





Abstract
In addition to known genes, much of the human genome is transcribed into RNA. Chance formation of novel open reading frames (ORFs) can lead to the translation of myriad new proteins. Some of these ORFs may yield advantageous adaptive de novo proteins. However, widespread translation of noncoding DNA can also produce hazardous protein molecules, which can misfold and/or form toxic aggregates. The dynamics of how de novo proteins emerge from potentially toxic raw materials and what influences their long-term survival are unknown. Here, using transcriptomic data from human and five other primates, we generate a set of transcribed human ORFs at six conservation levels to investigate which properties influence the early emergence and long-term retention of these expressed ORFs. As these taxa diverged from each other relatively recently, we present a fine scale view of the evolution of novel sequences over recent evolutionary time. We find that novel human-restricted ORFs are preferentially located on GC-rich gene-dense chromosomes, suggesting their retention is linked to pre-existing genes. Sequence properties such as intrinsic structural disorder and aggregation propensity—which have been proposed to play a role in survival of de novo genes—remain unchanged over time. Even very young sequences code for proteins with low aggregation propensities, suggesting that genomic regions with many novel transcribed ORFs are concomitantly less likely to produce ORFs which code for harmful toxic proteins. Our data indicate that the survival of these novel ORFs is largely stochastic rather than shaped by selection.
Although de novo emerged proteins have been identified in numerous organism, how they evolve and transition from chance transcriptional events to fully fledged proteins is little understood. Here we show that over the short time scale of primate evolution, the sequence properties (such as protein disorder and aggregation propensity) of expressed human open reading frames change little. This suggests that the retention of de novo gene precursors in the genome is primarily a stochastic process and not driven by selection on structural properties.
Introduction
Taxon-restricted genes have been discovered in numerous clades, including animals (Begun et al. 2007; Knowles and McLysaght 2009; Wissler et al. 2013; Palmieri et al. 2014; Prabh and Rödelsperger 2016), fungi (Carvunis et al. 2012; Vakirlis et al. 2018), and plants (Campbell et al. 2007; Zhang et al. 2019). Many of these genes are thought to have evolved de novo from ancestrally noncoding DNA rather than from the duplication and divergence of pre-existing protein-coding genes (Schmitz and Bornberg-Bauer 2017; Van Oss and Carvunis 2019; Vakirlis et al. 2020). Despite their recent origin, de novo proteins can acquire important biological roles, including adaptations to novel environments, and may even become essential components of existing cellular processes or physiological systems (Gubala et al. 2017; Baalsrud et al. 2018; Xie et al. 2019). In humans, de novo proteins have functional roles in the brain (Wu et al. 2011), as well as involvement in diseases, such as cancer (Samusik et al. 2013; Papamichos et al. 2015; Guerzoni and McLysaght 2016).
Analyses of the human genome and annotated protein-coding genes suggest that several dozen human-proteins arose de novo and indicate that de novo proteins are added to the genome at a slow and stable rate (Knowles and McLysaght 2009; Wu et al. 2011; Guerzoni and McLysaght 2016). However, transcriptomic data show the existence of many thousands of human-restricted transcripts (Ruiz-Orera et al. 2015). Species-restricted transcripts, which could serve as the raw material for novel protein-coding or RNA genes, are abundant suggesting that there is a rapid turnover in their creation and loss (Chen et al. 2015; Ruiz-Orera et al. 2015; Neme and Tautz 2016; Schmitz et al. 2018). How species-restricted transcripts transition into protein-coding genes is unclear. At a minimum, transcripts would require the formation of a sufficiently long open reading frame (ORF) in order to be translated into proteins (we use proteins here in a broad sense to refer amino-acid translations of nucleic acids irrespective of folding or functional information). Evidence from ribosome-profiling experiments indicates that many taxon-restricted sequences bind to ribosomes and are translated into proteins which evolve neutrally (Wilson and Masel 2011; Schmitz et al. 2018; Ruiz-Orera et al. 2018). Thus, eukaryotic genomes are likely home to a shifting population of novel transcripts containing ORFs with members regularly being gained and lost. Whether retention of these novel ORFs is driven largely by selection, on specific properties of the proteins, or is predominately stochastic and nonadaptive, is unknown.
Transcriptomics studies show the pervasive transcription of virtually the entire human genome (Clark et al. 2011; Hangauer et al. 2013). However, the extent to which novel ORFs are present in these transcripts, and to what degree they are translated into proteins is uncertain. Translation of these novel, previously noncoding, mRNAs may expose the cell to numerous potentially toxic proteins (Ángyán et al. 2012).
Many transcripts appear to be species- or taxon-restricted, suggesting that there is a high turnover of novel transcripts, either by stochastic loss of neutral sequences or the active purging from the genome of deleterious sequences (Chen et al. 2015; Ruiz-Orera et al. 2015, 2018; Neme and Tautz 2016; Schmitz et al. 2018). Thus, although much of the genome has the potential to produce novel de novo genes distinct from existing protein-coding genes, only a small fraction of this is realized. Whether certain genomic regions are more amenable to the production of new de novo genes, or if their base composition influences the sequence properties of newly emerging de novo genes, is unclear.
