





Abstract
Clostridium difficile causes pseudomembranous colitis and is responsible for many cases of nosocomial antibiotic-associated diarrhea. Major virulence factors of C. difficile are the glucosylating exotoxins A and B. Both toxins enter target cells in a pH- dependent manner from endosomes by forming pores. They translocate the N-terminal catalytic domains into the cytosol of host cells and inactivate Rho guanosine triphosphatases by glucosylation. The crystal structure of the catalytic domain of toxin B was solved in a complex with uridine diphosphate, glucose, and manganese ion, exhibiting a folding of type A family glycosyltransferases. Crystallization of fragments of the C-terminus of toxin A, which is characterized by polypeptide repeats, revealed a solenoid-like structure often found in bacterial cell surface proteins. These studies, which provide new insights into structure, uptake, and function of the family of clostridial glucosylating toxins, are reviewed.
Introduction
Clostridium difficile is the major cause of pseudomembranous colitis and has recently emerged as the most frequently identified cause of nosocomial antibiotic-associated diarrhea (AAD) in developed countries (Borriello 1998; Kelly and LaMont 1998; Bartlett 2002). In the United States, annual incidence of AAD are more than three million cases with estimated health care costs exceeding US$1.1 billion (Kyne et al. 2002). C. difficile causes disease by the release of two potent exotoxins, toxin A and toxin B, which induce diarrhea, inflammation, and damage of colonic mucosa (Kelly and LaMont 1998; Voth and Ballard 2005). About 6–10% of C. difficile strains produce besides toxins A and B, a binary actin-adenosine diphosphate (ADP) ribosylating toxin clostridium difficile transferase (CDT) (Perelle et al. 1997). However, the role of this toxin in C. difficile-induced diseases is not clear.
C. difficile toxins A and B belong to the family of large clostridial cytotoxins more appropriately called clostridial glucosylating toxins, because their toxic potency depends on their glucosyltransferase activity (Von Eichel-Streiber et al. 1996; Schirmer and Aktories 2004; Aktories and Barbieri 2005; Aktories and Just 2005; Voth and Ballard 2005). Toxin A consists of 2710 residues (308.0 kDa) and toxin B of 2366 residues (269.6 kDa) (Figure 1). Toxins A and B are encoded by the genes tcdA and tcdB, which are located in a 19.6 kb pathogenicity locus together with three additional genes (tcdC, tcdD, and tcdE) encoding negative (tcdC) and positive (tcdD) regulators as well as a holin-like pore-forming protein (tcdE) (Hammond and Johnson 1995; Hundsberger et al. 1997; Mani and Dupuy 2001). Variations in the structure of the pathogenicity locus (Torres 1991; Rupnik et al. 1997, 1998) are the basis for grouping C. difficile strains in more than 20 toxinotypes. Accordingly, several toxin variants have been reported, especially from toxin B (Rupnik et al. 1998; Chaves-Olarte et al. 1999; Mehlig et al. 2001). Notably, a recently emerging highly virulent C. difficile strain NAP1/027 is characterized by deletion in the tcdC locus by high toxins A and B production and by presence of the binary toxin CDT (McDonald et al. 2005).
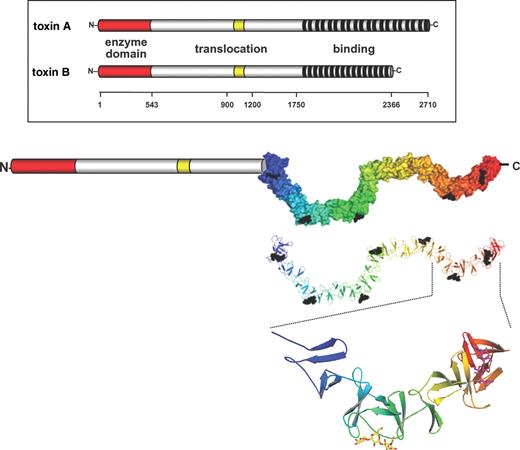
Structure of toxin A and B from C. difficile. Toxins A and B consist of three major domains. The N-terminus harbors the enzyme activity (see Figure 4) and the C-terminus is involved in receptor binding. The middle part is suggested to be involved in the translocation of the toxin into the cytosol. Recently, small parts of the C-terminal polypeptide repeats have been crystallized exhibiting a solenoid structure capable of binding of carbohydrates (Greco et al. 2006). From this crystallized fragment of the C-terminus the entire C-terminus was modeled. Here, the putative structure of the C-terminus is given with multiple carbohydrate units (Galα1-3Galβ1-4GlcNAc carbohydrates) bound. Structure of the C-terminus was modified from (Greco et al. 2006).
