





Abstract
Lycopodiophyta—consisting of three orders, Lycopodiales, Isoetales and Selaginellales, with different types of shoot apical meristems (SAMs)—form the earliest branch among the extant vascular plants. They represent a sister group to all other vascular plants, from which they differ in that their leaves are microphylls—that is, leaves with a single, unbranched vein, emerging from the protostele without a leaf gap—not megaphylls. All leaves represent determinate organs originating on the flanks of indeterminate SAMs. Thus, leaf formation requires the suppression of indeterminacy, that is, of KNOX transcription factors. In seed plants, this is mediated by different groups of transcription factors including ARP and YABBY.
We generated a shoot tip transcriptome of Huperzia selago (Lycopodiales) to examine the genes involved in leaf formation. Our H. selago transcriptome does not contain any ARP homolog, although transcriptomes of Selaginella spp. do. Surprisingly, we discovered a YABBY homolog, although these transcription factors were assumed to have evolved only in seed plants.
The existence of a YABBY homolog in H. selago suggests that YABBY evolved already in the common ancestor of the vascular plants, and subsequently was lost in some lineages like Selaginellales, whereas ARP may have been lost in Lycopodiales. The presence of YABBY in the common ancestor of vascular plants would also support the hypothesis that this common ancestor had a simplex SAM. Furthermore, a comparison of the expression patterns of ARP in shoot tips of Selaginella kraussiana (Harrison CJ, etal. 2005. Independent recruitment of a conserved developmental mechanism during leaf evolution. Nature 434(7032):509–514.) and YABBY in shoot tips of H. selago implies that the development of microphylls, unlike megaphylls, does not seem to depend on the combined activities of ARP and YABBY. Altogether, our data show that Lycopodiophyta are a diverse group; so, in order to understand the role of Lycopodiophyta in evolution, representatives of Lycopodiales, Selaginellales, as well as of Isoetales, have to be examined.
Introduction
All aboveground organs of land plants originate from the shoot apical meristem (SAM). Three main types of SAMs exist (fig. 1). Angiosperms and Gnetopsida have duplex SAMs whose apical initials (AIs) are located in the central part of the two to three outermost cell layers (the tunica) and divide anticlinally to form the epidermis and the outer cortex (Gifford and Foster 1989; Steeves and Sussex 1989). The initials of the layers below the tunica, the corpus, divide in various planes to form the ground and vascular tissues. Therefore, in these plants tunica and corpus are not clonally related (Newman 1965; Philipson 1990; Popham 1951; fig. 1A). Most monilophytes (i.e., ferns s.l.) and some lycophytes (Selaginellales) have monoplex SAMs with a single tetrahedral AI in the superficial cell layer of the apex, which is the ultimate source of all shoot apex cells (Bierhorst 1977; Gifford 1983). Simplex SAMs with few to several AIs in the superficial layer exist in other lycophytes (Lycopodiales and Isoetales), in some monilophytes as well as in most gymnosperms (Ambrose and Vasco 2016; Buvat 1989). Here, the AIs divide both anticlinally and periclinally, thereby producing peripheral and subsurface cells of the SAM, respectively.
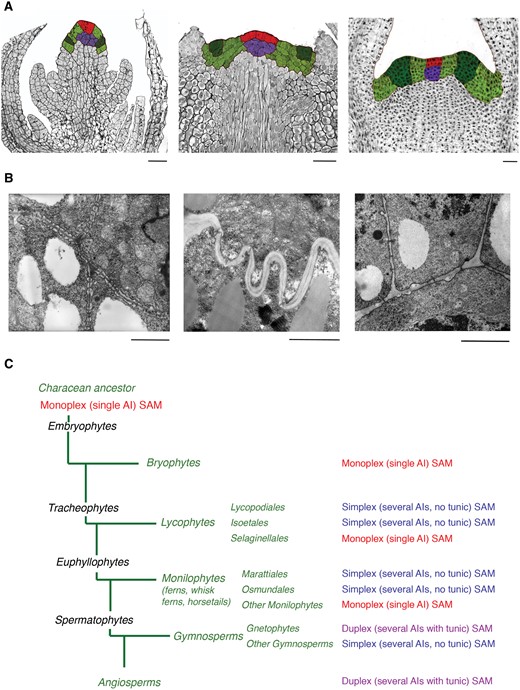
—Shoot apical meristems (SAMs) in land plants. (A) Types of the SAM. From left to right: monoplex (single Apical Initial, AI) SAM of Selaginella kraussiana, simplex (several AIs in the surface layer, that divide both anticlinally and periclinally) of Huperzia selago, duplex (several AIs in two or three outermost layers that divide exclusively anticlinally, also known as tunica-corpus type) in Syringa vulgaris. AIs are shown in red, subapical cells in purple, peripheral cells in light green, and cells of leaf primordia in dark green. (B) Morphology and density of plasmodesmata in different SAM types: numerous unbranched (simple) plasmodesmata in the walls between the apical cell and its immediate derivatives in S. kraussiana; scarce, mainly branched H-shaped and Y-shaped plasmodesmata in the basal walls of the peripheral AIs in H. selago; simple rare plasmodesmata in the walls between cells of layers L1 and L2 of the tunica in the duplex SAM of potato. (C) Distribution of the SAM types throughout the main groups of higher plants (Kenrick and Crane, 1997; Judd etal. 2002): This scheme infers that all SAMs evolved from a plesiomorphic monoplex precursor, with independent origins of simplex SAMs in lycophytes and gymnosperms, respectively. Bar: 50 µm (A); 1 µm (B).
The three types of SAMs differ in origin and distribution of the plasmodesmata (PD) connecting their cells. In plants, PD between cells can be primary or secondary with regard to their origin. Primary PD develop during cytokinesis and connect “sister cells” from the same cell lineage. Secondary PD arise de novo, that is, postcytokinetically, between cells, which belong to different cell lineages. All cells within a monoplex SAM are clonally related and connected exclusively by primary PD (Gunning 1978; Imaichi and Hiratsuka 2007); indeed, a hypothesis has been put forward that the presence of a single AI in plants with monoplex SAMs is a consequence of the inability of the cells to form secondary PD (Cooke etal. 1996). This might explain the presence of a gradient of PD density in monoplex SAMs where PD abundance gradually decreases from the AI towards the distant cells in the apex (Gunning 1978; Imaichi and Hiratsuka 2007). In simplex and duplex SAMs, cells can be connected by both primary and secondary PD, and no PD density gradients have been observed (Imaichi and Hiratsuka 2007). Altogether, the organization of the borders between SAM domains is drastically different in the three types of SAMs (Evkaikina etal. 2014; fig. 1B).
Interestingly, two types of SAMs can be found within the earliest branch of vascular plants, the lycophytes (i.e., Lycopodiophyta; fig. 1C), one of them (simplex) with clear seed plant-like traits such as formation of secondary PD between cells and the presence of multiple AIs. The type of SAM in lycophytes shows no correlation with homo-/heterospory; lycophytes with simplex SAMs include homosporous Lycopodiales and heterosporous Isoetales, whereas the heterosporous Selaginellales have monoplex SAMs. Neither does the type of SAM correlate with leaf type since all members of the Lycopodiophyta are inferred to have the same fundamental leaf type, which contrasts with that of other vascular plants. These leaf types have long been assumed to have evolved independently in the two major groups of vascular plants: microphylls in the Lycopodiophyta and megaphylls in euphyllophytes (monilophytes and seed plants). Microphylls are always simple in shape and contain a single vascular trace, whereas megaphylls develop complex vasculature and variable shape. Although the evolutionary origin of megaphylls as a result of gradual modifications of leafless axes, or telomes of rhyniophytes sensu lato, is widely accepted, the pattern of microphyll origin is still controversial. Three different concepts of their origin are under discussion: concept 1) that microphylls, like megaphylls, evolved by gradual reduction of lateral telomes (Zimmerman 1952); 2) the de novo origin of microphylls as stem tissue outgrowths (enations; e.g., Bower 1908, p. 552–554); and 3) the sterilization theory which sees microphylls as modified sporangia (Kenrick and Crane 1997).
