Robinson et al. 2011). Loci were reconstructed using HaploMake.py from the HaploSync tool suite v1.0 (https://github.com/andreaminio/HaploSync; accessed: 2022 March 1).
Haplotype sequence comparison
Pairwise alignments were performed using NUCmer from MUMmer v.4.0.0 (Marçais et al. 2018) and the --mum option. Alignments with at least 90% identity are shown in Fig. 2. Structural variants (SVs; >50 bp), SNPs, and INDELs (<50 bp) were called using show-diff and show-snps, respectively, from MUMmer v.4.0.0 (Marçais et al. 2018). The potential impact of SNPs on amino acid content was predicted using SnpEff v.4.3t (Cingolani et al. 2012).
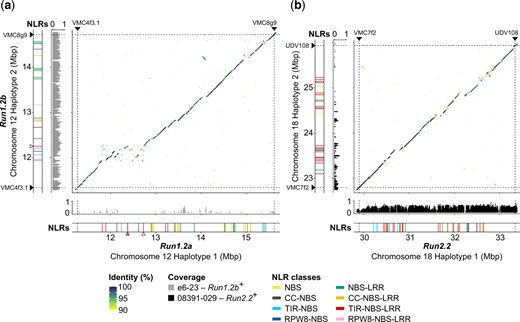
Haplotype comparison and NLR content at Run1.2 and Run2.2 in M. rotundifolia Trayshed. Whole-sequence alignments of the reconstructed haplotypes of Run1.2 (a) and Run2.2 (b) loci. Normalized median DNA-seq coverage per 10 kb of e6-23 (Run1.2b+) and 08391-029 (Run2.2+) on the diploid genome of M. rotundifolia Trayshed was used to identify Run1.2b and Run2.2 on the Haplotype 2 of chromosome 12 and Haplotype 1 of chromosome 18, respectively. Only DNA-seq reads aligning perfectly on the diploid genome of M. rotundifolia Trayshed were used for base coverage analysis. Chromosomal position of the Run1.2- and Run2.2-associated genetic markers is indicated by black triangles and dashed lines. Chromosomal positions of the TIR-NBS-LRR genes whose predicted proteins cluster with G52’s MrRUN1 and MrRPV1 in the phylogenetic tree of Fig. 3a are indicated by gray and white triangles, respectively.
Annotation of NLR genes
Potential NLR genes were identified using NLR-annotator with default parameters (Steuernagel et al. 2020). The intron–exon structure of genes within the R loci was evaluated using RNA-seq alignments. RNA-seq reads from Trayshed leaves (Cochetel et al. 2021), e6-23 (Run1.2b+), and 08391-029 (Run2.2+) were aligned onto the diploid M. rotundifolia Trayshed genome using HISAT2 v.2.1.0 (Kim et al. 2015) and the following settings: –end-to-end –sensitive -k 50. Alignments were visualized using IGV v.2.4.14 (Robinson et al. 2011). Gene models were manually refined when the alignments of RNA-seq reads indicated a different intron–exon structure than the ab initio structural annotation.
Predicted proteins were scanned with hmmsearch from HMMER v.3.3.1 (http://hmmer.org/) and the Pfam-A Hidden Markov Models (HMM) database (El-Gebali et al. 2019; downloaded on 2021 January 29). Protein domains corresponding to Pfam domains including NB-ARC (PF00931.23), LRR (PF00560.34, PF07725.13, PF12799.8, PF13306.7, PF13516.7, PF13855.7), TIR (PF01582.21, PF13676.7), and RPW8 (PF05659.12), with an independent E-value <1.0, and an alignment covering at least 50% of the HMM were selected (Supplementary Table 3). CC domains were identified using COILS (Lupas et al. 1991).