Quite early, a tripartite structure of the toxins was proposed comprising a biological active N-terminal domain, a middle part with a small hydrophobic stretch possibly involved in toxin translocation, and a C-terminal receptor binding domain (Von Eichel-Streiber et al. 1992, 1996). This structure model resembles AB toxins, which consist of a biologically active domain, and a binding and translocation domain.
The C-terminal binding domain of toxins A and B
The C-termini of toxins A and B consists of oligopeptide repeats of 21-, 30-, or 50 amino acids residues (Dove et al. 1990; Von Eichel-Streiber and Sauerborn 1990; Von Eichel-Streiber et al. 1992; Moncrief and Wilkins 2000). Because oligopeptide repeats of C. difficile toxins are similar to the carbohydrate-binding regions of glycosyltransferases from Streptococcus mutans, it was suggested that this toxin part is involved in binding to cell surface carbohydrate structures (Von Eichel-Streiber et al. 1992). Recently, the crystal structure of a fragment of this repetitive structure of the C-terminus of toxin A (toxinotype VI), covering residues 2582–2709, has been solved (Ho et al. 2005; Greco et al. 2006). This analysis showed that the C-terminus of toxin A has a solenoid-like structure, which consists of 32 short repeats with 15–21 residues and seven long repeats with 30 residues (Figure 1). The short and long repeats have a β-hairpin structure. In short repeats the β-hairpins are connected by loops of 7–10 amino acid residues. In long repeats 18 residues connect the β-hairpins. Each hairpin has contact to the neighboring hairpin structure but is rotated by 120° resulting in a screw-like structure. Highly similar to these solenoid-like structure of the C-terminal repeats of toxin A is the pneumococcal virulence factor LytA (Fernandez-Tornero et al. 2001). Solenoid structures are often found in bacterial surface proteins. Their extended surface allows protein–protein or protein–carbohydrate interactions. In line with this hypothesis is the idea that the repetitive units of toxin A bind to cell-surface carbohydrate receptor. In fact, toxin A binds to Galα1-3Galβ1-4GlcNAc carbohydrates (Krivan et al. 1986). Also carbohydrate antigens like Lex and Ley (Tucker and Wilkins 1991) components in human milk and carbohydrates from brush border membranes of hamster ileum (Rolfe and Song 1993) have been shown to bind toxin A. However, Teneberg and coworkers (1996) suggested that glycosphingolipids interact with toxin A. Recently, the crystal structure of toxin A was solved in complex with a synthetic carbohydrate consisting of the Galα1-3Galβ1-4GlcNAc structure (Greco et al. 2006). This analysis revealed that the carbohydrates bind to a specific region in the C-terminus of toxin A that is formed by long peptide repeats and the hairpin turn of the following short peptide repeat. However, this carbohydrate structure is most likely not part of the intestinal receptors in man, because they do not have a functional α-galactosyltransferase and, therefore, cannot form α-galactosyl bonds (Koike et al. 2002). Whether other glycans are essential for toxin binding remains to be tested. From the structures available it is feasible that even a weak affinity of a single glycan binding site may increase the binding avidity of the toxin if multivalent binding occurs.
Our knowledge about the cell surface receptor of toxin B receptor is even more scarce. However, experimental data suggest that toxin A and B use different types of receptors. The receptor for toxin B appears to be basolateral located on colon carcinoma T-84 target cells, whereas the receptor for toxin A is on the apical site (Stubbe et al. 2000).
