





Abstract
Elucidation of the rice genome will not only broaden our understanding of genetic characterization of the agronomic characteristics but also facilitate the rice genetic improvement through marker assisted breeding. However, the genome resources of aromatic rice varieties are largely unexploited. Therefore, the whole genome of two elite aromatic traditional japonica rice landraces in North Vietnam, Tam Xoan Bac Ninh (TXBN), and Tam Xoan Hai Hau (TXHH), was sequenced to identify their genome-wide polymorphisms. Overall, we identified over 40,000 novel polymorphisms in each aromatic rice landrace. Although a discontinuous 8-bp deletion and an A/T SNP just upstream the 5-bp deletion in exon 7 of BADH2 gene were present in both rice landraces, the number of SNP high resolution regions of TXBN was six times higher than that of TXHH. Furthermore, several hot spot regions of novel SNPs and indels were found in both genomes, providing their potential gene pools related to aroma formation. The genomic information of two aromatic rice landraces described in this study will facilitate the identification of fragrance-related genes and the genetic improvement of rice.
Introduction
The fragrance of aromatic rice is one of the most valuable grain quality traits that determines the economic value in rice market worldwide. Although the demand for aromatic rice is increasing in global markets (Phing Lau et al. 2016), there is a big gap in the genomic resources and the improvement of aromatic varieties.
The recent discoveries of genes conferring important agronomic traits by using either whole-genome sequencing-based approaches (Yano et al. 2016) or by exploiting traditional rice varieties (Gamuyao et al. 2012) have renewed the interest in genetic resources of traditional cultivars and landraces of rice. Therefore, the genetic characterization of elite aromatic rice cultivars is required for the identification of genes related to aroma formation that has been poorly understood.
Tam Xoan Bac Ninh (TXBN) and Tam Xoan Hai Hau (TXHH) are the two most popular aromatic traditional rice landraces cultivated in the Red River Delta region of North Vietnam. Due to their strong aromatic intensity and excellent cooking and nutritional qualities (Buu 2000), these landraces were granted geographical identification in 2004 (GTZ 2006). Aiming to study the genomic basis of aromatic quality of these landraces, we performed deep whole genome sequencing and analysis of DNA polymorphisms across their entire genomes. We found the islands of novel SNPs and indels with higher density in the chromosomes 8 and 11 in both aromatic rice landraces. We believe that our data provide not only a significant step forward in understanding the genetic determinants of fragrance and its utilization in improving aromatic varieties, but also a welcome resource for data mining by others.
Materials and Methods
Sample Collection
The two traditional aromatic japonica rice landraces, Tam Xoan Bac Ninh (VNPRC_314) and Tam Xoan Hai Hau (VNPRC_1048) were provided by the Plant Resource Center (Hanoi, Vietnam) and originated, respectively, from Bac Ninh Province (106°04′24″E, 21°11′15″N) and Nam Dinh Province (106°16′18″E, 20°7′23″N) in the Red River Delta region of North Vietnam. This region has been recognized as one of the richest genetic diversity centers of Asian cultivated rice (Fukuoka et al. 2003).
Library Preparation and Sequencing
Total DNA of each landrace was extracted from young leaf tissue using Qiagen DNeasy kit (Qiagen, Germany). The library preparation and whole-genome sequencing were performed using Illumina’s paired-end sequencing technology on Hiseq 2000 systems according to Illumina pipeline 1.9 at the Genome Analysis Centre (TGAC), UK. The 100-bp paired-end reads generated from all the genotypes were deposited in the NCBI sequence read archive (SRA) under the SRA study accession number SRP105436.
Read Alignment and Variant Identification
Raw Illumina paired-end reads were filtered for adapter sequences and low quality bases using Trimmomatic (Bolger et al. 2014). The processed reads were aligned to the reference genome for Japonica Nipponbare rice (IRGSP-1.0 pseudomolecule/MSU7) using Burrows-Wheeler Aligner (BWA) V0.7.8 (Li and Durbin 2010). To estimate the genome size, the whole-genome consensus sequences were generated using samtools mpileup and bcftools.