Phylogenetic analysis
Predicted NLR protein sequences from Trayshed’s Run1.2 and Run2.2 and G52’s Run1/Rpv1 (Feechan et al. 2013) were aligned using MUSCLE (Edgar 2004) in MEGAX (Kumar et al. 2018). Resistance gene analogs (RGAs) from Run1/Rpv1 (Feechan et al. 2013) were retrieved from GenBank using the following accession numbers: RGA1, AGC24025; RGA2, AGC24026; RGA4, AGC24027; MrRPV1 (RGA8), AGC24028; RGA9, AGC24029; MrRUN1 (RGA10), AGC24030; RGA11, and AGC24031. Phylogenetic analyses of the proteins were done with MEGAX (Kumar et al., 2018) using the Neighbor-Joining method (Saitou and Nei 1987) and 1,000 bootstrap replicates.
Gene expression analysis
Transcript abundance was evaluated with Salmon v.1.5.1 (Patro et al. 2017) and these parameters: --gcBias --seqBias --validateMappings. The transcriptome index file was built using a kmer size of 13, the combined transcriptomes of M. rotundifolia Trayshed, V. vinifera cv. Cabernet Sauvignon (Massonnet et al. 2020) and E. necator C-strain (Jones et al. 2014), and with their genomes as decoys. Quantification files were imported using an R package, tximport v.1.20.0 (Soneson et al. 2015). DESeq2 v.1.16.1 (Love et al. 2014) was used to assess differential gene expression.
Results
SVs between Trayshed’s Run1.2 haplotypes affect NLR content
The boundaries of Run1.2 were assigned by aligning the primer sequences of Run1.2-associated simple sequence repeats (SSR) markers on the 2 complete copies (Haplotype 1 and Haplotype 2) of chromosome 12 of M. rotundifolia Trayshed (Cochetel et al. 2021). To distinguish Run1.2a and Run1.2b, we sequenced the genome of the V. vinifera backcross e6-23 (Run1.2b+), into which Run1.2b was introgressed by crossing with M. rotundifolia Trayshed and backcrossing with V. vinifera (Fig. 1, Supplementary Tables 1 and 4). Short-sequencing reads from the Run1.2b+ accession covered and aligned perfectly (i.e. with no mismatches) to most of Run1.2 on chromosome 12 Haplotype 2 (Supplementary Fig. 1), and coverage gaps in Run1.2 on Haplotype 2 were complemented by coverage at Run1.2 on Haplotype 1. This indicates that haplotype switching occurred during the assembly and phasing of Trayshed’s genome. To correct this, Run1.2b was reconstructed using only sequences supported with DNA sequencing reads from the Run1.2b+ accession and Run1.2a was reconstructed using alternative sequences (Fig. 2a). The 2 reconstructed Run1.2 haplotypes, Run1.2a and Run1.2b, were 4.34 and 3.38 Mb long, respectively. Differences in length between the 2 haplotypes were associated with several large SVs (>50 bp). For instance, the region of Run1.2b from ∼12 to 12.3 Mb corresponds to a ∼800-kb region in the Run1.2a haplotype (Fig. 2a). In this case, their difference in length was due to several inserted sequences and duplication events in Run1.2a. We also found 32,704 SNPs and 7,150 INDELs between Run1.2a and Run1.2b.
To determine the effect of the heterozygous SVs and short polymorphisms on the gene content, we first refined the gene models for both Run1.2 haplotypes. A total of 78 protein-coding genes, including 22 NLR genes, were manually annotated (Table 1). Run1.2a contained 253 genes and Run1.2b contained 189 genes, indicating that SVs affect the gene content. There were 37 and 24 NLR genes in Run1.2a and Run1.2b, respectively, with both composed primarily of CC-NBS-LRR, TIR-NBS-LRR, and NBS-LRR genes (Fig. 2a; Supplementary Table 4). SVs between haplotypes affect the protein-coding sequences of 22 NLR genes in Run1.2a and 9 NLR genes in Run1.2b. These SVs resulted in the whole duplication of 4 and 2 NLR genes in Run1.2a and Run1.2b, respectively, and the partial duplication of 3 NLR genes in Run1.2a (Supplementary Table 4). In addition, SVs were found to cause the loss of functionality of 4 NLR-coding genes and the hemizygosity of a CC-NBS gene in Run1.2a relative to Run1.2b. Similarly, the LRR domain of 2 NLR genes from Run1.2b was lost compared to Run1.2a. We detected 32,704 SNPs and 7,150 INDELs between the 2 Run1.2 haplotypes (Run1.2a vs Run1.2b). Nonsynonymous SNPs were identified in 8 NLR genes in each haplotype.