At any rate, however, both microphylls and megaphylls are determinate organs originating on the flanks of indeterminate SAMs (Gifford and Foster 1989). Thus, in all vascular plants leaf formation involves the addition of a determinate growth program to the indeterminate growth program of the SAM. Class I KNOTTED1-like homeobox (KNOX) genes encode the key transcription factors that maintain the cells in an undifferentiated state (Byrne 2012; Kessler and Sinha 2004). They act in a noncell-autonomous manner, moving between cells via PD (Carles and Fletcher 2003; Gaillochet etal. 2015; Holt etal. 2014; Perales and Reddy 2012). The functions of KNOX I proteins are conserved among Selaginellales, ferns and angiosperms (Harrison etal. 2005). Chen etal. (2013, 2014) have suggested that the ability of KNOX I proteins to efficiently traffic through PD was acquired early in the evolution of land plants, that is, already in Bryophyta, which only contain primary PD. Simplex SAMs in Lycopodiales/Isoetales as well as duplex SAMs in angiosperms require secondary PD. There is evidence for differences in trafficking through primary and secondary PD (Ding etal. 1992; Hake and Freeling 1986).
Determinate leaf growth thus requires the repression of KNOX I activity in the progeny of stem cells destined to generate lateral organs. A complex genetic network is responsible for leaf lamina outgrowth and specification of adaxial–abaxial polarity. The most important members of this network are genes encoding MYB homologs (ASYMMETRIC LEAVES1, ROUGH SHEATH2 and PHANTASTICA, collectively called ARP), the adaxial determinants, and the YABBY genes, which were first proposed to represent abaxial determinants, are now thought to play a key role in regulating laminar outgrowth in response to adaxial–abaxial juxtaposition (reviewed by Yamaguchi etal. 2012).
Most studies have been conducted on angiosperms, but the genome sequence of a heterosporous lycophyte, Selaginella moellendorffii (Selaginellales), was published in 2011 (Banks etal. 2011) and has since then played an important role in comparative genomics and improved our understanding of land plant evolution. However, to progress in the field and obtain a better understanding of evolution of SAMs in land plants, more data and better taxonomic coverage are needed, not least from the Lycopodiophyta. We therefore sequenced the shoot tip transcriptome of a homosporous lycophyte, Huperzia selago (Lycopodiales).
Both the telome theory of leaf evolution and the sterilization hypothesis of microphyll evolution can be interpreted to mean a common origin of microphylls and megaphylls and are supported by the data of Harrison etal. (2005) and the gymnosperm transcriptome data of Finet etal. (2016). However, the enation hypothesis of microphyll evolution is supported by the data of Floyd and Bowman (2006) on HD-Zip III genes in Selaginella kraussiana. Similarly, the evolution of the different types of SAMs has not been resolved. Did the duplex SAM of angiosperms evolve from the simplex SAM of the common ancestor of vascular plants, with the monoplex SAMs in Selaginellaceae and monilophytes represent convergent evolution after independent losses? Or do the simplex SAMs of Isoetales/Lycopodiales and gymnosperms, which all contain secondary PD, represent cases of independent convergent evolution from monoplex SAMs, whereas the duplex SAMs of angiosperms evolved later from the simplex SAMs of gymnosperms? In this context, it is interesting that the evolution of angiosperms (with duplex SAMs) involved a large expansion of the KNOX gene family (Gao etal. 2015).
In order to see whether the evolution of simplex meristems in Lycopodiales led to a similar expansion, we analyzed the phylogeny of KNOX proteins in the H. selago transcriptome. As the down-regulation of KNOX I genes represents a key event in differentiation of leaf primordia in all plants, we set about to search for transcripts of the two major KNOX I repressors, ARP and YABBY, in the H. selago transcriptome.
Materials and Methods
Plant Material
All plants of Huperzia selago were collected near Kuznechnoe village at the shore cliffs of Ladoga Lake (Leningrad Region, Russian Federation). If necessary, H. selago plants were cultivated in a greenhouse at 19 °C and a light regimen of 8 h light/16 h dark with ca. 200 µMol photons m−2 sec−1 in pots using Kuznechnoye forest soil; otherwise, they were shock-frozen in a dry ice/EtOH mixture and stored at −80 °C. A voucher specimen prepared from cultivated material that had been harvested on June 16, 2012 has been deposited in the herbarium of the Swedish Museum of Natural History (S), leg. K. Pawlowski s.n. (S; Reg. No. S17-11258). Based on the study by Case (1943) describing the rhythmic pace of H.selago shoot growth and development and on our own observations, plant material for molecular studies was collected on June 11, 2011 (transcriptome), and on May 30, 2016 (RT-PCR), when the plants had completed a zone with sterile leaves and began to form fertile leaves with sporangia. Material for DNA isolations was harvested on July 24, 2016, when the fertile zone had developed further. Plants for shoot tips for in situ hybridization were harvested in September 2016; their uppermost region was sterile, followed by a completed fertile zone. Shoot tips were fixed after these plants had been cultivated in the greenhouse for a week.
For RNA isolation, ca. 4 mm long shoot tips were cut off with a razor; then the leaves were cut off under a stereomicroscope with razor blade, leaving shoot apices with shoot apical meristems and primordia of young leaves and sporangia. The material was divided into several samples of 100 mg fresh weight each, which were ground with mortar and pestle in liquid nitrogen upon addition of 100 mg Polyclar AT (Serva, Mannheim, Germany). The ground samples were transferred into Eppendorf tubes precooled in liquid nitrogen and stored at −80 °C until being used for RNA isolation. For DNA isolation, plant material was shock-frozen in liquid nitrogen and stored at −80 °C. For in situ hybridization, plants with soil were transferred to the laboratory and observed for 1 week to make sure they were still growing; then, shoot tips were fixed.
RNA Isolation and Transcriptome Sequencing
RNA was isolated using TRIzol reagent according to Chomczynski and Mackey (1995). The absence of contaminating DNA was confirmed by direct PCR for ubiquitin (Heidstra etal. 1994). After poly(A) selection, a strand-specific library was prepared using the TruSeq Stranded mRNA Sample Prep Kit (Illumina RS-122-2103; Illumina, San Diego, CA, USA). A bead based RoboCut size selection was performed using the 430 A and 280B buffers, resulting in fragments of ca. 280–430 bp with an average fragment size of ca. 350 bp. The library was sequenced in an Illumina MiSeq v2 flow cell to produce 21.29 million paired end reads of 250 bp (SciLifeLab, Stockholm, Sweden). The raw data were submitted to GenBank (BioProject PRJNA281995).
Transcriptome Assembly
We ran Trimmomatic v0.32 (Bolger etal. 2014) to trim the RNA-Seq reads, with a quality cut-off of 20. With these trimmed reads, we assembled a de novo transcriptome with Trinity r2013-08-14 (Grabherr etal. 2011; Haas etal. 2013), using default parameters. We discarded all the sequences whose length was <300 nucleotides. We then mapped back the same set of trimmed reads onto the Trinity assembly with bowtie (Langmead etal. 2009), and performed abundance estimation using RSEM (Li and Dewey 2011). For each Trinity component, we selected the most abundant transcript as representative sequence. We ended up with a data set of 63,908 transcript sequences.