As de novo proteins have minimal sequence similarity with pre-existing proteins, they may have radically different sequence properties and structures. Whether newly emerged de novo proteins have specific three-dimensional folds or are predominantly disordered in uncertain (Carvunis et al. 2012; Schmitz and Bornberg-Bauer 2017; Wilson et al. 2017; Vakirlis et al. 2018). Proteins with high levels of intrinsic structural disorder (ISD) lack well-defined three-dimensional structures and are less likely to misfold and form harmful aggregates or plaques (Tretyachenko et al. 2017). ISD level appears to be positively correlated with GC content of the nucleic-acid sequence and high ISD levels may be due to de novo genes emerging from GC-rich regions (Basile et al. 2017; Vakirlis et al. 2018). Indeed, GC-rich mRNA molecules may be more suitable de novo gene precursors as they are more stable and favorably translated into proteins than GC-poor RNA (Chen et al. 2015). How the structural properties of young proteins evolve over short timescales is little understood.
To bridge the gap between species-restricted transcripts and de novo proteins, we used transcriptomic data from human and other primates to focus on the fine-scale evolution of unannotated expressed ORFs. Using these data, we elucidate the rates at which novel ORFs emerge and are retained and infer which properties influence survival of nascent de novo proteins. We further categorize these ORFs based on their proximity to annotated coding-genes to determine if proximity to known genes has an influence on the rate of emergence or sequence properties of novel ORFs. Our results show that novel ORFs which map to coding regions are steadily added to the genome. However, ORFs emerging from intergenic and intronic regions are rapidly gained and lost with few surviving over longer evolutionary timescales. We find little evidence that sequence and structural properties of novel ORFs change as they age over time suggesting that their survival is nonadaptive rather than driven by selection.
Materials and Methods
Transcriptome Assembly
We assembled transcriptomes of six primate species; human (Homo sapiens), bonobo (Pan paniscus), chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), orang-utan (Pongo pygmaeus), and rhesus macaque (Macaca mulatta) and one rodent outgroup (mouse [Mus musculus]) using HISAT2 and StringTie following the protocol of Pertea et al. (2016).
We assembled transcriptomes using data from six tissue types (brain, cerebellum, heart, lung, kidney, and testes) for six of the seven species. For orang-utan only five tissues were used (RNA-seq data for testes was unavailable). Raw reads originally published by Brawand et al. (2011) were downloaded from the NCBI Sequence Read Archive (SRA). Before assembling the transcriptomes, we trimmed the reads of adapters and low quality bases (quality scores <15) using Trimmomatic (Bolger et al. 2014). For details on RNA-seq data and genomic GTF files used see supplementary table S2, Supplementary Material online.
The genomes of the seven species were indexed using HISAT2. All raw reads (after trimming and quality filtering) were then mapped to the corresponding genome using HISAT2 (for details see supplementary tables S3 and S4, Supplementary Material online). The resulting SAM files were converted to BAM files using SAMtools (version 1.6) (Li et al. 2009). The mapped reads were then assembled using StringTie (Pertea et al. 2016). Using StringTie, transcripts from each tissue sample for each species were merged to create a single transcriptome assembly for each of the seven species. For each assembled transcriptome, a FASTA file was created containing all transcript sequences. These FASTA files were used as the source of BLAST queries or BLAST databases.
Identifying Novel Transcribed ORFs and Assigning Conservation Levels
We predicted ORFs using the getorf program in the EMBOSS software suite (Rice et al. 2000). We selected ORFs that began with a start codon and ended with a stop codon. We used a threshold of 30 amino-acids for each ORF, that is, a minimum length of 90 nucleotides per ORF. We searched for ORFs in all six reading frames. To reduce the effects of transcripts with multiple copies and/or splice variants, we filtered out highly similar sequences. For sequences with high similarity (95% identity over 90% sequence length), we selected only the longest sequence.
To determine the approximate age of human ORFs, we used BLAST (Altschul et al. 1990) to search for homologous ORFs in the six other transcriptomes (i.e., bonobo, chimpanzee, gorilla, orang-utan, Rhesus macaque, and mouse) (BLASTp, cutoff of e-3). The divergence times for the six primate species were taken from Perelman et al. (2011). Divergence time for mouse and primates was taken from the “estimated divergence times” from timetree.org (Kumar et al. 2017). Thus, for each predicted human ORF, we could assign an approximate age to it, based on where in the primate phylogeny we identified corresponding homologous sequences. To reduce the risk that lowly expressed genes be incorrectly assigned to younger conservation levels, we filtered out all ORFs with expression <0.5 TPM.
To find annotation statuses of transcribed ORFs, we mapped all human transcribed ORFs to the GTF file (see supplementary table S2, Supplementary Material online) used to assemble the human transcriptomes. We categorized each transcribed human ORF into one of eight different categories based on proximity to annotation features. These were as follows: a minimum of 5 kb away from an annotated gene (class 0), within 5 kb of annotated gene but not overlapping (class 1), within 5 kb of annotated gene but on opposite strand (class 2), overlapping gene on same strand but not exon (class 3), overlapping gene on other strand but not exon (class 4), overlapping exon on same strand but out of frame (class 5), overlapping exon on other strand but out of frame (class 6), and overlapping exon in frame (class 7). For the sake of simplicity, we only show results of the analyses of certain annotation classes. Annotation classes shown are class 0 (corresponding to intergenic ORFs), class 3 corresponding to intronic ORFs on same strand as gene), and classes 5–7 combined and treated as a single new class (corresponding to exon overlap). A simplified representation of annotations statuses of transcribed human ORFs is shown in figure 1a.