For variant identification, duplicates were removed from alignment files using Picard tools, and variants were called using HaplotypeCaller and VariantFiltration with reference to the Nipponbare reference genome according to the up-to-date version of Genome Analysis Toolkit (GATK) best practices on Whole Genome Sequence (Van der Auwera et al. 2013). High-quality variants were retained for data analyses by filtering out raw calls with following stringent cutoffs: QUAL < 30, QD < 5.0, FS > 60.0, MQ, 40.0, MQRankSum < −12.5, ReadPosRankSum < −8.0 and SOR > 4.0 for SNPs and QD < 5.0, FS > 200.0, ReadPosRankSum < −20.0, and SOR > 10.0 for indels.
To obtain the novel SNPs and indels in the genome of two aromatic rice landraces, all available rice SNPs and indels data were downloaded from the dbSNP database (ftp://ftp.ncbi.nih.gov/snp/organisms/rice_4530/VCF/, 2017-03-26). Novel SNPs and indels were selected using SelectVariants and CombineVariants modules of the GATK.
Variant Analysis
Functional annotation and genetic consequences of filtered variants were done with SnpEff tool V4.3k (Cingolani et al. 2012) using the Nipponbare genome version 7.0 as a reference to annotate SNPs and indels. Genomic distribution of SNPs and indels was calculated in 100 kb nonoverlapping sliding window across all rice chromosomes using VCFtools V0.1.13 (Danecek et al. 2011) and displayed using Circos V0.69-4 (Krzywinski et al. 2009). Transition/transversion (Ts/Tv) rate was calculated in 1Mb window with the options −TsTv of VCFtools.
Results
Whole Genome Sequencing and Mapping
A total of 288 × 106 paired-end reads of 100 bp length were generated for the two aromatic rice landraces. Following low quality (QPhred < 30) and ambiguously mapped read removal, over 76% of reads were aligned to the Nipponbare reference genome. The coverage and the average read depths obtained across all the chromosomes, respectively, were 85.5% and 21.7X for TXBN and 87.8% and 24.8X for TXHH (table 1). The assembled genome size of TXBN and TXHH was estimated to be 312 Mb and 363 Mb, respectively.
Genome Features and Resources of TXBN and TXHH
| Genome Features/Resources | ||
|---|---|---|
| TXBN | TXHH | |
| NCBI bioproject ID | PRJNA384811 | |
| NCBI SRA accession No. | SRP105436 | |
| NCBI biosample ID | SAMN06848499 | SAMN06848500 |
| Total raw reads | 133,528,362 | 154,565,174 |
| Read length (bp) | 100 | 100 |
| Total clean reads | 118,282,878 | 131,916,386 |
| Clean reads percentage (%) | 77.47 | 78.18 |
| Clean reads mapped percentage (%) | 78.29 | 76.89 |
| Mapping depth (X) | 21.67 | 24.85 |
| Reference coverage (%) | 85.81 | 87.76 |
| Estimated genome size (Mb) | 312 | 319 |
| SNP | 1,330,823 | 1,160,772 |
| Intergenic | 1,014,897 | 872,046 |
| Genic | 315,926 | 288,726 |
| Intron | 181,622 | 165,595 |
| UTRs | 68,946 | 61,757 |
| CDS | 65,358 | 61,374 |
| Synonymous | 30,517 | 28,574 |
| Nonsynonymous | 34,841 | 32,800 |
| Indel | 291,206 | 290,883 |
| Intergenic | 223,335 | 223,102 |
| Genic | 67,871 | 67,781 |
| Intron | 48,841 | 48,777 |
| UTRs | 17,683 | 17,639 |
| CDS | 1,347 | 1,365 |
| Genome Features/Resources | ||
|---|---|---|
| TXBN | TXHH | |
| NCBI bioproject ID | PRJNA384811 | |
| NCBI SRA accession No. | SRP105436 | |
| NCBI biosample ID | SAMN06848499 | SAMN06848500 |
| Total raw reads | 133,528,362 | 154,565,174 |
| Read length (bp) | 100 | 100 |
| Total clean reads | 118,282,878 | 131,916,386 |
| Clean reads percentage (%) | 77.47 | 78.18 |
| Clean reads mapped percentage (%) | 78.29 | 76.89 |
| Mapping depth (X) | 21.67 | 24.85 |