Toxins' uptake
After binding to their receptors, the toxins are endocytosed (Florin and Thelestam 1983; Henriques et al. 1987; Barth et al. 2001) (Figure 2). However, the precise endocytic pathway has not been elucidated. Translocation into the cytosol occurs from an acidic endosomal compartment. This was recently verified by using bafilomycin to block the vesicular H+-adenosine triphophatase (Qa'Dan et al. 2000; Barth et al. 2001). Most likely, the low pH of early endosomes causes a conformational change of the protein and the exposure of hydrophobic regions of the toxins, which are previously masked within the protein (Qa'Dan et al. 2000). The crucial role of the low pH of endosomes can be mimicked by short term decrease of the pH of the cell culture medium (Barth et al. 2001). Thus, even in the presence of bafilomycin, which blocks acidification of endosomes, shift of the pH of the medium allows the uptake of toxin A and B. The uptake of the toxins is paralleled by the formation of pores. This was shown by prior loading cells with 86Rb ions. In the presence of toxin B, decrease of the extracellular pH of 86Rb+ loaded cells caused significant outflow of the radioactive cations (Barth et al. 2001). This effect was specifically blocked by addition of antibody against the toxins. An N-terminal deleted mutant of toxin B, comprising the C-terminus and the middle part of toxin B, was sufficient to induce pore formation (Barth et al. 2001). However, the N-terminus of the toxins, which harbors the catalytic domain, was not able to induce or prevent pore formation. The pore-forming effect of toxin A is largely cell type specific (e.g., pore formation occurs in Caco-2 cells but not in Chinese hamster ovary cells) and depends on the presence of cholesterol (Giesemann et al. 2006). Cholesterol depletion of membranes with methyl-beta-cyclodextrin inhibited 86Rb+ efflux and cholesterol repletion reconstituted pore-forming activity of toxin A (Giesemann et al. 2006). Similarly, in black lipid membranes, toxin A induces ion-permeable pores at low pH only in cholesterol-containing bilayers, whereas toxin B forms pores in the absence of cholesterol (Giesemann et al. 2006).
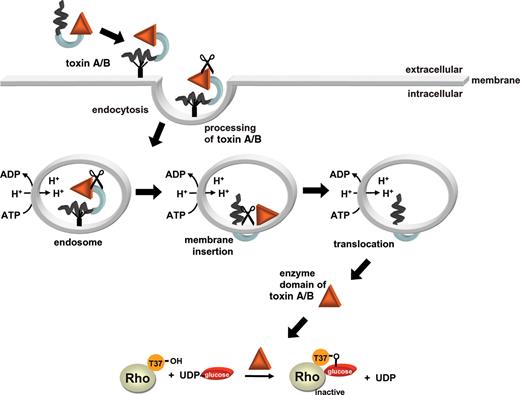
Model of the uptake of C. difficile toxins. Toxins A and B bind to receptors on the surface of target cells and are then endocytosed. Acidification of the toxins in endosomes exposes hydrophobic regions of the protein allowing their insertion into the membrane. The toxins form pores, and the N-terminal catalytic domain is translocated from an acidic endosomal compartment into the cytosol. The location of toxin processing (indicated by scissors) is unknown. In the cytosol, the toxins glucosylate Rho proteins by using UDP-glucose as a cosubstrate.
The precise mechanism of the toxin translocation is still unclear. Recent studies indicate that the toxins have to be processed to reach the cytosol. It is suggested that only the catalytic domain of toxin B is translocated into the cytosol (Pfeifer et al. 2003) (Figure 2). The enzyme domain but not other parts of toxin B is detected in the cytosol after treatment of cells with the holotoxin. Moreover, this cytosolic fraction, which contains glucosyltransferase activity and modifies RhoA, does not cause toxin effects when added to naive cells. Thus, the cytosol contains the catalytic domain but not the binding and translocation domains. Rupnik and coworkers (2005) demonstrated that the first 543 amino acids are cleaved to reach the cytosolic fraction. So far it is not clear, where the cleavage occurs and which processes are involved (Rupnik et al. 2005).