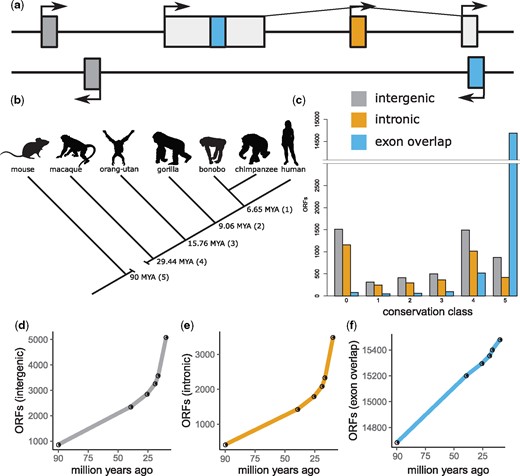
Novel transcribed human open reading frames (ORFs). (a) Cartoon showing annotation status of novel ORFs. Intergenic ORFs (dark gray) are located 5 kb away from annotated genes, intronic ORFs are located within intron of annotated gene, exon overlapping ORFs map to coding regions (either on same strand or other strand as annotated gene). (b) Cladogram of species studied showing divergence times with conservation level in brackets (conservation level 0 corresponds to human-specific ORFs). (c) Counts of ORFs found at each conservation level. (d–f) Rate at which new intergenic, intronic, and exon overlapping ORFs are added to the genome respectively.
Synteny Analysis
As BLAST-based homology detection may miss highly diverged sequences, we verified our findings by predicting homologous transcribed ORFs independent of sequence similarity using synteny information. We followed the methodology used Ruiz-Orera et al. (2015). In short, we predicted all ORFs longer than 30 codons using getorf (Rice et al. 2000). We used the LiftOver tool from USCS Genome Browser (Lee et al. 2020) to convert all ORF coordinates in chimpanzee, gorilla, and orangutan to human genome (GRCh38) coordinates. We then considered ORFs homologous if any human ORF overlapped with an ORF in one of the other species by at least one base. This gave us the proportion of shared ORFs at three conservation levels. We repeated this process using the chimpanzee as the focal species to determine the proportion of chimpanzee ORFs which are shared with the other great apes.
Analyses of ORF Sequence Properties
We predicted and/or calculated properties of each transcribed ORF using several software tools. ISD was predicted using the short disorder predictor setting of the program IUPred (Dosztányi et al. 2005). Aggregation propensity was measured using TANGO (Fernandez-Escamilla et al. 2004; Monsellier et al. 2008). Armomaticity, isoelectric point, codon adaptation index, and hydropathy were measured using EMBOSS (Rice et al. 2000). We predicted transmembrane domains using Phobius (Käll et al. 2004; Käll et al. 2007) and TMHMM (Krogh et al. 2001). For each sequence property measured, we calculated an effect size of the difference between the youngest and oldest ORFs (i.e., human-specific ORFs and human ORFs with homologs in mouse) using Cohen’s d in R (R Core Team 2014) using the package effsize (version 0.7.1) (Torchiano 2018).
Genomic Context
Genomic context of novel transcribed ORFs was determined as follows.
We predicted repetitive elements present in the transcribed ORFs using the program RepeatMasker (Smit et al. 2015). We predicted repetitive elements for each age category but only in the human sequences.
We measured GC content for each ORF (for the human homolog). We also calculated GC content for each human chromosome (genome assembly GRCh 38). We divided the number of human-restricted transcribed ORFs found on each chromosome by the length of the chromosome in million base pairs to calculate the number of human-restricted transcribed ORFs per chromosome per million base pair.
Selection Pressure Analyses
To see if transcribed ORFs were under purifying selection, we calculated dN/dS (ω) values for each human ORF and its chimpanzee homolog. We aligned the translated human ORFs with the homologous sequence from the chimpanzee using Muscle (Edgar 2004). Human-restricted ORFs were not analyzed as, by definition, they lack homologs in other species. Using the amino-acid alignments, we aligned the corresponding nucleotide sequences using pal2nal.pl (Suyama et al. 2006). We used the program codeml from the PAML (version 4.0) suite (Yang 2007) to calculate dN/dS (ω) scores for each alignment using two models: one in which sequences evolved neutrally (m0) and another in which ω could vary (m1a). We used a likelihood ratio test to select between the two models. We adjusted P values using the FDR method to control for multiple testing using the stats package in R (R Core Team 2014). To determine whether older ORFs were more likely to be under purifying selection, we used a χ2 test to compare the number of younger ORFs (conservation levels 1–4) which had ω below a threshold value (0.25 and 0.5) against older sequences (conservation level 5).
