




Abstract
Mutations in the immunoglobulin mu binding protein-2 ( Ighmbp2 ) gene cause motor neuron disease and dilated cardiomyopathy (DCM) in the neuromuscular degeneration ( nmd ) mouse and spinal muscular atrophy with respiratory distress (SMARD1) in humans. To investigate the role of IGHMBP2 in the pathogenesis of DCM, we generated transgenic mice expressing the full-length Ighmbp2 cDNA specifically in myocytes under the control of the mouse titin promoter. This tissue-specific transgene increased the lifespan of nmd mice up to 8-fold by preventing primary DCM and showed complete functional correction as measured by ECG, echocardiography and plasma creatine kinase-MB. Double-transgenic nmd mice expressing Ighmbp2 both in myocytes and in neurons display correction of both DCM and motor neuron disease, resulting in an essentially wild-type appearance. Additionally, quantitative trait locus (QTL) analysis was undertaken to identify genetic modifier loci responsible for the preservation of cardiac function and a marked delay in the onset of cardiomyopathy in a CAST/EiJ backcross population. Three major CAST-derived cardiac modifiers of nmd were identified on chromosomes 9, 10 and 16, which account for over 26% of the genetic variance and that continue to suppress the exacerbation of cardiomyopathy, otherwise resulting in early death, as incipient B6.CAST congenics. Overall, our results verify the tissue-specific requirement for IGHMBP2 in cardiomyocyte maintenance and survival and describe genetic modifiers that can alter the course of DCM through cardiac functional adaptation and physical remodeling in response to changes in load and respiratory demand.
INTRODUCTION
Cardiomyopathies are morphologically and hemodynamically classified as dilated cardiomyopathy (DCM) (the most common form in humans) and hypertrophic and/or restrictive ( 1 ). Regardless of apparent etiology, however, the ultimate sequela is congestive heart failure (CHF) and premature death. The mutation in the mouse immunoglobulin mu binding protein-2 ( Ighmbp2 ) gene was cloned over 7 years ago, followed by several reports of mutations and polymorphisms in its human counterpart, but so far, its exact biological function is unknown ( 2 – 4 ). We recently reported the tissue-specific rescue of neuromuscular degeneration in nmd mice harboring a full-length Ighmbp2 cDNA driven by the rat neuron-specific enolase ( Eno2 ) promoter, C57BL/6J-Tg( Eno2-Ighmbp2 ), hereafter referred to as TgNI- nmd ( 5 ). This successful rescue of neurogenic atrophy in nmd mice established, for the first time, its cell autonomous nature in motor neuron survival. Likewise, we also described the mapping of a major wild-type CAST-derived modifier locus on mouse chromosome 13 ( Mnm ) that specifically halted motor neuron degeneration ( 2 , 5 ). Despite these genetic interventions, however, the course and progression of DCM and CHF remained. Long-established mechanisms leading to cardiomyopathy include impaired force generation from defective sarcomeres ( 6 ), aberrant intracellular signaling and/or nuclear membrane disorders ( 7 , 8 ), mitochondrionopathy and deficits in cardiac energetics ( 9 , 10 ), facilitated cell death by apoptosis or necrosis ( 11 ) and/or inappropriate cellular response to growth, maturation and differentiation ( 12 , 13 ). IGHMBP2 is novel in that it is a predominantly cytoplasmic member of the DEXDc DEAD-like super family of DNA/RNA helicases possibly involved in RNA transport or translation and also implicated in the regulation of transcription and mRNA splicing ( 2 , 14 ). Therefore, mutant nmd mice, which express ∼20% of normal IGHMBP2 levels due to a splice donor mutation ( 2 ), provide a valuable tool for gaining insight into the mechanisms of neuromuscular degeneration and DCM ( 5 ).