Sequence length, protein-coding gene content, and NLR gene content of Run1.2 and Run2.2 reconstructed haplotypes.
| Loci | Run1.2a | Run1.2b | Run2.2 | Chr18 Hap2 |
|---|---|---|---|---|
| Sequence length (bp) | 4,340,059 | 3,379,591 | 3,445,914 | 3,137,389 |
| Protein-coding gene loci | 253 (44) | 189 (34) | 207 (59) | 179 (43) |
| Total NLR genes | 37 (16) | 24 (6) | 39 (33) | 29 (22) |
| NBS genes | 3 (1) | 2 | 6 (6) | 3 (3) |
| CC-NBS genes | 2 (1) | 2 | 0 | 0 |
| RPW8-NBS genes | 2 (2) | 0 | 0 | 0 |
| TIR-NBS genes | 1 (1) | 2 (1) | 9 (7) | 3 (1) |
| NBS-LRR genes | 8 (2) | 9 | 4 (4) | 2 (2) |
| CC-NBS-LRR genes | 13 (6) | 3 (1) | 0 | 0 |
| RPW8-NBS-LRR genes | 0 | 2 | 0 | 0 |
| TIR-NBS-LRR genes | 8 (3) | 4 (4) | 20 (16) | 21 (16) |
| Loci | Run1.2a | Run1.2b | Run2.2 | Chr18 Hap2 |
|---|---|---|---|---|
| Sequence length (bp) | 4,340,059 | 3,379,591 | 3,445,914 | 3,137,389 |
| Protein-coding gene loci | 253 (44) | 189 (34) | 207 (59) | 179 (43) |
| Total NLR genes | 37 (16) | 24 (6) | 39 (33) | 29 (22) |
| NBS genes | 3 (1) | 2 | 6 (6) | 3 (3) |
| CC-NBS genes | 2 (1) | 2 | 0 | 0 |
| RPW8-NBS genes | 2 (2) | 0 | 0 | 0 |
| TIR-NBS genes | 1 (1) | 2 (1) | 9 (7) | 3 (1) |
| NBS-LRR genes | 8 (2) | 9 | 4 (4) | 2 (2) |
| CC-NBS-LRR genes | 13 (6) | 3 (1) | 0 | 0 |
| RPW8-NBS-LRR genes | 0 | 2 | 0 | 0 |
| TIR-NBS-LRR genes | 8 (3) | 4 (4) | 20 (16) | 21 (16) |
Numbers in parentheses correspond to the genes with a structure manually refined.
Sequence length, protein-coding gene content, and NLR gene content of Run1.2 and Run2.2 reconstructed haplotypes.
| Loci | Run1.2a | Run1.2b | Run2.2 | Chr18 Hap2 |
|---|---|---|---|---|
| Sequence length (bp) | 4,340,059 | 3,379,591 | 3,445,914 | 3,137,389 |
| Protein-coding gene loci | 253 (44) | 189 (34) | 207 (59) | 179 (43) |
| Total NLR genes | 37 (16) | 24 (6) | 39 (33) | 29 (22) |
| NBS genes | 3 (1) | 2 | 6 (6) | 3 (3) |
| CC-NBS genes | 2 (1) | 2 | 0 | 0 |
| RPW8-NBS genes | 2 (2) | 0 | 0 | 0 |
| TIR-NBS genes | 1 (1) | 2 (1) | 9 (7) | 3 (1) |
| NBS-LRR genes | 8 (2) | 9 | 4 (4) | 2 (2) |
| CC-NBS-LRR genes | 13 (6) | 3 (1) | 0 | 0 |
| RPW8-NBS-LRR genes | 0 | 2 | 0 | 0 |
| TIR-NBS-LRR genes | 8 (3) | 4 (4) | 20 (16) | 21 (16) |
| Loci | Run1.2a | Run1.2b | Run2.2 | Chr18 Hap2 |
|---|---|---|---|---|
| Sequence length (bp) | 4,340,059 | 3,379,591 | 3,445,914 | 3,137,389 |
| Protein-coding gene loci | 253 (44) | 189 (34) | 207 (59) | 179 (43) |
| Total NLR genes | 37 (16) | 24 (6) | 39 (33) | 29 (22) |
| NBS genes | 3 (1) | 2 | 6 (6) | 3 (3) |
| CC-NBS genes | 2 (1) | 2 | 0 | 0 |
| RPW8-NBS genes | 2 (2) | 0 | 0 | 0 |
| TIR-NBS genes | 1 (1) | 2 (1) | 9 (7) | 3 (1) |
| NBS-LRR genes | 8 (2) | 9 | 4 (4) | 2 (2) |
| CC-NBS-LRR genes | 13 (6) | 3 (1) | 0 | 0 |
| RPW8-NBS-LRR genes | 0 | 2 | 0 | 0 |
| TIR-NBS-LRR genes | 8 (3) | 4 (4) | 20 (16) | 21 (16) |
Numbers in parentheses correspond to the genes with a structure manually refined.