Evidence for Translation of Transcribed ORFs
As a proxy for coding probability, we calculated hexamer scores for each ORF using CPAT (Wang et al. 2013). To see if transcribed ORFs bind to ribosomes, we downloaded human ribosome-profiling data from gwips.ucc.ie (see supplementary table S1, Supplementary Material online for data sources). We found the overlap between our transcribed ORFs and ribosome-profiling reads based on their genomic coordinates. ORFs which overlapped with genomic regions which also overlapped with ribosome-profiling reads were assumed to bind to ribosomes. To determine ribosome-release score (RRS), we calculated the ratio of ribosome-profiling reads which overlapped the ORF to those which overlapped the 350 nucleotides following the stop codon of the ORF (Guttman et al. 2013). As the ORFs did not have annotated 3′ UTRs, we used the 350 nucleotides after the ORF stop codon as a proxy 3′ UTR as this is the typical length of eukaryote intronless proteins (Lynch and Marinov 2015).
We also looked for evidence of ORF translation from mass-spectrometry based experiments. First, we downloaded all human small proteins with mass spectrometry data from the SmProt database (Hao et al. 2017). Next, we used BLASTP to search the downloaded peptides using the transcribed primate ORFs as query sequences. Mass spectrometry data from human testes and cell culture were analyzed using PeptideShaker (Vaudel et al. 2015) (see supplementary table S1, Supplementary Material online for data used). Peaks were identified using X! Tandem search tool. Reversed sequences of the transcribed ORFs were used as decoy sequences to detect false positives. Peptides were validated at a 1.0% False Discovery Rate (FDR) which was estimated using the decoy hit distribution. Further identification parameters were as follows: Trypsin as cleavage enzyme, with maximum of two missed cleavages, Fragment Ion Types: b and y, Precursor m/z Tolerance: 10 ppm, Fragment m/z Tolerance: 0.5 Da, Precursor Charge: 2–4, Isotopes: 0–1. We selected all ORFs that had at least one uniquely mapping peptide-spectrum. For details of ORFs evidence of translation from mass spectrometry experiments see supplementary table S8, Supplementary Material online.
Results
Using transcriptomic data from six primate species, we identified 29,751 transcribed human ORFs arising from intergenic, intronic, and exonic regions of the human genome (fig. 1 and supplementary table S5, Supplementary Material online). We used BLAST-based similarity searches to infer homologous ORFs transcribed in other primate transcriptomes and thus, get approximate ages and conservation levels for each ORF. The majority (94.74%) of transcribed ORFs mapping to exons have mouse homologs indicating that they are very ancient (i.e., at least 90 million years old). Transcribed ORFs arising from intergenic or intronic DNA, however, often lacked homologs in older conservation levels and are more likely to be restricted to more recent conservation levels (e.g., 82.9% of intergenic and 88.69% of intronic ORFs are primate-restricted). In total, we found 2,749 human-restricted transcribed ORFs, of which 1,512 are intergenic, 1,158 are intronic, and 79 map to annotated exons. A further 5,378 transcribed ORFs were primate-restricted transcribed ORFs consisting of 2,738 intergenic, 1,923 intronic, and 717 ORFs mapping to exons. We found that ORFs mapping to exons arise at a steady rate. In contrast, intergenic and intronic ORFs emerge at a higher rate than ORFs mapping to exons at more recent timescales but at a lower rate over more distant timescales (fig. 1). Additionally, ORFs arising from intronic regions were almost twice as common as ORFs arising from intergenic regions (9,167 intronic compared with 5,101 intergenic) despite introns accounting for a smaller proportion of the human genome than intergenic DNA (see supplementary table S5, Supplementary Material online for the number of ORFs of each conservation level and annotation class and supplementary fig. S8, Supplementary Material online for the number of ORFs expressed in the same tissue in different species).
To verify that our findings were not due to biases in BLAST-based homology detection (e.g., missing highly diverged homologs, see supplementary fig. S6, Supplementary Material online for sequence identity of inferred homologs), we predicted homology independently of sequence similarity. Using synteny information, we found that both the human and chimpanzee genomes contain a large proportion of species-restricted transcribed ORFS (see fig. 2). As we searched more distantly related genomes, we found fewer shared ORFs. Note, for the synteny-based homology inference, we did not filter our highly similar sequences as we had done in the BLAST-based analysis and, thus, the absolute numbers of shared ORFs differs between the two analyses.
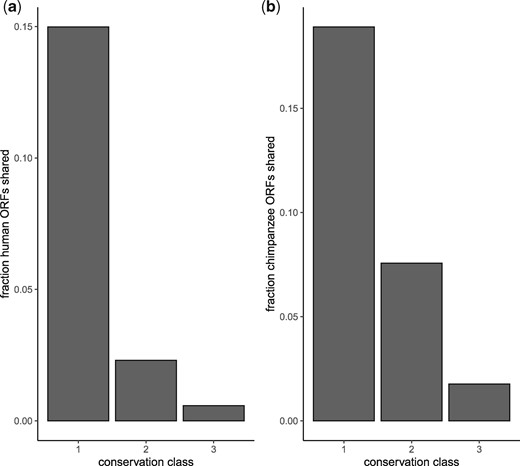
Syntenic transcribed open reading frames (ORFs) in great apes. (a) Fraction of human ORFs with syntenic homologs in other conservation levels. (b) Fraction of chimpanzee ORFs with syntenic homologs in other conservation levels. Conservation levels correspond to those used in figure 1b.