Rho proteins are targets of C. difficile toxins
Toxin targets in the cytosol are Rho proteins. Rho proteins are molecular switches involved in numerous signal processes, including actin cytoskeleton regulation, cell cycle progression, gene transcription, and control of the activity of many enzymes like protein and lipid kinases, phospholipases, and nicotanimide adenine dinucleotide-oxidase (Etienne-Manneville and Hall 2002; Burridge and Wennerberg, 2004). In respect to their role in host–pathogen interactions, Rho proteins essentially participate in epithelial barrier functions and cell–cell contact, in immune cell migration, phagocytosis, cytokine production, wound repair, immune cell signaling, and superoxide anion production (Figure 3). Rho proteins are regulated by a guanosine triphosphatase (GTPase) cycle. They are inactive in the guanosine diphosphate (GDP)-bound form. Activation occurs after GDP/GTP exchange, which is induced by guanine nucleotide exchange factors (GEFs). In the active form, the Rho proteins interact with numerous effectors and adaptors. The active state is terminated by GTP hydrolysis, which is facilitated by GTPase activating proteins (GAPs). In addition, Rho proteins are regulated by guanine nucleotide dissociation inhibitors (GDIs). These proteins keep the inactive form of Rho proteins soluble in the cytosol and appear to be involved in the transport of the Rho proteins to the subcellular compartment, where they are supposed to act.
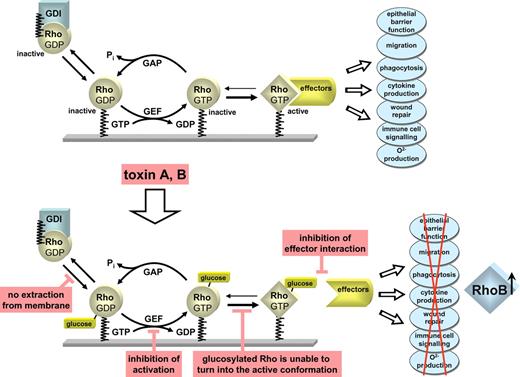
Regulation of Rho GTPases. Rho GTPases are regulated by a GTPase cycle. They are inactive in the GDP-bound form and active with GTP bound form. Three families of proteins regulate the activity state of Rho GTPases. Guanine nucleotide exchange factors (GEFs) cause activation of Rho GTPases. GAPs facilitate the inactivation of Rho proteins by stimulating their GTP hydrolyzing activity. GDIs keep the inactive GDP-bound form of Rho proteins in the cytosol. The active form of Rho interacts with multiple effectors, including protein and lipid kinases, phospholipases, and various adaptor proteins to control various immune and defense functions of target cells, including epithelial barrier function, immune cell signaling, phagocytosis, O2− -production, wound healing, immune cell signaling. C. difficile toxins A and B glucosylate Rho GTPases at Thr35/37. This inhibits effector interaction, blocks activation by GEFs and prevents cytosol membrane cycling. Glucosylated Rho GTPases are no longer able to turn into their active conformation. Moreover, toxins induce increased expression of RhoB.
Substrates of toxins A and B are RhoA, B, C, Rac1-3, RhoG, Cdc42, and TC10 but not RhoE or RhoD. All Rho proteins are modified by toxins A and B at the same amino acid residue, which is Thr37 in the case of RhoA and Thr35 in Rac and Cdc42 (Just, Selzer et al. 1995, Just, Wilm et al. 1995). The related C. sordellii lethal toxin and toxin B isoforms (e.g., toxin B 1470), which have a substrate specificity similar to lethal toxin, modify Rac and Ras proteins but not RhoA, also glucosylate Ras proteins at the equivalent position, namely Thr35 (Just et al. 1995a, 1996; Popoff et al. 1996; Chaves-Olarte et al. 1999). Thr37/35 is highly conserved within the family of small GTPases and essential for nucleotide binding and coordination of the divalent cation magnesium. Moreover, this residue is located in the switch-I region of the GTPase. In this region, major conformational changes occur subsequent to the activation of GDP/GTP exchange (Vetter and Wittinghofer 2001).