Run2.2 is mainly composed of TIR-NBS-LRRs
A similar approach was applied to identify and reconstruct Run2.2 in Haplotype 1 of chromosome 18 of M. rotundifolia Trayshed using short-sequencing reads from the genotype 08391-029 (Run2.2+; Supplementary Tables 1 and 4; Supplementary Fig. 2). The reconstructed Run2.2 was 3.45 Mb long, slightly longer than its alternative on Haplotype 2 (3.14 Mb). We manually refined the models of 102 protein-coding genes in the 2 haplotypes, including 55 NLR genes (Table 1). More genes were annotated at Run2.2 (207) than at its alternative (179). There were 39 NLR-coding genes at Run2.2 and 29 NLR genes in its alternative. The 2 haplotypes were mainly composed of TIR-NBS-LRR genes, with 20 genes in Run2.2 and 21 genes in its alternative (Supplementary Table 4). Unlike Run1.2, no NLR genes with a CC or RPW8 N-terminal domain were found at Run2.2. Interestingly, the NLR genes occurred in 2 clusters in each haplotype (Fig. 2b; Table 1).
Run2.2 and its alternative contained 456 SVs between them, with an average length of 2.1 ± 3.6 kb. These SVs affected 21 and 16 NLR genes in Run2.2 and its alternative, respectively. SVs were found responsible for the partial duplication of 4 and 2 NLR genes in Run2.2 and its alternative haplotype, respectively (Supplementary Table 4). Furthermore, large deletions encompassed the complete coding sequence of 3 and 4 NLR-coding genes of Run2.2 and its alternative haplotype, respectively. We also identified 24,128 SNPs and 5,773 INDELs between Run2.2 and its alternative, and nonsynonymous SNPs were detected in 16 and 18 NLR genes, respectively.
Run1.2 and Run2.2 loci contain distinct sets of NLRs
Predicted protein sequences of the NLR genes identified in Run1.2a, Run1.2b, Run2.2, and Run2.2’s alternative on chromosome 18 Haplotype 2 were compared by constructing a phylogenic tree (Fig. 3a). In addition, we compared Trayshed’s NLRs with the TIR-NBS-LRRs at Run1/Rpv1 in M. rotundifolia G52 (Barker et al. 2005; Feechan et al. 2013). Run1/Rpv1 is the only R locus characterized in grapes and is an alternative version of Run1.2. Two distinct groups of NLRs were discovered, distinguished by the presence or absence of a TIR domain. A similar clustering pattern was observed when phylogeny was built using NBS domain sequences only (Supplementary Fig. 3), as previously observed in other plants (Seo et al. 2016; Prigozhin and Krasileva 2021). NLRs also tended to cluster by R locus, indicating an allele relationship between haplotypes for 74.4% of the NLRs (Supplementary Table 4). Regarding the TIR-containing proteins, we found the TIR-NBS-LRRs from Run1/Rpv1 clustering with the TIR-NBS-LRR proteins from Run1.2. MrRPV1 from M. rotundifolia G52 clustered with 2 TIR-NBS-LRRs, one from each Run1.2 haplotype, and MrRUN1 clustered with a TIR-NBS-LRR from Run1.2a (Fig. 3a; Supplementary Table 4). Clustering of TIR-NBS-LRRs of Run1.2 and Run1/Rpv1 support an allelic relationship between them. However, the number of LRR motifs in their LRR domain was different (Fig. 3b), suggesting some allelic diversity. In addition, differences in LRR domains suggest that these TIR-NBS-LRRs might be specific to different effectors and/or pathogens (McHale et al. 2006).