| Reference coverage (%) | 85.81 | 87.76 |
| Estimated genome size (Mb) | 312 | 319 |
| SNP | 1,330,823 | 1,160,772 |
| Intergenic | 1,014,897 | 872,046 |
| Genic | 315,926 | 288,726 |
| Intron | 181,622 | 165,595 |
| UTRs | 68,946 | 61,757 |
| CDS | 65,358 | 61,374 |
| Synonymous | 30,517 | 28,574 |
| Nonsynonymous | 34,841 | 32,800 |
| Indel | 291,206 | 290,883 |
| Intergenic | 223,335 | 223,102 |
| Genic | 67,871 | 67,781 |
| Intron | 48,841 | 48,777 |
| UTRs | 17,683 | 17,639 |
| CDS | 1,347 | 1,365 |
Genome Features and Resources of TXBN and TXHH
| Genome Features/Resources | ||
|---|---|---|
| TXBN | TXHH | |
| NCBI bioproject ID | PRJNA384811 | |
| NCBI SRA accession No. | SRP105436 | |
| NCBI biosample ID | SAMN06848499 | SAMN06848500 |
| Total raw reads | 133,528,362 | 154,565,174 |
| Read length (bp) | 100 | 100 |
| Total clean reads | 118,282,878 | 131,916,386 |
| Clean reads percentage (%) | 77.47 | 78.18 |
| Clean reads mapped percentage (%) | 78.29 | 76.89 |
| Mapping depth (X) | 21.67 | 24.85 |
| Reference coverage (%) | 85.81 | 87.76 |
| Estimated genome size (Mb) | 312 | 319 |
| SNP | 1,330,823 | 1,160,772 |
| Intergenic | 1,014,897 | 872,046 |
| Genic | 315,926 | 288,726 |
| Intron | 181,622 | 165,595 |
| UTRs | 68,946 | 61,757 |
| CDS | 65,358 | 61,374 |
| Synonymous | 30,517 | 28,574 |
| Nonsynonymous | 34,841 | 32,800 |
| Indel | 291,206 | 290,883 |
| Intergenic | 223,335 | 223,102 |
| Genic | 67,871 | 67,781 |
| Intron | 48,841 | 48,777 |
| UTRs | 17,683 | 17,639 |
| CDS | 1,347 | 1,365 |
| Genome Features/Resources | ||
|---|---|---|
| TXBN | TXHH | |
| NCBI bioproject ID | PRJNA384811 | |
| NCBI SRA accession No. | SRP105436 | |
| NCBI biosample ID | SAMN06848499 | SAMN06848500 |
| Total raw reads | 133,528,362 | 154,565,174 |
| Read length (bp) | 100 | 100 |
| Total clean reads | 118,282,878 | 131,916,386 |
| Clean reads percentage (%) | 77.47 | 78.18 |
| Clean reads mapped percentage (%) | 78.29 | 76.89 |
| Mapping depth (X) | 21.67 | 24.85 |
| Reference coverage (%) | 85.81 | 87.76 |
| Estimated genome size (Mb) | 312 | 319 |
| SNP | 1,330,823 | 1,160,772 |
| Intergenic | 1,014,897 | 872,046 |
| Genic | 315,926 | 288,726 |
| Intron | 181,622 | 165,595 |
| UTRs | 68,946 | 61,757 |
| CDS | 65,358 | 61,374 |
| Synonymous | 30,517 | 28,574 |
| Nonsynonymous | 34,841 | 32,800 |
| Indel | 291,206 | 290,883 |
| Intergenic | 223,335 | 223,102 |
| Genic | 67,871 | 67,781 |
| Intron | 48,841 | 48,777 |
| UTRs | 17,683 | 17,639 |
| CDS | 1,347 | 1,365 |
Variant Identification and Annotation
Applying stringent filtering criteria, a total of 1,622,029 (1,330,823 SNPs and 291,206 indels) and 1,451,655 (1,160,772 SNPs and 290,883 indels) variants were identified in TXBN and TXHH, respectively (table 1). The genetic signature of aromatic rice, a discontinuous 8-bp deletion and an A/T SNP just upstream the 5-bp deletion in exon 7 of BADH2 gene (Amarawathi et al. 2008), was obtained in both rice landraces. Interestingly, the number of SNP high resolution regions (≥1000 SNPs/100 kb) of TXBN was six times higher than that of TXHH (fig. 1), indicating the higher phylogenomic divergence of TXBN from Japonica rice (McNally et al. 2009). In contrast, there was high similarity in both number and structural annotation of indels between TXBN and TXHH (table 1), suggesting that these differences might arise from the same functional selection pressure. Overall, the DNA variations were mainly distributed in the noncoding regions, probably due to the relaxed selection pressure of these regions in evolution.