Toxin catalyzed modification of Rho and Ras proteins has several functional consequences (Figure 3). First, after glucosylation Rho/Ras proteins are no longer able to interact with their effectors (Herrmann et al. 1998; Sehr et al. 1998). Second, glucosylation inhibits the activation of small GTPases by GEFs (Herrmann et al. 1998; Sehr et al. 1998). Third, glucosylated Rho proteins are associated with the cell membrane and the membrane-cytosol cycle is blocked (Genth et al. 1999). However, the most important structural consequence of glucosylation is probably inhibition of the change into the active conformation of the GTPase. This was shown for glucosylated Ras but so far not for Rho GTPases (Vetter et al. 2000; Geyer et al. 2003). All the above mentioned effects caused by glucosylation of Rho/Ras proteins would inhibit Rho/Ras dependent signaling. A more recent study resulted in a surprising turn in the functional consequences of the action of toxins A and B, because it was observed that the toxins increase the expression of RhoB in target cells (Gerhard et al. 2005). And this effect depends on the glucosyltransferase activity of the toxins. Even more exciting, a large portion of upregulated RhoB remains in the activated state, and is able to escape modification by C. difficile toxins, toxin A or B. This may explain some inflammatory effects of the toxins, which cannot be ascribed solely to Rho-inactivating activity of the glucosyltransferases.
Structure of the catalytic domain
Recently, the crystal structure of the catalytic domain (residues 1–543) of toxin B, which is translocated into the cytosol of target cells, has been solved (Reinert et al. 2005). The overall folding of the catalytic core turned out to be a mixed α/ß-structure with mostly parallel ß-strands as the central part (Figure 4A). Altogether, the protein fragment consists of 11 ß-strands and 21 α-helices. The catalytic core of toxin B consists of 234 residues but possesses more than 300 additional residues. These additional residues are mainly helices. Especially prominent are the 4 N-terminal helices, which appear to be an independent domain. The catalytic core is very similar to other glycosyltransferases like Neisseria meningitidis galatosyltransferase LgtC (Persson et al. 2001), Bacillus subtilis glycosyltransferase SpsA (Charnock and Davies 1999), glycogenin (Gibbons et al. 2002) and bovine galactosyltransferase alpha3GalT (Gastinel et al. 2001), which all belong to type A family of glycosyltransferases (Unligil and Rini 2000; Coutinho et al. 2003; Breton et al. 2006).
![Crystal structure of the catalytic domain of toxin B. (A) The catalytic core, which is similar to that of type-A glycosyltransferases is in blue. Residues, which are specific for clostridial glucosylating toxins, but not found in other glycosyltransferases, are shown in red. UDP-glucose and Mn2+ are shown in yellow and red, respectively [structure is from (Reinert et al., 2005)]. (B) The 286DXD288 motif and the essential tryptophan102 residue are magnified. (C) Function of the DXD motif in coordination of UDP-glucose and manganese ion and of Trp102 in positioning of the uracil ring of UDP is indicated.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/glycob/17/4/10.1093/glycob/cwm004/2/m_cwm00404.jpeg?Expires=1716513050&Signature=y~kzLaQvx9zSOAlRj46CkDDTpIpVtOKiwitCsJsyPkmpYW5A~uxoqbtXiqvfquwRlbW0kiYZTQA579xqTtyYHyiQO0eHa0MRUYkv9jKJrwC9FpX58o4xX~WNKWwKZ4j83e4IM2zMSjhY-8QGaZEKNSA7PN0ybh~RNopsHF3xByOJw6D4oSG3dLTUL9Qz8fpyb0~MmsMGaEWm69OrYOBY9aTM~9S1nbiMvRN9NDKIvtDVZzO~MwSmm61ge-u80KkLLAe181K2RDeyfbVMtyn9kenof8B0QCz9EY5CxB56TA8wXxADSb6qTzH4Rbxhs5flKYyQvGzuzrmOqVLBuSzkCw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Crystal structure of the catalytic domain of toxin B. (A) The catalytic core, which is similar to that of type-A glycosyltransferases is in blue. Residues, which are specific for clostridial glucosylating toxins, but not found in other glycosyltransferases, are shown in red. UDP-glucose and Mn2+ are shown in yellow and red, respectively [structure is from (Reinert et al., 2005)]. (B) The 286DXD288 motif and the essential tryptophan102 residue are magnified. (C) Function of the DXD motif in coordination of UDP-glucose and manganese ion and of Trp102 in positioning of the uracil ring of UDP is indicated.