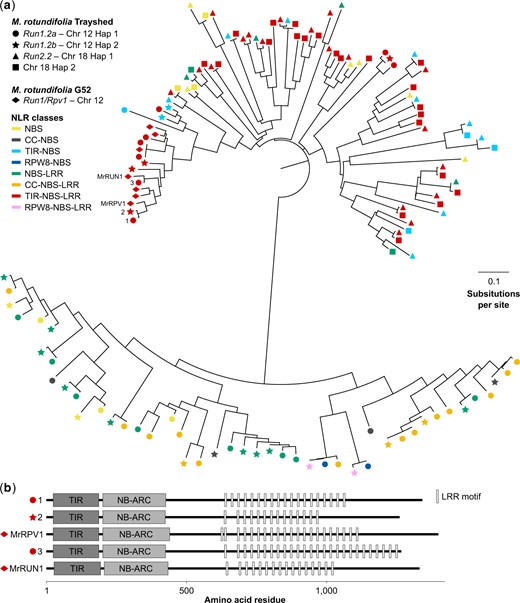
Comparison of the NLRs composing Run1.2 and Run2.2 in M. rotundifolia Trayshed. (a) Neighbor-joining clustering of the predicted protein sequences of the NLRs composing Run1.2 and Run2.2 haplotypes, and Run1/Rpv1 (Feechan et al. 2013). (b) Domain diagram of Trayshed’s TIR-NBS-LRRs clustering with G52’s MrRUN1 and MrRPV1. Proteins are reported using same number assigned in (a). LRR motifs were identified using the consensus sequence LxxLxLxx, with L indicating a leucine residue and x indicating any amino acid (Kajava and Kobe 2002).
Most of the NLR genes at Run1.2b and Run2.2 are constitutively expressed
Constitutive NLR gene expression is essential for disease resistance (Michelmore et al. 2013). To identify expressed NLR genes that are potentially responsible for PM resistance, we measured gene expression in Run1.2b+ and Run2.2+ leaves 1 and 5 dpi with either E. necator C-strain or a mock solution using RNA-seq (Fig. 4).
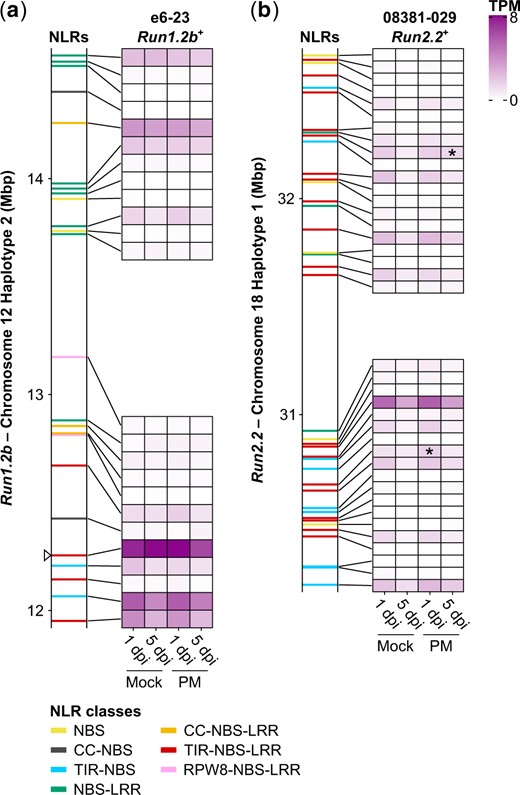
Transcript abundance of NLR genes in Run1.2b (a) and Run2.2 (b). Gene expression was monitored in Run1.2b+ and Run2.2+ at 1 and 5 dpi with E. necator conidia (PM) or a mock solution (Mock). Transcript abundance is shown as the mean of TPM. n = 3. NLR genes differentially expressed in response to PM are indicated by an asterisk (P ≤ 0.05). White triangle indicates the chromosomal position of the TIR-NBS-LRR gene whose predicted protein clusters with G52’s MrRPV1 in Fig. 3a.