To test if transcribed certain ORFs were more likely to be retained in the genome, we predicted sequence properties (ISD, ORF length, aggregation propensity, GC-content, aromaticity, isoelectric point, hydropathy, codon adaptation index, and presence of transmembrane domains) for all transcribed ORFs at each conservation level (fig. 3, supplementary figs. S9–S11, Supplementary Material online). For intergenic and intronic ORFs, we found little change in ISD, aggregation propensity, and GC-content, aromaticity, isoelectric point, hydropathy, codon adaptation index of, and presence of transmembrane domains in ORFs over time. The length of ORFs, however, did increase with time. Exonic ORFs did change over time slightly in terms of ISD, GC-content, codon adaptation index, isoelectric point, and length (see supplementary table S6 and fig. S10, Supplementary Material online).
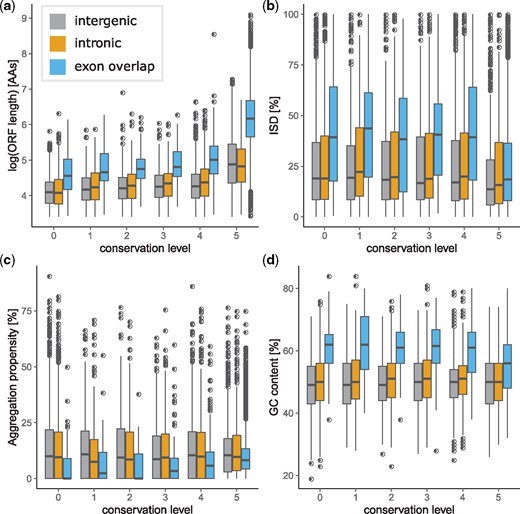
Sequence properties of transcribed human open reading frames (ORFs). (a) Log ORF length. (b) Intrinsic structural disorder (ISD). (c) Aggregation propensity. (d) GC-content.
GC content has an influence on ISD of young proteins (Basile et al. 2017). To determine whether ISD of our candidate genes were dependent on GC content of the sequence, we compared GC content of each ORF with its predicted ISD. ISD was positively correlated with each GC content for ORFs of each annotation class but was stronger for intergenic and intronic ORFs (fig. 4 intergenic: R = 0.422, intronic: R = 0.481, Coding: R = 0.288).
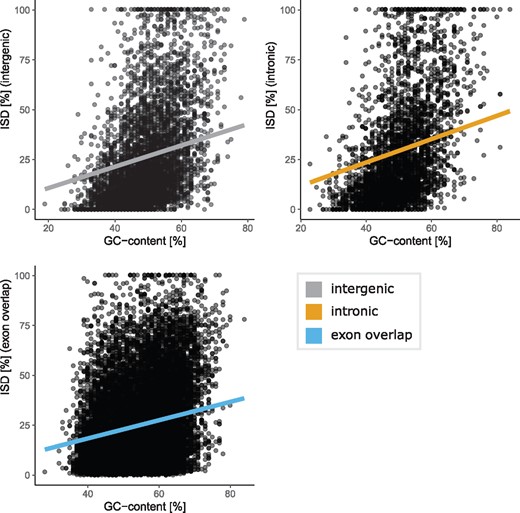
Correlation between GC content of ISD. Top left: intergenic ORFs. Top right: intronic ORFs. Bottom left: ORFs overlapping exons.
Young ORFs (i.e., human-restricted) have GC content intermediate between mean chromosomal GC and old ORFs (i.e., those with homologs in mouse, fig. 5a). Chromosomes with higher GC content contained more human-restricted ORFs than chromosomes with lower GC content when controlling for chromosome length (Rho = 0.84, P = 2.05e–06, Spearman correlation) (fig. 5b).
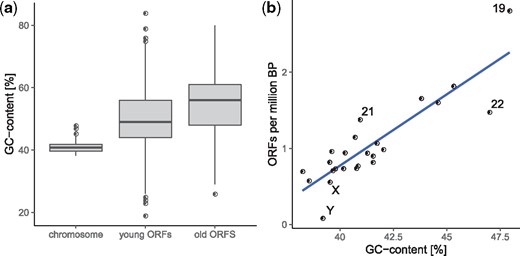
Novel transcribed human open reading frames (ORFs) and chromosomal GC-content. (a) GC-content of chromosomes, young ORFs (conservation class 0), and old ORFs (conservation class 5). (b) GC-content of chromosomes and number of novel ORFs (conservation class 0) per million base pairs.
To determine how primate-restricted ORFs were evolving, we calculated pair-wise ω values for human–chimpanzee homologs (fig. 6). Median ω values of younger conservation classes suggest that these ORFs are evolving neutrally whereas older ORFs (conservation level 5) were evolving under purifying selection. Approximately 31% (2,091/6,778) of primate specific ORFs had ω values below 0.5, 12% (824/6,778) had ω values under 0.25. In contrast, 73% (11,825/16,199) of transcribed ORFs in the oldest conservation level had ω values below 0.5 and 49% (7,958/16,199) were under 0.25. ORFs of the oldest conservation level were significantly more likely to have ω < 0.5 (χ2 = 3392.5632, df = 1, P < .00001) or 0.25 (χ2 = 2647.5017, df = 1, P < .00001). For further details of χ2 tests performed see supplementary table S7, Supplementary Material online. We did not find ω to be correlated with the number of tissues each ORF was expressed in (supplementary fig. S7, Supplementary Material online) and most ORFs share similar tissue expression (supplementary fig. S8, Supplementary Material online).