Transcriptome Analysis
We ran a blastx search of our representative transcriptome against all the plant protein sequences in the nr (nonredundant) database using blast 2.2.29+ (Camacho etal. 2009) and kept the 20 best hits for each transcript. We then used Blast2GO V.2.8.0 (Conesa etal. 2005) to retrieve the corresponding GO IDs associated with these hits. Following the Gene Ontology Consortium guidelines (Gene Ontology Consortium 2001), we removed the GO IDs that should not be used for direct annotations. Five transcripts of KNOX genes were found in the transcriptome (GenBank accession KX761181 - KX761185), as well as one transcript of YABBY (GenBank accession KX761186).
Molecular Cloning
Molecular cloning of YABBY cDNAs from fresh shoot tips of H. selago was performed by PCR amplification. Total RNA was isolated via the method of Chomczynski and Mackey (1995) using TRIzol reagent. First strand cDNA was synthesized using Maxima First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA). Full length H. selago YABBY cDNA was amplified using the gene-specific primers 5′-GAGCGTACTTGCTACAAATCATGTCATCC-3′ and 5′-GGATGAACTTTCTGGTGGAAGGAGTTACA-3′ and 1.25 U of Taq DNA polymerase (Syntol, Moscow, Russia) according to the manufacturer’s instructions. The absence of contaminating DNA was confirmed by direct PCR for ubiquitin (Heidstra etal. 1994). PCR conditions were: 40 cycles of 95°C for 30 s, 63 °C for 30 s, and 72 °C for 1 min. PCR products were cloned in pJET1.2 (Thermo Scientific) and sequenced. The genomic sequence of YABBY was amplified using H. selago DNA as a template with the same gene-specific primers and 0.4 U of Phusion High-Fidelity DNA Polymerase (Thermo Scientific). DNA was extracted according to Doyle and Doyle (1990). The PCR protocol was 98 °C for 5 min, followed by 35 cycles of 98 °C for 10 s, 72 °C for 2.5 min. PCR products were cloned in pJET1.2 (Thermo Scientific) and sequenced (GenBank accession MF175244).
In situ RNA–RNA Hybridization and Cytology
Full length H. selago YABBY cDNA in pJET1.2 was used for both sense and antisense probe synthesis. Plasmids containing the cDNA in different orientations were linearized with NcoI (Thermo Scientific) according to the manufacturer’s instructions, followed by extraction with phenol/chloroform 1:1 (v/v). 0.5 µg of each linearized plasmid were used for invitro transcription with T7 RNA polymerase in the presence of DIG-11-UTP according to the manufacturer’s instructions (Roche, Mannheim, Germany) in the presence of RNase inhibitor (Syntol). After invitro transcription, the reaction mix was treated with 1 Unit of DNase (Thermo Scientific) for 15 min at 37 °C, then 2 µl of 200 mM EDTA (pH 8.0) were added, followed by 10 min incubation at 65 °C. Probes were hydrolyzed in 60 mM Na2CO3/40 mM NaHCO3, pH 10.2 for 19 min at 60 °C to obtain fragments of 200–300 nucleotides (Roche). Hydrolysis was stopped using 3 M sodium acetate (pH 6.0), followed by EtOH precipitation. Unincorporated nucleotides were removed using an RNA probe purification kit (E.Z.N.A., Omega bio-tek, Norcross, GA, USA).
Huperzia selago shoot apices were fixed with 4% paraformaldehyde in microtubule-stabilizing buffer (MTSB; 80 mM PIPES pH 6.8, 1 mM MgCl2, 5 mM EGTA plus 0.2% Tween-20 and 0.2% Triton X-100) or in 3% paraformaldehyde in 1/3 MTSB (containing 0.2% Tween-20, 0.2% Triton X-100 and additionally 10% DMSO) overnight at 4 °C after vacuum infiltration. Both treatments yielded the same results; the results depicted in this manuscript are from the fixation in 4% PFA Afterwards, the tissue was dehydrated in a graded ethanol series and embedded in paraffin with 60 °C melting temperature following Karlgren etal. (2009; Arabidopsis embedding version).
In situ hybridizations were performed according to Coen etal. (1990) and Eisel et al. (2008) with some modifications. Results were documented using an Olympus BX51 light microscope (Olympus, Hamburg, Germany) equipped with a Colorview II camera (Olympus).
For the analysis of the leaf structure of H. selago, plant material was fixed in FAA (Formalin-Acetic Acid-Alcohol), dehydrated with a graded ethanol series, embedded in paraffin, cross-sectioned with Accu-Cut SRM 200 Rotary Microtome (Sakura), deparaffinized in xylenes, rehydrated and stained with 0.1% Safranin O solution according to standard plant anatomy protocols (Ruzin 1999).
Plant Evolution: Data, Sequence Alignment, and Phylogenetic Analyses Using Parsimony
Data from the newly produced transcriptome of Huperzia was analyzed along with data from GenBank (accessions are given in supplementary table S1; Supplementary Material online). Sequences of three chloroplast markers were utilized, rbcL, atpB and rps4. Alignment was performed using ClustalX (Chenna etal. 2003) with minor subsequent manual editing. The aligned data matrix comprised 105 taxa representing the major clades of land plants, and a total of 3,359 characters (1,344 from rbcL, 1,398 from atpB, and 613 from rps4). Parsimony analyses were performed in Paup* (Swofford 2002). A starting tree for divergence time analyses (see below) was produced by using the heuristic search option, 100 random sequence additions, TBR branch swapping. Bootstrap analysis was conducted, performing 1,000 bootstrap replicates with ten random sequence additions in each. Trees were rooted using liverworts (Marchantia, Plagiochila, and Pellia) as outgroup, and Euphyllophytes were enforced as monophyletic according to generally accepted views of land plant phylogeny (e.g., Pryer etal. 2001; Qiu etal. 2007; Rydin etal. 2002).
Estimation of Divergence Times of Clades
Phylogeny and absolute divergence times of clades were estimated using a Bayesian approach as implemented in software BEAST v. 1.8 (Drummond etal. 2012). The analyses were set up using software BEAUti v. 1.8 (Drummond etal. 2012). A full description of the methodological approach is given in the Supplementary Material Online (supplementary file S1). Two identical analyses were run, each for 100 million generations, with parameters logged every 10,000 generations. In addition a third analysis was run, identical to the other two but with sampling from priors only. Values sampled for different parameters and convergence of runs were examined and assessed in Tracer v. 1.5 (Rambaut and Drummond 2007). Results were summarized using LogCombiner v. 1.8 and Tree Annotator v. 1.8 (Drummond etal. 2012). Graphic output was produced in FigTree (http://tree.bio.ed.ac.uk/software/figtree/; last accessed June 8, 2014).
Gene Trees
The H. selago transcriptome data set was cleaned and translated with Transdecoder (Haas etal. 2013). It was then merged with the predicted proteomes of Amborella trichopoda (AmbTri), Arabidopsis thaliana (AraTha; Arabidopsis Genome Initiative 2000), Chlamydomonas reinhardtii (ChlRei; Merchant etal. 2007), Oryza sativa (OrySat; Goff etal. 2002), Ostreococcus lucimarinus (OstLuc; Palenik etal. 2007), Physcomitrella patens (PhyPat; Rensing etal. 2008), Populus trichocarpa (PopTri; Tuskan etal. 2006), Selaginella moellendorffii (SelMoe; Banks etal. 2011), Vitis vinifera (VitVin; Jaillon etal. 2007) and Zea mays (ZeaMay; Schnable etal. 2009), downloaded from Phytozome v. 11 (Goodstein etal. 2012); and the genomes of Pinus taeda (PinTae) (v. 1.0, downloaded from http://dendrome.ucdavis.edu/ftp/Genome_Data/genome/pinerefseq/Pita/v1.0/; last accessed August 18, 2017) and Picea abies (PicAbi) (v. 1.0, downloaded from http://congenie.org/start; last accessed August 18, 2017) (Nystedt etal. 2013). We used blastp (v. 2.2.28; Camacho etal. 2009) to find similar sequences; AT1G62360.1 (Arabidopsis STM) was used as the query for KNOX and AT1G08465.1 (Arabidopsis YAB2) as the query for YABBY. To achieve better coverage of the Lycopodiophyta, transcriptome data sets for species that contained both class I and class II KNOX transcripts were obtained from the OneKP database, namely, Lycopodium deuterodensum, Isoetes tegetiformans, Selaginella apoda, and Selaginella kraussiana (Matasci etal. 2014; Wickett etal. 2014).