In Run1.2b+ leaves, nearly all NLR genes at Run1.2b (23/24) were expressed (Fig. 4a). Nine of them had an expression level higher than 1 transcript per million (TPM) in at least one condition (TPM > 1), while 15 NLR genes were expressed at a lower level (TPM ≤ 1). The most highly expressed genes in Run1.2b across all conditions (mean TPM > 4 TPM) included 2 TIR-NBS-LRRs and a TIR-NBS at the 5′-end of the locus. In addition, the gene with the most elevated expression was the TIR-NBS-LRR gene which predicted protein clustered with MrRPV1 in the phylogenic tree (Fig. 3; Supplementary Table 4). PM inoculation was not found to significantly impact the expression of any NLR gene composing Run1.2b.
In Run2.2+, we identified 11 NLR genes with a transcript abundance greater than 1 TPM, 28 lowly expressed (TPM ≤ 1), and 4 with no expression (Fig. 4b). A TIR-NBS-LRR gene at the 5′-end of Run2.2 was the most highly expressed across conditions. Seven other TIR-NBS-LRR genes in the locus had moderate expression levels. Only 2 NLR genes at Run2.2 were modulated in response to PM, including one at 1 dpi and another at 5 dpi.
These RNA-seq data show that most of the NLR genes composing Trayshed’s loci are constitutively expressed, although they exhibit different levels of expression in Run1.2b+ and Run2.2+ genotypes. Expressed NLR genes are candidate genes involved in PM resistance associated with Run1.2b and Run2.2. All candidate genes, their coordinates, expression, and relationship to Run1/Rpv1-associated NLRs are reported in Supplementary Table 4.
Discussion
By combining a diploid assembly of Trayshed’s genome and DNA sequencing data generated from 2 backcrossed V. vinifera genotypes, we distinguished, phased, and reconstructed the 4 complete haplotypes of Run1.2 and Run2.2. To our knowledge, this is the first report of complete, haplotype-resolved R loci for grapes. The Run1/Rpv1 locus of M. rotundifolia G52 was sequenced prior, but its assembly was fragmented and haploid (Feechan et al. 2013). The same approach used in this study could be applied to resolve the entire Run1/Rpv1 locus. Additional Run1 haplotypes could be compared to better understand the evolution of the structure of the locus across muscadines and its impact on the NLR gene content.
Trayshed was previously defined as homozygous at Run2.2 based on amplicon size (Riaz et al. 2011). However, in silico PCR of Trayshed’s genome showed 2 amplicon sizes for the UDV108 marker: 225 bp on chromosome 18 Haplotype 1 and 323 bp on chromosome 18 Haplotype 2 (Supplementary Table 4). The numerous SVs and small polymorphisms between the 2 haplotypes indicate heterozygosity between the 2 haplotypes. Although Run2.2 locus on chromosome 18 Haplotype 1 confers resistance to PM (Riaz et al. 2011), evaluation of PM susceptibility and sequencing of backcrossed individuals possessing the region on chromosome 18 Haplotype 2 will be necessary to determine whether this haplotype is associated with PM resistance. Complete downy mildew resistance, Rpv2, was associated with the same genomic region as Run2.2 on chromosome 18 of Trayshed (Wiedemann-Merdinoglu et al. 2006). If both Run2.2 haplotypes were characterized, then resistance genes effective against Plasmopara viticola could be identified.