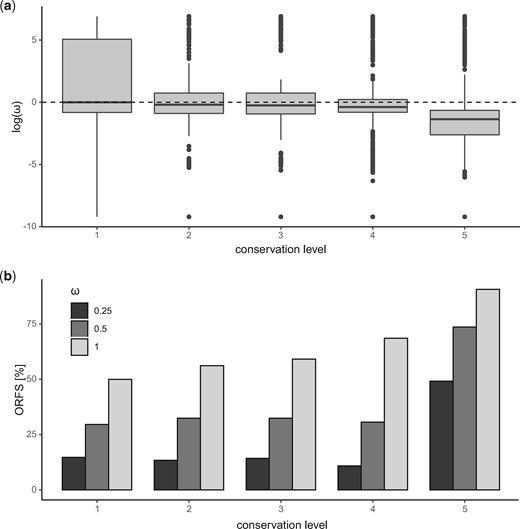
dN/dS omega (ω) values of transcribed human–chimpanzee homologous open reading frames (ORFs). (a) Log transformed scores. (b) Proportion of ORFs with ω below thresholds of 1, 0.5, and 0.25.
We used two methods to infer protein-coding status of transcribed ORFs. First, we calculated RRS for all human ORFs with human ribosome profiling data. RRS above 1 (or 0 in log transformed data as in fig. 7a) are likely to be under active translation as there are more ribosome reads overlapping the ORF compared with the 3′ UTR. RRS scores indicative of translation were found in approximately one quarter of ORFs with ribosome binding evidence. ORFs mapping to annotated exons are more likely to have RRS scores indicative of protein-coding status than intergenic or intronic ORFs (fig. 7a and b). The second method used to identify likely protein-coding sequences was to calculate hexamer scores for each transcribed ORF. Hexamer scores above 0 indicate protein-coding sequences. ORFs which arise from exons are more likely to have hexamer scores above 0 than intergenic or intronic ORFs. Even so, ∼37% (2,812/7,624) of primate restricted ORFs mapping to intergenic or intronic DNA have hexamer scores above 0 indicating that a proportion of these novel intergenic and intronic transcribed ORFs resemble protein-coding sequences (fig. 7c and d). Furthermore, we found evidence from mass spectrometry studies for 70 primate specific proteins by searching the SmProt database (Hao et al. 2017). By reanalyzing mass spectrometry data, we found evidence for at least one peptide from the mass spectrometry data mapping to each of a further five primate-specific ORFs from our data set.
Additionally, we found that many human and primate restricted ORFs overlap with repetitive elements. Alu-elements were especially common with 1,845 (14%) of primate-restricted ORFs containing Alu-elements (see supplementary fig. S1, Supplementary Material online for details).
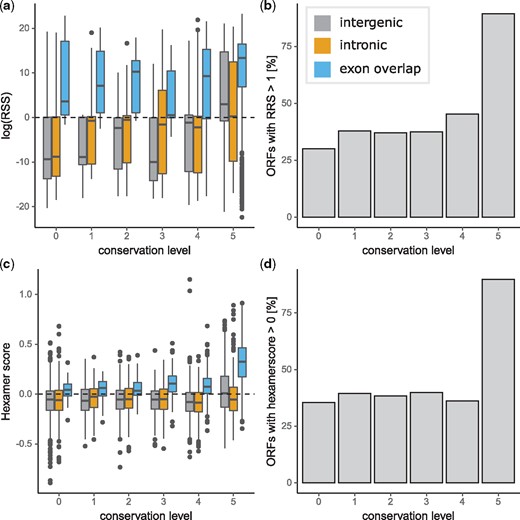
Translation of transcribed human open reading frames (ORFs). (a) Log Ribosome Release Score. Values above 0 indicate coding sequence. (b) Proportion of ORFs with RRS above 1 indicating coding potential. (c) Hexamer score of novel ORFs. Scores above 0 indicate coding sequences. (d) Proportion of sequences with hexamer scores above 0 indicating coding sequences.
Discussion
Widespread transcription and translation of the human genome can lead to the expression of a multitude of potentially beneficial or hazardous novel RNA and protein molecules. Here, we used transcriptomic data from six primate species to investigate the early emergence and transcription of novel ORFs and the properties which contribute to their conservation over time. We found that the rate at which novel transcribed ORFs are recruited into the genome varies with their proximity to annotated protein-coding genes. ORFs which have arisen from intergenic or intronic regions are rapidly gained and lost. However, we find that ORFs which arise from pre-existing exons (e.g., through a frame-shift) are gained at stable rate, confirming previous findings showing that de novo proteins are added to the genome of great apes at a constant rate (Guerzoni and McLysaght 2016). Transcriptomics studies indicate that novel taxon-restricted transcripts, which may or may not have selected biological functions, are rapidly gained and lost (Neme and Tautz 2016). Our results show that many transcribed ORFs are species-restricted (e.g., ∼15% of human ORFs, see fig. 1) and suggest that there is a similar rapid turnover of novel ORFs, with the vast majority not surviving over longer evolutionary times. This suggests that the formation of both novel transcripts and ORFs is common, but their long-term conservation is rare.