Although uridine diphosphate (UDP)-glucose, the cosubstrate of clostridial glucosylating toxins, was cleaved during crystallization by the enzyme activity of the catalytic domain, the fact that UDP and glucose were found in the catalytic domain of toxin B allows major insights into the catalytic mechanism and structure–function relationships of the toxin. In addition, Mn2+, which is essential for the enzyme activity, was found in the structure. The catalytic cleft, which binds UDP-glucose is formed by helices α12 and α18, and the connecting loop of β2/α5, β5/β6, β9/β10, and the loop between β11 and α21. Previously, the so-called DXD motif was identified to be typical for many type A glycosyltransferases (Busch et al. 1998; Busch and Aktories 2000). The structure of toxin B reveals that DXD (Asp286 and Asp288) is involved in Mn2+, UDP and glucose complexation (Figure 4B and C). Whereas Asp288 binds directly to manganese, the interaction of Asp286 with the cation occurs via a water molecule. Asp286 also interacts with the 3′-hydroxyl group of the UDP-ribose and with the 3′-hydroxyl group of glucose. Therefore, it has major functions in the tight binding of UDP-glucose in the catalytic cleft of the enzyme. In addition, earlier studies showed that tryptophan 102 is essential for enzyme activity (Busch et al. 2000). This residue stabilizes the uracil ring of UDP by aromatic stacking.
So far the roles of the helical subdomains which surround the catalytic core have not been elucidated. Recently, it was shown that the first 18 N-terminal residues of C. sordellii toxin mediate the preferential binding to phosphatidylserine at the intracellular membrane (Mesmin et al. 2004). This binding was important for the glucosylation of Rac and the biological activity of the toxin. Although toxin B does not share the phosphatidylserine binding site with lethal toxin, the domain-like structure of the N-terminal helices are in line with a putative function of the toxins interaction with membranes.
Catalytic mechanism and cosubstrate specificity
Glycosyltransferases can be classified as either inverting or retaining enzymes depending on conservation of the anomeric configuration of the cosubstrate (UDP-glucose in the case of toxins A and B) (Figure 5). Crystal structure analysis and nuclear magnetic resonance studies of Ras, which was glucosylated by C. sordellii lethal toxin, proposed that the modification of the GTPases by clostridial glucosylating toxins results in an α-anomeric configuration of the attached glucose, indicating that the toxins are retaining glucosyltransferases (Vetter et al. 2000; Geyer et al. 2003). Two possibilities are discussed for the molecular mechanism: a double displacement reaction that involves formation and subsequent breakdown of a covalent intermediate is one possibility (Davies 2001). For this reaction a nucleophile is required, which must be in the vicinity of the C1 position of the glucose to form a β-anomeric intermediate. Subsequently, a general base is responsible for the deprotonation of the reactive hydroxyl group of the protein substrate to allow the second SN2 reaction on the C1 carbon, which then retains the original α-linkage of the glucose. However, in the structure of toxin B no nucleophile that can attack the ß-site of C1 was found in the appropriate position. Therefore, a SNi (internal return) mechanism has been suggested. This mechanism is characterized by a short-lived oxocarbenium-like intermediate. Recent studies with the oxocarbenium intermediate inhibitor gluconolactone support this view (Geyer et al. 2003).