Protein domain analysis was performed with Pfam-A (Finn etal. 2016) and hmmer (v. 3.1b2) (Mistry etal. 2013). All YABBY orthologs contained the characteristic YABBY domain whereas the most strict requirement for KNOX protein demanded that they are full-length homologs—that they have two KNOX domains, a homeobox and ELK domain (supplementary fig. S1, Supplementary Material online). Another, less strict set of requirements allowed for any sequence with at least one of the two KNOX domains or a homeobox domain (supplementary fig. S1, Supplementary Material online). Protein sequences were aligned with mafft (v7.123b; parameter genafpair, maximum number of cycles of iterative refinement = 1000; Katoh and Standley 2013), columns with >20% gaps were removed with trimAl (version 1.4.rev15; Capella-Gutiérrez etal. 2009) and trees were inferred with RAxML (v. 8.2.4, parameters: rapid bootstrap analysis and search for the best-scoring ML tree in one program run (-f a), 100 bootstraps, protein model JTTIG) (Stamatakis 2014). Protein model was determined using ProtTest 3 (Darriba etal. 2011). Trees were visualized with iTol v3 (Letunic and Bork 2016).
Results
The H. selago Shoot Tip Transcriptome
RNA was isolated from shoot tips of field samples of H. selago. After reduction of one isoform per gene, and removal of contigs <300 bp, the assembly contained 78,760 contigs. When the sequences with an RPKM value of 0 were removed, 63,908 contigs were left. For annotation, blastx searches were run in May 2014. From 63,908 sequences, only 22,212 yielded a hit against NCBI NR (plants) and/or Swissprot. For those contigs that got a hit in the Gene Ontology database with Blast2GO, the associated GO terms for biological processes and molecular functions are shown in figure 2. TransDecoder (v 2.0.1; Haas etal. 2013) was run on the 41,696 transcripts that did not get a blastx hit against NCBI NR plant. Among them, 7,681 sequences have a coding region of at least 75 aa long.
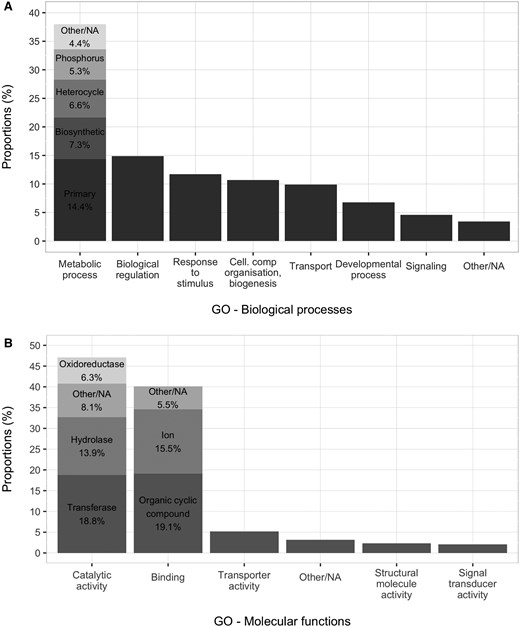
—Gene ontology (GO) mapping results for H. selago contigs. (A) Biological processes, (B) Molecular functions. The chosen GO categories are at the same “high level” in the GO hierarchy. The best represented categories, especially for molecular functions, are similar to those found in a whole plant transcriptome of Selaginella moellendorfii (Weng etal. 2005).
However, only ca. 35% of the assembled cDNAs showed similarity with database sequences, or in other words, the transcriptome contained ca. 65% of orphans. Such a high percentage of orphans is unusual, but not unheard of; a functional transcriptome of the lone star tick had been shown to contain 71% of orphans, a fact ascribed to taxonomic isolation (Gibson etal. 2013). Still, this was a very high number of orphans. Since a field sample had been used for isolating RNA for sequencing, blobtools (version 0.9.19; Kumar etal. 2013) was used to examine the transcriptome for potential contaminants/endophytes. Blobtools was run based on four blast (Version 2.4.0+) searches: a blastn megablast on the NCBI nucleotide database, a blastx on the SwissProt database, a blastn megablast on the SILVA database (RNAcentral SILVA collection) and a blastx search against NR (NCBI). The reads were remapped on the final transcriptome (63,908 contigs). The read_cov plot (supplementary fig. S2; Supplementary Material Online) shows that 19% of the reads mapped to transcripts annotated as “no hits”, whereas 75% of the reads mapped to transcripts annotated as belonging to the Streptophyta clade. Almost no reads mapped to known sequences belonging to other organisms from various clades (bacteria, chordata, ascomycota). The percentage of “no hits” reads is not surprising given the high number of transcripts annotated as orphans. These orphans likely represent lineage-specific genes that are currently absent from the databases used for the searches, rather than unknown contaminants. Thus, a problem of contamination by known organisms can be excluded.
To ascertain the quality of the assembly, the 63,908 contigs were compared with the BUSCO plant set (Simão etal. 2015, at the time only available on request). This set contains single-copy genes from the Embryophyta clade, based on the OrthoDB database of orthologs (http://www.orthodb.org/; last accessed October 31, 2016). Of 956 BUSCO groups, 780 were found complete in the transcriptome (590 groups were found once and 190 more than once). Altogether, 81.5% of the BUSCO groups were complete, 5.3% were fragmented and 13.2% were missing. This compares well with other transcriptomes analyzed using BUSCO (spinach, Xu etal. 2015; Pinus patula, Visser etal. 2015; several sea grasses, Lee etal. 2016).
We next examined the phylogenetic position of H. selago to determine whether it could explain the high percentage of orphans.
Phylogenetic Analysis of the Position of H. selago Relative to Selaginella sp. and to Angiosperms
In order to provide a context of the high amount of orphans in the transcriptome, the evolutionary position of H. selago was examined phylogenetically using Bayesian inference. The topological results (fig. 3) are generally well supported and consistent with accepted views of land plant phylogeny. Effective age prior distributions (as assessed by a prior only analysis) were consistent with those specified (table 1) for all nodes. Huperzia diverged from its closest relative Lycopodium about 93 Ma, from the heterosporous lycopods (Isoetes and Selaginella) 376 Ma, and the most recent common ancestor of Huperzia and the angiosperm Arabidopsis dates to 419 Ma. All node ages and confidence intervals are given in table 1. In summary, the divergence of Selaginellales and Lycopodiales within the lycopods took place in the Late Devonian before the evolution of megaphylls, and Huperzia and Lycopodium diverged during the Late Cretaceous, during the early radiation of angiosperms (Beerling etal. 2001; Pires and Dolan 2012).