The onset, progression and outcome of cardiomyopathy in humans are highly variable and subject to diverse modifications either intrinsic (genetic) or extrinsic (environmental) or both ( 15 ). Elucidating the etiology of these factors and their roles in the pathogenesis of DCM and CHF is pivotal to the successful clinical management of this complex disease ( 16 ). In a backcross with the wild-type derived CAST/EiJ inbred strain, we observed a large variation in the maximum lifespan of nmd mice with altered onsets and severity of DCM. A genome-wide scan of affected N2 Cardiac modified nmd ( Cmn ) mice using lifespan as the quantitative trait identified three major quantitative trait loci (QTLs) (CAST-derived cardiac modifiers) on mouse chromosomes 9, 10 and 16 that significantly delayed the onset of DCM and heart failure. Two similar studies looking at genetic modifiers affecting survival and cardiac function in a transgenic model of cardiomyopathy reported significant linkage on chromosomes 2 and 3 ( 17 ) and 4 and 18 to survival ( 18 ) as well as QTLs on chromosomes 2, 13 and 18 linked to ‘fractional shortening’ and left ventricular end diastolic dimension (LVEDD) ( 18 ). Although both studies mapped QTLs on chromosome 2, none of these loci were identified in our study, highlighting the complexity and multigenic nature of cardiovascular remodeling in nmd mice. In addressing the cell-autonomous role of Ighmbp2 in the pathogenesis of DCM, we show here that nmd mice expressing a full-length Ighmbp2 cDNA under the control of the muscle-specific mouse titin promoter C57BL/6J-Tg( Ttn-Ighmbp2 ), hereafter referred to as TgMI, are rescued from fatal DCM. Furthermore, despite the presence of progressive neuromuscular degeneration, TgMI- nmd mice lived significantly extended lifespan, confirming the indispensable role of Ighmbp2 in the survival of cardiomyocytes.
RESULTS
Muscle-specific rescue of DCM in nmd mice
As the Ighmbp2 gene is ubiquitously expressed and motor neuron degeneration in nmd mice was rescued by expressing a full-length Ighmbp2 cDNA transgene (TgNI) in neurons ( 5 ), we reasoned that myocytes might similarly have a tissue-specific requirement for IGHMBP. To address the role of Ighmbp2 expression in cardiac and skeletal myocytes, we generated transgenic nmd mice expressing the full-length Ighmbp2 cDNA under the control of the mouse titin ( Ttn ) promoter (TgMI). A schematic representation of the transgenic construct is shown in Figure 1 A. RT–PCR analysis revealed that TgMI-derived Ighmbp2 mRNA expression was limited to the heart and skeletal muscles in two independent transgenic lines (Fig. 1 B) (data not shown). To assess whether the expression of the Ighmbp2 transgene could rescue cardiomyocyte degeneration, B6-TgMI from lines 45 and 108 was independently crossed with the heterozygous B6.BKS- +/Ighmbp2 nmd-2J (hereafter referred to as B6- +/nmd ) mice to produce TgMI- nmd mutant mice. Physical examination and longitudinal evaluation of overall health were compared between TgMI- nmd , double transgenic TgMI+NI- nmd and their sib-controls. In Figure 1 C, the absence of neuron-specific expression of the Ighmbp2 cDNA in both non-transgenic nmd and TgMI- nmd mice led to severe skeletal muscle neurogenic atrophy—precluding the analysis of the potential role of skeletal myocytes in the progression of cardiomyopathy and consequent survival. In contrast, double transgenic TgMI+NI- nmd mice are indistinguishable from age-matched controls, reflecting the combinatorial effects of the transgenes in preventing motor neuron disease and cardiomyopathy. The growth rate for TgMI- nmd mice, as measured by mean bodyweight, is comparable to that for non-transgenic nmd mice from 2 weeks of age onward and is nearly half the mean bodyweight of the double transgenic nmd mice or their sib-controls (Fig. 1 D). Thus, the loss of spinal motor neurons and the resulting skeletal muscle atrophy seen in mutant nmd mice account for the reduced bodyweight in TgMI- nmd mice ( 5 ). Additionally, both male and female transgenic TgMI- nmd mice from lines 45 and 108 displayed equivalent growth rates, average lifespan (data not shown) and performance scores in functional tests; therefore, mice from these two independent lines were combined for all subsequent analyses. Double transgenic nmd mice carrying both neuron and muscle-specific transgenes (TgMI+NI-nmd) displayed growth rates indistinguishable from their control littermates (+/+ or +/ nmd ) at all time points (Fig. 1 D).