Sixty percent of the NLR genes identified among the 4 haplotypes were manually annotated (Table 1). This was made possible by correctly phasing haplotypes and highlights the necessity of meticulously dissecting complex genomic regions if candidate trait-associated genes are sought (Massonnet et al. 2020). SVs and short polymorphisms affecting NLR genes were discovered by comparing the haplotypes of each resistance locus. These polymorphisms could be used to design haplotype-specific markers which would accelerate the development of PM-resistant cultivars through marker-assisted selection.
The NLRs in Trayshed’s Run1.2 and Run2.2 loci differ. All 3 classes of NLRs, CC-NBS-LRRs, RPW8-NBS-LRRs, and TIR-NBS-LRRs were found in Run1.2 haplotypes, but no CC or RPW8 domains were identified among NLRs at Run2.2. The only characterized NLR gene associated with grape PM resistance, MrRUN1, is a TIR-NBS-LRR (Feechan et al. 2013). Based on phylogeny, only Run1.2a possesses a TIR-NBS-LRR clustering with MrRUN1. This suggests that the mechanisms of PM resistance associated with Trayshed’s Run1.2a and the haplotype Run1 of M. rotundifolia G52 might be more similar compared to Run1.2b. However, their number of LRR motifs differ, suggesting that they might be specific to different effectors (McHale et al. 2006). Comparison of the leaf transcriptome of Run1.2a+, Run1.2b+, and Run1+ accessions in response to PM would help evaluate the commonality in defense-related mechanisms between the 3 haplotypes. Furthermore, the functional characterization of the NLR genes composing Trayshed’s R loci would identify the NLR(s) responsible for PM resistance. This would help determine whether the NLR class is a decisive factor for grape PM resistance, and if stacking resistance genes from the 2 haplotypes of Run1.2 enhances the level of PM resistance and/or its durability. Fine mapping and generation of sequencing data (DNA-seq and RNA-seq) from recombinants could be used to narrow down the list of candidate NLR genes to test.
Most of the NLR genes composing Run1.2b and Run2.2 were expressed at a low level whether or not the pathogen was present. Constitutive low expression of NLR genes is common in plants (Michelmore et al. 2013; Zhang et al. 2020), supporting a constitutive ability to sense pathogens. In contrast, some plant NLR genes were found to be more highly expressed during pathogen infection, indicative of induction of the defense-related surveillance in response to biotic stress (Mohr et al. 2010; Sagi et al. 2017; Zhang et al. 2020). No NLR gene at Run1.2b was found differentially expressed in response to E. necator C-strain in the Run1.2b+ genotype, suggesting that PM resistance associated with Run1.2b likely relies on constitutive expression. On the other hand, 2 NLR genes were differentially expressed in response to PM in the backcrossed V. vinifera genotype possessing Run2.2. Additional expression profiling experiments could be done to determine whether the gene expression modulation of these NLR genes in response to E. necator plays a role in Trayshed’s PM resistance. Assessing gene expression level and transcriptional modulation of the NLR genes composing Run1.2a from accessions carrying only this haplotype (e.g. e1-78; Feechan et al. 2015) would help identify candidate NLR genes for this haplotype. In addition, it would be interesting to profile the gene expression of the candidate NLRs in different individuals carrying Trayshed’s PM-associated loci to evaluate the effect of the genetic background on NLR gene expression. Monitoring NLR gene expression in response to additional E. necator strains would help determine whether the NLR genes composing the 2 PM-associated loci exhibit a strain-specific gene expression and/or transcriptional modulation. Finally, the approach used in this study could be applied to other R loci to discover haplotype-specific markers for breeding PM-resistant cultivars.
Data availability
Sequencing data are accessible through NCBI under the BioProject ID PRJNA780568. New genome assembly of M. rotundifolia Trayshed and annotation files is available at Zenodo under the DOI:10.5281/zenodo.5703495 and at www.grapegenomics.com.
Supplemental material is available at G3 online.
Funding
This work was partially funded by the American Vineyard Foundation grant #2017–1657, the US Department of Agriculture (USDA)-National Institute of Food and Agriculture (NIFA) Specialty Crop Research Initiative award #2017-51181-26829, the National Science Foundation (NSF) grant #1741627 and partially supported by funds to DC from the Louis P. Martini Endowment in Viticulture.
Conflicts of interest
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}