Age Priors, and Posterior Distributions and Probabilities for Selected Clades
| Nodes | Prior Source (distribution) | Monophyly Enforced | Prior Ages: Median (95% CI) | Posterior Ages: Median (95% HPD) | Posterior Prob. of Clades | Prior Setting References (see text for details) |
|---|---|---|---|---|---|---|
| Land plants (root) | Fossila (lognormal) | — | 480.2 (472.8–497.1) | 487 (471–532) | 1 | Taylor etal. (2009), Kenrick (2003) |
| Land plants except Marchantiophyta | Tree prior only | yes | — | 477 (446–527) | 1 | — |
| Anthocerophyta and Tracheophyta | Tree prior only | — | — | 445 (427–468) | 0.94 | — |
| Tracheophyta | Fossila (lognormal) | yes | 423.1 (412.4–459.8) | 419 (410–433) | 1 | Kidston and Lang (1920), Hao (1988), Kenrick and Crane (1997) |
| Lycopodiophyta | Tree prior only | — | — | 376 (354–399) | 1 | — |
| Lycopodiaceae | Tree prior only | — | — | 93 (41–181) | 1 | — |
| Selaginellaceae and Isoetaceae | Fossila (lognormal) | yes | 346.1 (335.4–382.8) | 341 (332–355) | 1 | Bateman and DiMichele (1994), Korall etal. (1999), Rowe (1988) |
| Euphyllophyta | Fossila (lognormal) | yes | 401.5 (396.2–419.9) | 402 (395–413) | 1 | Granoff etal. (1976), Kenrick and Crane (1997), Hoffman and Tomescu (2013) |
| Monilophyta | Tree prior only | — | — | 342 (306–373) | 1 | — |
| Spermatophyta | Fossila (lognormal) | yes | 329.6 (319.8–352.2) | 327 (317–343) | 1 | Hilton and Bateman (2006), Doyle (2008), Taylor etal. (2009) |
| Angiospermae | Fossila (lognormal) | yes | 146.3 (138.3–173.9) | 161 (146–181) | 1 | Friis etal. (2011) |
| Eudicots | Fossila (exponential) | yes | 126.7 (126.1–129) | 127 (126–129) | 1 | Friis etal. (2011) |
| Cycadophyta | Tree prior only | — | — | 57 (28–121) | 1 | — |
| Pinaceae | Tree prior only | — | — | 48 (27–78) | 1 | — |
| Gnetales | Tree prior only | — | — | 139 (97–182) | 1 | — |
| Cupressophyta | Tree prior only | — | — | 184 (136–231) | 1 | — |
| Nodes | Prior Source (distribution) | Monophyly Enforced | Prior Ages: Median (95% CI) | Posterior Ages: Median (95% HPD) | Posterior Prob. of Clades | Prior Setting References (see text for details) |
|---|---|---|---|---|---|---|
| Land plants (root) | Fossila (lognormal) | — | 480.2 (472.8–497.1) | 487 (471–532) | 1 | Taylor etal. (2009), Kenrick (2003) |
| Land plants except Marchantiophyta | Tree prior only | yes | — | 477 (446–527) | 1 | — |
| Anthocerophyta and Tracheophyta | Tree prior only | — | — | 445 (427–468) | 0.94 | — |
| Tracheophyta | Fossila (lognormal) | yes | 423.1 (412.4–459.8) | 419 (410–433) | 1 | Kidston and Lang (1920), Hao (1988), Kenrick and Crane (1997) |
| Lycopodiophyta | Tree prior only | — | — | 376 (354–399) | 1 | — |
| Lycopodiaceae | Tree prior only | — | — | 93 (41–181) | 1 | — |
| Selaginellaceae and Isoetaceae | Fossila (lognormal) | yes | 346.1 (335.4–382.8) | 341 (332–355) | 1 | Bateman and DiMichele (1994), Korall etal. (1999), Rowe (1988) |
| Euphyllophyta | Fossila (lognormal) | yes | 401.5 (396.2–419.9) | 402 (395–413) | 1 | Granoff etal. (1976), Kenrick and Crane (1997), Hoffman and Tomescu (2013) |
| Monilophyta | Tree prior only | — | — | 342 (306–373) | 1 | — |
| Spermatophyta | Fossila (lognormal) | yes | 329.6 (319.8–352.2) | 327 (317–343) | 1 | Hilton and Bateman (2006), Doyle (2008), Taylor etal. (2009) |
| Angiospermae | Fossila (lognormal) | yes | 146.3 (138.3–173.9) | 161 (146–181) | 1 | Friis etal. (2011) |
| Eudicots | Fossila (exponential) | yes | 126.7 (126.1–129) | 127 (126–129) | 1 | Friis etal. (2011) |
| Cycadophyta | Tree prior only | — | — | 57 (28–121) | 1 | — |
| Pinaceae | Tree prior only | — | — | 48 (27–78) | 1 | — |
| Gnetales | Tree prior only | — | — | 139 (97–182) | 1 | — |
| Cupressophyta | Tree prior only | — | — | 184 (136–231) | 1 | — |
Absolute ages of fossil strata are inferred based on stratigraphic interpretations in The Geological Time Scale (Gradstein etal. 2012).
Age Priors, and Posterior Distributions and Probabilities for Selected Clades
| Nodes | Prior Source (distribution) | Monophyly Enforced | Prior Ages: Median (95% CI) | Posterior Ages: Median (95% HPD) | Posterior Prob. of Clades | Prior Setting References (see text for details) |
|---|---|---|---|---|---|---|
| Land plants (root) | Fossila (lognormal) | — | 480.2 (472.8–497.1) | 487 (471–532) | 1 | Taylor etal. (2009), Kenrick (2003) |
| Land plants except Marchantiophyta | Tree prior only | yes | — | 477 (446–527) | 1 | — |
| Anthocerophyta and Tracheophyta | Tree prior only | — | — | 445 (427–468) | 0.94 | — |
| Tracheophyta | Fossila (lognormal) | yes | 423.1 (412.4–459.8) | 419 (410–433) | 1 | Kidston and Lang (1920), Hao (1988), Kenrick and Crane (1997) |
| Lycopodiophyta | Tree prior only | — | — | 376 (354–399) | 1 | — |
| Lycopodiaceae | Tree prior only | — | — | 93 (41–181) | 1 | — |
| Selaginellaceae and Isoetaceae | Fossila (lognormal) | yes | 346.1 (335.4–382.8) | 341 (332–355) | 1 | Bateman and DiMichele (1994), Korall etal. (1999), Rowe (1988) |
| Euphyllophyta | Fossila (lognormal) | yes | 401.5 (396.2–419.9) | 402 (395–413) | 1 | Granoff etal. (1976), Kenrick and Crane (1997), Hoffman and Tomescu (2013) |
| Monilophyta | Tree prior only | — | — | 342 (306–373) | 1 | — |
| Spermatophyta | Fossila (lognormal) | yes | 329.6 (319.8–352.2) | 327 (317–343) | 1 | Hilton and Bateman (2006), Doyle (2008), Taylor etal. (2009) |
| Angiospermae | Fossila (lognormal) | yes | 146.3 (138.3–173.9) | 161 (146–181) | 1 | Friis etal. (2011) |
| Eudicots | Fossila (exponential) | yes | 126.7 (126.1–129) | 127 (126–129) | 1 | Friis etal. (2011) |
| Cycadophyta | Tree prior only | — | — | 57 (28–121) | 1 | — |
| Pinaceae | Tree prior only | — | — | 48 (27–78) | 1 | — |
| Gnetales | Tree prior only | — | — | 139 (97–182) | 1 | — |
| Cupressophyta | Tree prior only | — | — | 184 (136–231) | 1 | — |
| Nodes | Prior Source (distribution) | Monophyly Enforced | Prior Ages: Median (95% CI) | Posterior Ages: Median (95% HPD) | Posterior Prob. of Clades | Prior Setting References (see text for details) |
|---|---|---|---|---|---|---|
| Land plants (root) | Fossila (lognormal) | — | 480.2 (472.8–497.1) | 487 (471–532) | 1 | Taylor etal. (2009), Kenrick (2003) |
| Land plants except Marchantiophyta | Tree prior only | yes | — | 477 (446–527) | 1 | — |
| Anthocerophyta and Tracheophyta | Tree prior only | — | — | 445 (427–468) | 0.94 | — |
| Tracheophyta | Fossila (lognormal) | yes | 423.1 (412.4–459.8) | 419 (410–433) | 1 | Kidston and Lang (1920), Hao (1988), Kenrick and Crane (1997) |
| Lycopodiophyta | Tree prior only | — | — | 376 (354–399) | 1 | — |
| Lycopodiaceae | Tree prior only | — | — | 93 (41–181) | 1 | — |
| Selaginellaceae and Isoetaceae | Fossila (lognormal) | yes | 346.1 (335.4–382.8) | 341 (332–355) | 1 | Bateman and DiMichele (1994), Korall etal. (1999), Rowe (1988) |
| Euphyllophyta | Fossila (lognormal) | yes | 401.5 (396.2–419.9) | 402 (395–413) | 1 | Granoff etal. (1976), Kenrick and Crane (1997), Hoffman and Tomescu (2013) |
| Monilophyta | Tree prior only | — | — | 342 (306–373) | 1 | — |
| Spermatophyta | Fossila (lognormal) | yes | 329.6 (319.8–352.2) | 327 (317–343) | 1 | Hilton and Bateman (2006), Doyle (2008), Taylor etal. (2009) |
| Angiospermae | Fossila (lognormal) | yes | 146.3 (138.3–173.9) | 161 (146–181) | 1 | Friis etal. (2011) |
| Eudicots | Fossila (exponential) | yes | 126.7 (126.1–129) | 127 (126–129) | 1 | Friis etal. (2011) |
| Cycadophyta | Tree prior only | — | — | 57 (28–121) | 1 | — |
| Pinaceae | Tree prior only | — | — | 48 (27–78) | 1 | — |
| Gnetales | Tree prior only | — | — | 139 (97–182) | 1 | — |
| Cupressophyta | Tree prior only | — | — | 184 (136–231) | 1 | — |
Absolute ages of fossil strata are inferred based on stratigraphic interpretations in The Geological Time Scale (Gradstein etal. 2012).