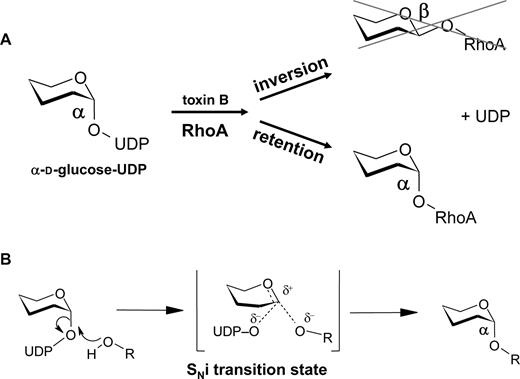
Mechanism of toxin B-catalyzed glucosylation of RhoA. (A) Toxin B is a retaining glucosyltransferase. The α-anomeric configuration of UDP-glucose is retained in the glucose-RhoA linkage after modification of the GTPase. (B) The mechanisms of reaction is most likely an internal return stereospecific SN2-like-mechanism (SNi-mechanism), which depends on an oxocarbenium ion-like transition state.
Various clostridial glucosylating toxins mainly differ in their protein substrate specificity. However, the α-toxin from Clostridium novyi differs in its cosubstrate specificity. Whereas α-toxin accepts UDP-N-acetylglucosamine (UDP-GlcNAc) as a cosubstrate (Selzer et al. 1996), all the other toxins use UDP-glucose. The crystal structure of toxin B was the first hint to explain this difference (Figure 6). Ile-383 and Glu-385 of toxin B are replaced by serine and alanine in α-toxin. These residues appear to limit the space of the catalytic cleft for binding of the cosubstrate. Exchange of these residues also changes the cosubstrate specificities of the toxins. Thus, the toxin B double mutant Ile383Ser and Gln385Ala accepts UDP-GlcNAc as a cosubstrate and causes N-acetylglucosaminylation of RhoA (Jank et al. 2005). Accordingly, the Km value for UDP-GlcNAc of toxin B decreases by about 40-fold. On the other hand, change of Ser385 and Ala387 to isoleucine and glutamine, respectively, turns α-toxin into a UDP-glucose accepting transferase.
Cosubstrate specificity of toxin B. (A) Toxin B from C. difficile, which utilizes UDP-glucose as a sugar donor, has Ile and Gln residues in the catalytic pocket. In contrast, α-toxin from C. novyi, which uses UDP-GlcNAc as a sugar donor, has Ser and Ala residues at the equivalent positions. Therefore, Ile383 and Gln385 in toxin B were changed to Ser and Ala, respectively. In the double mutant the cosubstrate specificity was changed from UDP-glucose to UDP-GlcNAc.
Conclusion
The structural data for the N-terminus of toxin B and the C-terminus of toxin A, which are now available, allow novel insights into the architecture of the catalytic core and the binding domain of the toxins. These data mean a big step forward in understanding the structure–function relationship and the mode of action of the entire family of clostridial glucosylating toxins. Results from crystal structure analyses may have major impact for the identification of compounds, which are capable of inhibiting the catalytic activity of the toxins or the binding of toxins to their target membranes. Those compounds would be of high value for the development of novel therapeutic drugs for treatment of toxin-induced pseudomembranous colitis or antibiotics-associated diarrhea. However, the complete structure of the toxins is not yet available. Most likely, elucidation of the role and function of the enigmatic middle part of the toxins will provide us with new excitements and many surprises. In addition, solid data on the nature of the toxins' receptors are urgently required for further understanding of the mode of action of this important family of protein toxins.
Conflict of interest statement
None declared.
Abbreviations
- AAD
antibiotic-associated diarrhea
- ADP
adenosine diphosphate
- CDT
clostridium difficile transferase
- GAP
GTPase activating proteins
- GDI
guanine nucleotide dissociation inhibitors
- GEF
guanine nucleotide exchange factor
- GTP
guanosine triphosphate
- UDP
uridine diphosphate
- UDP-GlcNAc
UDP-N-acetylglucosamine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}