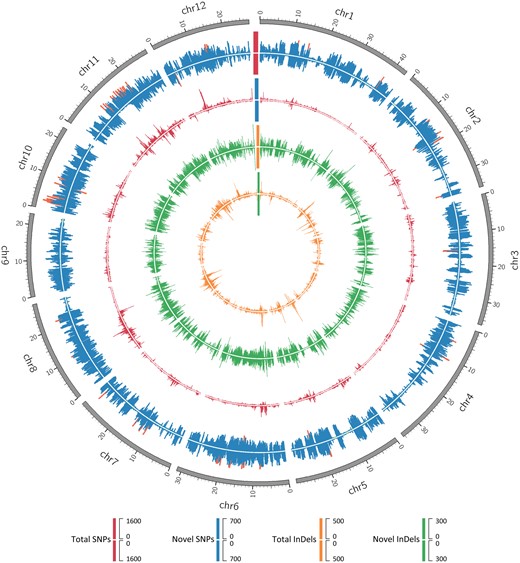
Polymorphism comparisons between TXBN and TXHH rice cultivars. Tracks from inner to outer circles indicate the distribution of total SNP number (blue) with SNP high resolution regions highlighted (orange), unique SNP number (red), indel number (green) and unique indel number (orange) on all 12 rice chromosomes in the 100 kb sliding windows. The chromosome structure in the scale of Mb is in gray. For each track, the outer and inner layers indicate TXBN and TXHH data, respectively.
We observed that the transitional SNPs (C/T and G/A) were more frequent than transversional ones (C/G, T/A, A/C and G/T) in both aromatic rice genomes (supplementary table S1, Supplementary Material online), similar to the findings from previous studies (Huang et al. 2009; McNally et al. 2009; Trinh et al. 2017). Interestingly, the distributions of transition/transversion (Ts/Tv) ratio across the genome of two landraces were extremely similar (supplementary fig. S1, Supplementary Material online), while their SNP density patterns were different (fig. 1). Since the Ts/Tv ratio could be affected by natural selection (Li et al. 1985; Yang and Bielawski 2000), the high similarity of this ratio in TXBN and TXHH suggested that they might have evolved under the same selection pressures.
As of March 2017, a total of 8.2% (40,973 variants) and 7% (42,491 variants) of SNPs and indels in TXBN and TXHH, respectively, were differed from the dbSNP database. These polymorphisms are differential distributed along 12 chromosomes with the present of the hot-spot regions of novel SNPs and indels (fig. 1). Importantly, the chromosomes 8 and 11 of both rice landraces contained higher number and islands of novel variants in comparison to other chromosomes. Importantly, the known SNPs and novel SNPs in both traditional aromatic varieties had the same Ts/Tv ratio of approximately 2.4 (supplementary table S1, Supplementary Material online), indicating that both types of variants could have been subjected to purifying selection for similar evolutionary time-frames. Therefore, further validation and functional studies of the novel polymorphic islands equally distributed in both aromatic landraces may lead to the identification of fragrance-related genes.
Together, the whole genome sequence and variant annotation of two traditional rice landraces harboring strong aroma intensity are of immense value for further studies looking for the genetic determinants of fragrance and genetic improvement of rice.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Author Contributions
X-H.N. and H.T.K. designed research; X-H.N., T.K.N., H.B.T.K., D.K.T., and D.X.T. analyzed data; D.K.T., T.K.N., T.D.N., and H.T.K. performed research; and X.-H.N. wrote the manuscript.
Acknowledgments
We would like to thank all the colleagues to provide significant contributions to the sample collection and Dr Mai-Huong To for insightful comments to the manuscript.
Funding
Authors would like to thank the Ministry of Science and Technology (MOST), Vietnam and the Agricultural Biotechnology Program of Ministry of Agriculture and Rural Development (MARD) to support the projects codes: 26/2014/HD-NDT, 04/First/2a/AGI and 164/HD-KHCN-CNSH. This study is also supported by the BBSRC Newton Fund (BB/N013735/1 to Dr Mario Caccamo).
Literature Cited
Author notes
Associate editor: Howard Ochman
Data deposition: The 100-bp paired-end reads generated from this study has been deposited at the NCBI sequence read archive (SRA) under the accession SRP105436.

{kind=link}