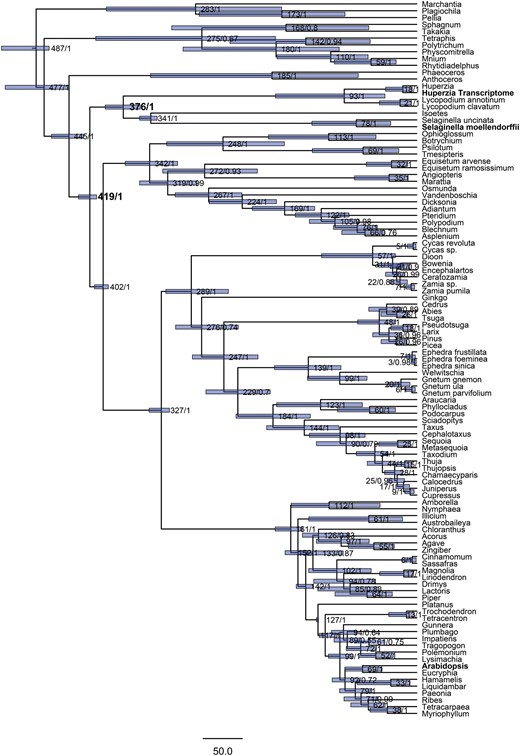
—Dated phylogeny inferred from analysis of three chloroplast genes (atpB, rbcL, and rps4), using a Bayesian framework and a relaxed molecular clock. Values represent node ages (median heights), followed by posterior probabilities of clades. Blue bars illustrate the node height 95% HPD intervals. The most recent common ancestor of Huperzia and Selaginella is estimated to have existed about 376 Ma, and that of Huperzia and Arabidopsis about 419 Ma. Accession numbers of all sequences used in the phylogeny are given in supplementary table S1, Supplementary Material online.
Phylogeny of a Family of Noncell-Autonomous Transcription Factors: KNOX
To trace the evolution of the KNOX protein family in H. selago we inferred a KNOX gene tree with orthologs from a wide range of plant species with sequenced genomes (see Material and methods for an overview). We supplemented these with transcriptome sequences from the OneKP project to more precisely determine the time of duplication events. The gene tree shows a clear distinction between KNOX I and KNOX II genes (fig. 4). In both groups, Lycopodiophyta sequences represent sister groups to the seed plant sequences. No class II KNOX protein was found in the Ostreococcus lucimarinus genome (Palenik etal. 2007) which is consistent with the conclusion of Gao etal. (2015) that algal KNOX proteins have higher similarity with KNOX I.
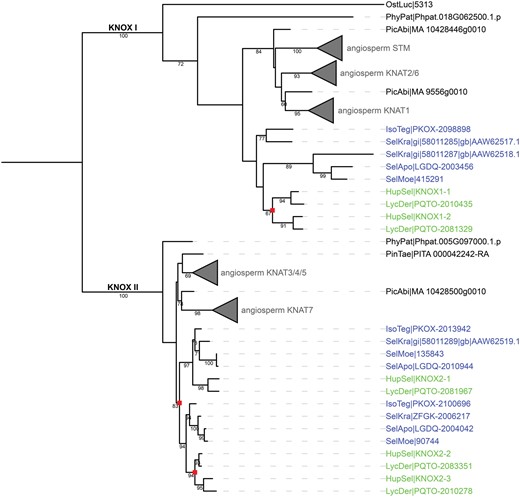
—Gene tree of KNOX proteins. The gene tree of KNOX proteins shows a clear separation of sequences into KNOX I (top branch) and KNOX II (bottom branch) classes, with Lycopodiophyta sequences in both classes. Lycopodiophyta sequences are highlighted; species from class Isoetopsida are shown in blue (Selaginella sp. and Isoetes tegetiformans) whereas sequences from class Lycopodiopsida (Huperzia selago and Lycopodium deuterodensum) are shown in green. Duplication nodes for Lycopodiophyta are marked with red squares. Bootstrap support is only shown for values >60. Angiosperm sequences are collapsed for clarity. Complete trees are shown in supplementary figure S1, Supplementary Material online. All sequences used are listed in supplementary table S1, Supplementary Material online.
The KNOX tree was rooted based on the duplication event that took place before the last common ancestor of the chlorophytes and streptophytes. The transcriptome of H. selago contains five KNOX I and KNOX II genes. Three KNOX gene duplications took place in the ancestor of Lycopodiophyta, one in class I and two in class II (fig. 4; supplementary fig. S1, Supplementary Material online). The duplication in class I and one of the duplications in class II took place within the Lycopodiales, represented in the tree by H. selago and Lycopodium deuterodensum.
The H. selago Transcriptome Does Not Contain an ARP Homolog
No ARP homologs were found in the H. selago transcriptome using the Arabidopsis protein sequence (GenBank accession O80931.1) and tBlastN. To confirm the lack of of ARP homologs in the H. selago transcriptome, we mapped the Huperzia reads on the ARP mRNA sequence from S. kraussiana (GenBank accession number AAW62520.1). Two different mapping programs, bwa-mem (Li and Durbin 2009) and Stampy (Lunter and Goodson 2011), were used, but despite the high conservation of the first 110 amino acids even between S. kraussiana and Arabidopsis (for comparison on the protein and RNA level, see supplementary fig. S1; Supplementary Material online) no reads mapped to the ARP sequence (data not shown). Hence, no ARP gene seems to be expressed in shoot tips of H. selago.