We find roughly twice as many intronic ORFs as intergenic ORFs even though introns account for far less of the human genome (see supplementary table S5, Supplementary Material online). This may be due to the higher transcriptional rate of introns compared with intergenic regions and suggests that certain portions of the genome may be more prone to de novo gene emergence than others.
Closely related taxa share more novel sequences than more distantly related taxa (Neme and Tautz 2016). As we used primate taxa with relatively recent divergence times, we were able to identify a large number (∼29%) of novel transcribed ORFs shared between multiple primate taxa (see fig. 1 and supplementary table S5, Supplementary Material online). The relatively high number of ORFs at each conservation level (i.e., >1,000 ORFs) and of each annotation category allowed us to confidently compare changes in sequence properties of novel ORFs over time and infer how they influence the survival of novel ORFs. Several protein properties, such as ISD and aggregation propensity, have been suggested to play a role in the survival of nascent de novo genes (table 1). The interpretation of some trends in the evolution of sequence properties of young sequences may be compounded because of biases inherent to using BLAST-based methods to assign homology (Moyers and Zhang 2018). However, reanalyses of published data suggest that the trends reported are not solely due to biases of using BLAST (Domazet-Lošo et al. 2017). In our study, we used closely related taxa to minimize BLAST biases (Moyers and Zhang 2018; Vakirlis et al. 2020), as well as evaluating our finding on a subset of our data not likely to be error-prone (see supplementary figs. S2–S4, Supplementary Material online). We find that predicted ISD and aggregation propensity of intergenic and intronic ORFs do not change significantly over time, in agreement with previous findings in mouse and other mammals (Schmitz et al. 2018). Young ORFs which map to exons do appear to have slightly elevated ISD levels compared with older exonic ORFs. This observation may be because the majority of these young ORFs map to alternative reading frames of GC-rich sequences, which, due to their higher GC-content, have higher ISD (Casola 2018).
Comparison of Sequence Properties Between Youngest and Oldest ORFs Investigated in this and Other Studies
| Study | Focal Species | GC Content | ISD | Aggregation | Length |
|---|---|---|---|---|---|
| Ruiz-Orera et al. (2015) | Human/chimpanzee | Lower | — | — | Shorter |
| Chen et al. (2015) | Human | Higher | — | — | Shorter |
| Schmitz et al. (2018) | Mouse | — | Same | Same | Shorter |
| Xie et al. (2019) | Mouse | — | Lower | — | Shorter |
| This study/intergenic | Human | Same | Same | Same | Shorter |
| This study/intronic | Human | Same | Same | Same | Shorter |
| This study/CDS | Human | Higher | Higher | Lower | Shorter |
| Study | Focal Species | GC Content | ISD | Aggregation | Length |
|---|---|---|---|---|---|
| Ruiz-Orera et al. (2015) | Human/chimpanzee | Lower | — | — | Shorter |
| Chen et al. (2015) | Human | Higher | — | — | Shorter |
| Schmitz et al. (2018) | Mouse | — | Same | Same | Shorter |
| Xie et al. (2019) | Mouse | — | Lower | — | Shorter |
| This study/intergenic | Human | Same | Same | Same | Shorter |
| This study/intronic | Human | Same | Same | Same | Shorter |
| This study/CDS | Human | Higher | Higher | Lower | Shorter |
Comparison of Sequence Properties Between Youngest and Oldest ORFs Investigated in this and Other Studies
| Study | Focal Species | GC Content | ISD | Aggregation | Length |
|---|---|---|---|---|---|
| Ruiz-Orera et al. (2015) | Human/chimpanzee | Lower | — | — | Shorter |
| Chen et al. (2015) | Human | Higher | — | — | Shorter |
| Schmitz et al. (2018) | Mouse | — | Same | Same | Shorter |
| Xie et al. (2019) | Mouse | — | Lower | — | Shorter |
| This study/intergenic | Human | Same | Same | Same | Shorter |
| This study/intronic | Human | Same | Same | Same | Shorter |
| This study/CDS | Human | Higher | Higher | Lower | Shorter |
| Study | Focal Species | GC Content | ISD | Aggregation | Length |
|---|---|---|---|---|---|
| Ruiz-Orera et al. (2015) | Human/chimpanzee | Lower | — | — | Shorter |
| Chen et al. (2015) | Human | Higher | — | — | Shorter |
| Schmitz et al. (2018) | Mouse | — | Same | Same | Shorter |
| Xie et al. (2019) | Mouse | — | Lower | — | Shorter |
| This study/intergenic | Human | Same | Same | Same | Shorter |
| This study/intronic | Human | Same | Same | Same | Shorter |
| This study/CDS | Human | Higher | Higher | Lower | Shorter |
Proteins with high levels of ISD have been suggested to be better tolerated in the cell as they are less likely to form harmful aggregations and plaques (Monsellier et al. 2008; Ángyán et al. 2012; Tretyachenko et al. 2017; Wilson et al. 2017). ISD has also been shown to impact the emergence of novel protein extensions in yeast (Kleppe and Bornberg-Bauer 2018). However, studies of yeast transcriptomes showed that younger ORFs have lower rather than higher levels of disorder than older ORFs (Carvunis et al. 2012; Vakirlis et al. 2018), whereas in mouse disorder levels remain unchanged between old and young age classes (Schmitz et al. 2018). Our results show that young ORFs code for proteins with similar levels of ISD as older ORFs. In addition, young ORFs have similar aggregation propensities to older ORFs and are not more likely to form aggregations than pre-existing proteins. Novel ORFs which produce highly deleterious, aggregation prone, proteins are likely to be rapidly purged from the genome and thus, possibly missing from our analyses. Alternatively, regions of the genome predisposed to the emergence and transcription of novel ORFs may also be less likely to produce codons which code for aggregation prone proteins, thus the initial pool of novel ORFs may be more similar to pre-existing proteins than previously anticipated (Basile et al. 2017).