The H. selago Shoot Tip Transcriptome Contains a YABBY Homolog
YABBY transcription factors are thought to be specific to seed plants with one or two YABBY genes present in the last common ancestor of extant seed plants (Finet etal. 2016), although a precursor has been found in a marine alga, Micromonas (Worden etal. 2009). Surprisingly, we were able to identify a YABBY domain protein in the H. selago transcriptome. It has a clear similarity to the YABBY domain as found in Arabidopsis YABBY proteins (fig. 5). The genomic sequence corresponding to the cDNA was amplified and sequenced (GenBank accession MF175244); the intron–exon structure of HsYABBY is similar to that of Arabidopsis YABBY genes (Siegfried etal. 1999; Supplementary fig. S1 and table S1; Supplementary Material online). Phylogenetic analysis showed that HsYABBY is a sister to YABBY proteins from euphyllophytes (supplementary fig. S1 and table S1; Supplementary Material online). These results suggest that the last common ancestor of Lycopodiophyta and Euphyllophyta thus already contained a YABBY-domain protein that was likely lost in Sellaginellaceae.

—Identification of H. selago YABBY. (A) Alignment of H. selago YABBY with angiosperm YABBY proteins. Alignment of H. selago YABBY with Arabidopsis YABBY orthologs. The YABBY domain is marked in green. (B) A schematic tree of YABBY proteins. The full tree is available in supplementary figure S1, Supplementary Material online; all sequences used are listed in supplementary table S1, Supplementary Material online. The angiosperm orthologous groups are collapsed and named after the Arabidopsis gene. H. selago YABBY, belonging to the earliest branching species in the tree, is the outgroup to all YABBY orthologous groups.
HsYABBY Is Expressed in the Surface Cell Layer of the SAM and the Leaf Primordia
In situ RNA–RNA hybridization experiments showed that HsYABBY is expressed in the SAM with the strongest signal in the surface layer where the apical initials (AI) of the simplex SAM type are located, and weaker signal in their subsurface derivatives. However, much weaker or no expression was detected in the AI themselves (fig. 6A and C). In early stages of leaf development, HsYABBY expression could be detected in all cells of the incipient leaf primordia and of sporangia primordia, whereas in older leaf primordia its expression is confined to the surface cells of both the abaxial and adaxial sides (fig. 6A, C, and D). HsYABBY expression could also be detected in the cauline procambium as well as in the leaf trace procambium (fig. 6A).
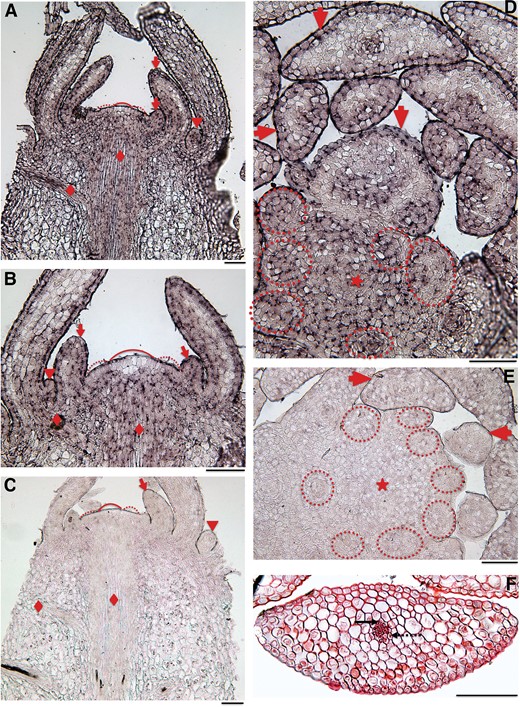
—YABBY expression in shoot tips H. selago analyzed by in situ hybridization. (A, B) Median longitudinal sections through the SAM were hybridized with antisense (A, B) and sense RNA (C). The structure of the H. selago SAM is outlined in figure 1A. HsYABBY mRNA can be detected in the SAM (peripheral parts indicated by curved dashed lines, central part indicated by curved line), with the strongest signal in the outermost cell layer and in the peripheral zone, incipient and young leaf primordia (LPs; arrows), and sporangia primordia (arrowheads) and in the axial and leaf trace procambium (diamonds). Considerably weaker signal is seen in the central cells of the outer layer, the apical initials (AIs, indicated by the curved line) and the subapical cells which are part of the meristem. (D, E) Transverse section of the shoot tip hybridized with antisense (D) and sense (E) RNA of HsYABBY. YABBY expression is uniform in the youngest LPs, whereas in older LPs it is confined to both the abaxial and the adaxial epidermis and to the procambium of the leaf trace. The meristem is marked by an asterisk, incipient LPs are outlined by dashed lines; older LPs are marked with arrows. (F) A transversal section through a H. selago leaf stained with Safranin shows its uniform mesophyll cells and amphicribral vascular bundle. An arrow points to the xylem, a dashed arrow to the phloem. Bars denote 100 µm.
Discussion
The sequencing of the shoot tip transcriptome of Huperzia selago revealed in the first place an unusually high amount of orphan sequences, which could be explained by taxonomic isolation. Our phylogenetic studies revealed that the two branching points in the evolution of Lycopodiophyta could be dated to 376 (Lycopodiales vs. Selaginellales/Isoetales) and 341 Ma (Selaginellales vs. Isoetales), respectively, that is, they preceded the separation between gymnosperms and angiosperms. Therefore, considerable diversity can be expected between Lycopodiophyta from different orders. This assumption is supported by fossil discoveries that document a substantial historical lycophyte diversity, including groups that had gone extinct already by the end of the Paleozoic (e.g., Kenrick and Crane 1997). Lycophytes were a common feature of Earth’s vegetation during the late Paleozoic, the large tree-lycopods of the Isoetales being one of the most well-known examples (see, e.g., Larsén and Rydin 2016 for a brief summary of isoetalean diversity). Thus, the treatment of Selaginella sp. as general model species of the Lycopodiophyta is likely to lead to erroneous conclusions.
Lycopodiales and Isoetales contain simplex SAMs whereas Selaginellales contain monoplex SAMs. Simplex SAMs require the formation of secondary plasmodesmata (PD) whereas monoplex SAMs require only primary PD. One aim of this study was to find out whether the family of class I KNOX genes encoding noncell autonomous transcriptional regulators expanded during the evolution of simplex SAMs. A considerable expansion of KNOX genes took place at the basis of angiosperm evolution (Banks etal. 2011; Gao etal. 2015). In angiosperms, at least two different subgroups of class I KNOX proteins exist, STM (only in eudicots), KNAT1 and KNAT2/6 (Gao etal. 2015; fig. 4; supplementary fig. S1, Supplementary Material online). The H. selago shoot tip transcriptome described in this study contained five KNOX genes, whereas four KNOX genes (two class I and two class II genes) had been found in the Selaginella moellendorfii genome (Gao etal. 2015). Our analysis showed a gene duplication of class I KNOXes in the genomes of Lycopodiales (fig. 4). The situation for Isoetales is not clear—either there was no gene duplication, or not all class I KNOX gene sequences of Isoetes sp. are available, which might be due to the fact that no shoot tip transcriptome was analyzed. No clear conclusion can be drawn with regard to the ability of lycophyte KNOX proteins to traffick through PD (supplementary fig. S1; Supplementary Material online).
Two groups of transcription factors can repress the expression of class I KNOX genes. The group of MYB orthologues collectively called ARP functions in the regulation of the proximal-distal leaf length by epigenetically repressing the expression of class I KNOX genes (Machida etal. 2015). The fact that no ARP transcripts could be found in the H. selago shoot tip transcriptome, might be taken to suggest that ARP, while present in Selaginellales, is missing in Lycopodiales. Conifer homologs of ARP can be found in the shotgun transcriptomes in GenBank using tBlastN (supplementary fig. S1; Supplementary Material online). Thus, although no ARP protein is annotated in GenBank for gymnosperms—which, like Lycopodiales, form simplex meristems—it should be pointed out that the absence of ARP genes, or ARP expression, cannot be a feature of plants with simplex meristems.
Transcription factors of the YABBY family have been shown to be involved in the initiation of the outgrowth of the lamina, the maintenance of polarity, and the establishment of the leaf margin. Like ARP proteins, YABBY can repress the expression of KNOX1 genes (Kumaran etal. 2002). So far, YABBY proteins have been believed to be restricted to Spermatophyta (Finet etal. 2016). In gymnosperms, YABBY proteins are grouped in four clades, whereas five clades are present in angiosperms. Based on phylogeny and expression patterns, Finet etal. (2016) concluded that either one or two YABBY genes were present in the last common ancestor of extant seed plants where they presumably were involved in the determination of polarity connected to the evolution of laminar leaves. Yet, we identified a YABBY homologue in the H. selago shoot transcriptome. Its expression pattern in shoot tips showed major differences with those of angiosperm YABBY genes (see e.g., Siegfried etal. 1999) in that HsYABBY transcripts are localized in the SAM with the strongest signal in the surface layer where the AIs of the simplex SAM are located, although not in the AIs themselves (fig. 6), whereas angiosperm YABBY transcripts are never present in the SAM. However, the expression pattern of HsYABBY showed striking similarity with that of ARP from Selaginella kraussiana (fig. 1J in Harrison etal. 2005), the expression of which in the SAM had been explained by its ancestral function being the control of shoot dichotomy. As Huperzia is characterized by dichotomous branching just like Selaginella (Gola and Jernstedt 2011), HsYABBY might have the same function here.
In incipient leaf primordia, HsYABBY expression was detected in all cells, whereas later its expression was confined to the surface layers on both the abaxial and the adaxial side. Again, this is different from the distinct abaxial expression of YABBY in angiosperms, but corresponds perfectly to the expression pattern of ARP in S. kraussiana (Harrison etal. 2005). A further similarity between the expression pattern of SkARP and HsYABBY is the presence of both mRNAs in the leaf trace procambium. HsYABBY is transcribed also in the cauline procambium which was not shown for SkARP. The expression of both SkARP and HsYABBY in procambium cells, where angiosperm ARP or YABBY genes are not expressed, might be linked to the different mechanisms employed for vascularization in lycophytes versus angiosperms (Floyd and Bowman 2006).
Mature microphylls of both Selaginella and Huperzia lack internal polarization into adaxial palisade and abaxial spongy mesophyll; they are composed of uniform spongy cells (Chu 1974; Harrison etal. 2007; fig. 6E). For H. selago microphylls, there is no difference between the upper and lower epidermis regarding stomata count (Chu 1974). Moreover, vascular bundles of both species are concentric and amphicribral (Smith 1938; fig. 6E). This is also the case for the symmetric “abaxialized” leaves of an Antirrhinum majus ARP (PHAN) loss-of-function mutant (Waites and Hudson 1995). Similarly, a gain-of-function mutant of Arabidopsis that constitutively represses the expression of a member of the YABBY family (fil; McConnell and Barton 1998) has symmetric “adaxialized” leaves with a concentric vascular bundle (reviewed by Bowman etal. 2002). In summary, the resemblance between Selaginella sp. and Huperzia sp. microphylls on the one hand, and angiosperm leaves lacking dorsoventral polarization through combined action of ARP and YABBY on the other hand, suggests that either one of the two suppressors of KNOX expression is sufficient for the switch between determinacy and indeterminacy that results in microphyll development.
In summary, either YABBY evolved convergently in both Lycopodiales and seed plants, or the common ancestor of both must have contained both ARP and YABBY genes, both of which encode transcription factors able to suppress the expression of class I KNOX genes. Given the absence of YABBY in Selaginella and the apparent absence of ARP in Huperzia, and the strong similarity of their expression patterns in shoot tips, we propose that in Lycopodiales, the ARP gene or its expression in shoot apices was lost, while in Selaginellales, the YABBY gene was lost. And if YABBY genes evolved in the common ancestor of vascular plants and are expressed in the primordia of both leaves and sporangia of Huperzia, one possible scenario is that like KNOX1 and ARP genes they were part of a sporangium-specific developmental program that was recruited for leaf development in lycophytes, monilophytes and seed plants (Worsdell 1905; Vasco etal. 2016).
Based on the fact that the SAM-like structure in Chara resembles a monoplex SAM (Graham etal. 2000) and that most seedless land plants analyzed have monoplex SAMs, it has been traditionally assumed that the monoplex SAM is plesiomorphic for land plants (Gifford and Foster 1989; Kenrick and Crane 1997). Kidston and Lang (1920) had observed several initials in the SAMs of Rhyniophytes, but due to poor tissue preservation this was not considered reliable proof of simplex SAM ancestry. However, stronger support of the plesiomorphy of SAMs with several, not single, apical initials came from research of Cooke etal. (1996) and Imaichi and Hiratsuka (2007) on the symplastic structure of SAMs in an evolutionary context. As mentioned earlier, monoplex SAMs (SAMs with a single AI) are characterized by containing exclusively primary PDs, whereas SAMs with several AIs have both primary and secondary PDs. Both groups of authors interpreted this as support for a scenario where the loss of the ability to form secondary PDs led to the development of monoplex SAMs from simplex SAMs. This would have happened independently in microphyllous Selaginella and most euphyllous monilophytes. Furthermore, recent molecular evidence (Frank etal. 2015) supports the idea that the simplex SAM might indeed be ancestral for the sporophytes of terrestrial plants and later was lost in the lycophyte Selaginella and in most monilophytes. The existence of YABBY homologues in Spermatophyta as well as in the shoot tips of H. selago, and therefore probably in the common ancestor of vascular plants, also suggests that it might be more parsimonious to assume that simplex SAMs, and thus also secondary PDs, were present in the common ancestor of vascular plants.
Conclusions
The common ancestor of lycophytes and euphyllophytes is likely to have contained two types of transcription factors that could repress the transcription of class I KNOX genes, ARP as well as YABBY. This supports the new paradigm of leaf evolution proposed by Vasco etal. (2016) that supposes a deep homology of all types of leaves. In due course, YABBY was lost in Selaginellales, whereas our data suggest that ARP may have been lost in Lycopodiales.
The presence of YABBY in the common ancestor of vascular plants would also support the hypothesis that this common ancestor had a simplex SAM and thus also had to have evolved secondary PD. If this was the case, secondary PD in Lycopodiales and seed plants should be homologous, and lycophyte KNOX proteins should be able to traffick through secondary PD.
The fact that analyses of the H. selago transcriptome could change our views on different hypotheses about the ancestral SAM type in land plants shows that more genomic/transcriptomic data from plants from different ancestral lineages have to be studied.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Acknowledgments
This study was supported by the institutional research projects no. 01201255613 and no. 0126-2016-0001 of the Komarov Botanical Institute RAS and by the Russian Foundation for Basic Research (grants #14-04-01397 and #17-04-00837 to M.A.R.). The Core Facility Center “Cell and Molecular Technologies in Plant Science” at the Komarov Botanical Institute RAS and the Research Resource Centre for Molecular and Cell Technologies of Saint-Petersburg State University are gratefully acknowledged for technical support. Bioinformatics support by BILS (Bioinformatics Services for Swedish Life Science) is gratefully acknowledged. We would also like to thank the two anonymous reviewers who improved the manuscript with their constructive criticism.
Literature Cited
Author notes
Associate editor: Susanne S. Renner
These authors contributed equally to this work.
Data deposition: Raw sequence reads used for the de novo transcriptome assembly were deposited at NCBI GenBank (accession PRJNA281995). Huperzia selago phylogenetic markers: KX761189, KX761188, KX761187 KNOX cDNAs: KX761181 - KX761185 YABBY cDNA: KX761186 YABBY genomic: MF175244.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}