Several studies indicate that younger protein-coding genes have shorter coding sequences and contain fewer introns than older genes, although little is known about the rate of ORF extension or addition of introns (Carvunis et al. 2012; McLysaght and Guerzoni 2015; Villanueva-Cañas et al. 2017; Klasberg et al. 2018; Schmitz et al. 2018; Vakirlis et al. 2018). Our results agree with these findings and show that young intergenic, intronic, and exonic ORFs are all shorter than older ORFs in their respective categories. Short ORFs (typically <100 codons in length) code for small protein products which are able to fold correctly more rapidly and efficiently than larger more complex proteins and are therefore less likely to form toxic aggregations (Hartl et al. 2011). Small proteins can also have important biological functions and activities and many conserved microproteins have been found in numerous taxa (Andrews and Rothnagel 2014; Mackowiak et al. 2015; Ruiz-Orera and Albà 2019). Additionally, the extension of an ORF into noncoding DNA can yield novel de novo protein domains (Klasberg et al. 2018).
It seems likely that novel ORFs may remain in the human genome for millions of years before being selected for specific functions. Indeed, data in great apes show the existence of hundreds of novel multi-exon transcripts evolving neutrally, indicating that young de novo may evolve gene-like properties prior to acquiring biological functions (Ruiz-Orera et al. 2015). In our data, which included single exon transcribed ORFs, pairwise ω values show that the majority of primate-restricted ORFs are evolving neutrally. However, over 30% of primate-restricted ORFs (2,091/6,778) with chimpanzee homologs tested have ω values below 0.5 indicating that they are under mild purifying selection and suggesting that they may be translated into functional protein products. We find that a similar proportion of primate-restricted transcribed ORFs have hexamer scores indicative of protein-coding genes and RRS suggesting active translation of the ORF. We found little evidence of translation from mass spectrometry data; however, the data used were not explicitly generated to detect short or lowly expressed proteins. Studies specifically searching for de novo proteins have returned far higher quantities (Zhang et al. 2019). Thus, future studies of de novo proteins may benefit from targeted mass spectrometry experiments aimed at short or lowly expressed proteins.
We find that young ORFs tend to be preferentially located on gene-dense GC-rich chromosomes (fig. 5b and supplementary fig. S5, Supplementary Material online). This may be due to the greater transcriptionally activity in GC-rich regions of the genome compared with GC-poor regions leading to the expression of more ORFs (Versteeg et al. 2003). Moreover, as stop codons are AT-rich, GC-rich DNA is more likely to contain long uninterrupted ORFs. However, Illumina sequencing data (such as those used here) can be biased against reporting especially GC-rich or GC-poor sequences (Benjamini and Speed 2012; Ross et al. 2013). Thus, our analyses may have missed ORFs from regions with extremely high or low GC. Additionally, we filtered out very short ORFs (<30 codons) which are likely to be prevalent in AT-rich regions. Specifically, using transcriptomics data that account for the heterogeneity in GC content of the human genome may be needed to accurately determine whether certain genomic regions are more likely to give rise to novel ORFs.
Our results support previous findings which indicate that each species is host to a myriad of novel transcribed sequences. Fine-scale analyses, using closely related taxa, have allowed for the study of the evolution of protein properties in insects (Heames et al. 2020) and fish (Schmitz et al. 2020) but, until now, not mammals. Our use of recently diverged primate taxa allows us to trace the fine-scale evolution of expressed ORFs over recent time scales in a mammalian order. We find that novel ORFs are frequently formed and transcribed from intergenic and intronic regions. Properties of these transcribed ORFs, such as ISD, aggregation propensity, and proximity to annotated genes; do not change with increasing level suggesting that their chance of survival over time and long term retention are largely stochastic rather than driven by selection.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Acknowledgments
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—281125614/GRK2220. The animal silhouettes used in figure 1b were taken from phylopic.org and are in the public domain except the chimpanzee (by T. Michael Keesy and Thomas Hisget) and orang-utan (Gareth Monger) which are used on CC BY 3.0 license (creativecommons. org/licenses/by/3.0).
Data Availability
The data underlying this article are available in Zenodo at: https://dx.doi.org/10.5281/zenodo.4048343.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}