Transgenic rescue of DCM: morphological and functional assessments
Longitudinal investigation of cardiac rescue in TgMI- nmd mice was initially assessed using maximum lifespan (Fig. 2 A). When compared with non-transgenic nmd , the cumulative survival curve of TgMI- nmd mice is significantly greater, but also significantly less than that of double transgenic TgMI+NI- nmd mice or their sib-controls ( nmd versus TgMI- nmd , P <0.0001, χ 2 =77.9; TgMI- nmd versus TgMI+NI- nmd , P <0.0001, χ 2 =28.7; TgMI- nmd versus control, P <0.0002, χ 2 =14.4). To investigate the nature of these differences, routine necropsy and evaluation of not only the heart but also other organs were conducted. Mean ventricular free-wall thickness in TgMI- nmd or TgMI+NI- nmd mice was not significantly different from +/+ or +/ nmd transgenic sib-controls and was significantly thicker than those of B6- nmd hearts (Fig. 2 B) ( P <0.0001). Although the mean weight of nmd hearts with the neuron-specific transgene was significantly less than that of double transgenic nmd (* P =0.0126), it was not statistically different from that of TgMI- nmd or transgenic ( +/+ or +/nmd ) sib-control hearts (Fig. 2 C). Unexpectedly, the mean weights of double transgenic nmd hearts were greater than that of TgMI- nmd (** P =0.0031) or that of control (** P =0.0002). In fact, the mean weight of double transgenic nmd hearts was nearly 50% heavier (Fig. 2 C). In Figure 2 D, this apparent increase in double transgenic nmd average heart weight is proportionally distributed throughout all four chambers; such that individually, each weighed significantly more than that of TgMI- nmd or their sib-controls (* P <0.05–0.0005). This effect is unlikely to be due to an adverse effect of transgenic TgMI expression in the heart because the effect was not seen in single transgenic TgMI- nmd or TgMI- +/+ mice (Fig. 2 C) or in double-transgenic +/+ littermates (data not shown). Therefore, this may reflect an unmet tissue-specific need for IGHMBP2 in other vital tissues/organs (i.e. liver, kidney or adrenals) of nmd mice, indirectly altering heart size; or alternatively, this may reflect a compensatory increase in heart size due to the greater cardiac load and the demand in the rescued double transgenic nmd mice, relative to paralyzed and activity restricted TgMI- nmd mice.
Morphological evaluations of TgMI- nmd hearts (fixed in tetany) indicated normal macroscopic presentation with ventricular chambers devoid of blood, the papillary muscles are large and prominent and the atria are comparably small (Fig. 3 A–C). These observations are consistent with the absence of histopathologic changes in ventricular myocytes stained with H&E (Fig. 3 D–F), cardiac functional integrity illustrated by normal conduction patterns in electrocardiograms (ECG, Fig. 3 G and H) and normal echocardiographic parameters (LVEDD, LVESD, Fig. 3 I and J). Nevertheless, TgMI- nmd mice were still vulnerable to late-onset complications from progressive neuromuscular degeneration including anorexia/dysphagia (difficulty in mastication or deglutition) culminating in wasting and malnutrition. An apparent susceptibility to mega-esophagus as well as mastication and deglutition difficulties is completely eliminated in the presence of both neuron and muscle-specific transgenes with the concomitant rescue of neuromuscular degeneration and muscular dystrophy phenotypes ( 5 ). Hence, double transgenic TgNI+MI- nmd mice essentially lived greater than 2 years. Even with apparently enlarged hearts, they fail to develop cardiomyopathy. However, an increased incidence of dermatitis possibly due to a B6 background effect, often necessitated their premature culling.
QTL analysis identifies genetic modifiers of cardiomyopathy
Disease penetrance and variation in phenotypic manifestations are commonly attributed to genetic background polymorphisms ( 15 ). We have previously shown that the nmd motor neuron disease can be suppressed by a CAST-derived modifier ( Mnm C ) on Chr 13 ( 2 ). To determine whether the nmd cardiomyopathy is also amenable to genetic modification, we examined the maximum lifespan of 166 nmd mice from a segregating N2 backcross population (B6.CAST- Mnm C / Mnm C Ighmbp2 nmd-2J /+ ×CAST/EiJ)F1×(B6.CAST- Mnm C / Mnm C Ighmbp2 nmd-2J /+ ). The N2 mutant nmd mice showed a wide range of lifespan ranging from as short as 2 weeks to as long as 57 weeks of age (Fig. 4 A), with most mutants ultimately developing cardiomyopathy. However, variation in the clinical manifestations of CHF strongly suggested the presence of CAST-derived genetic modifiers of DCM. Using survival as a biological indicator of cardiac improvement, QTL analysis of short-lived versus long-lived N2 mutant mice was performed. As shown in Figure 4 B, following a genome-wide screen, long-lived nmd mice revealed similar segregation patterns for putative Cardiac modifier of nmd ( Cmn ) QTLs on chromosomes 9 ( Cmn1 ) and 10 ( Cmn2 ) and a QTL on chromosome 16 ( Cmn3 ) with a significant gender effect (Fig. 4 C) (Table 1 ). Confidence intervals (95%) of these three significant QTLs are shown in Figure 4 D, and two suggestive B6-derived QTLs on chromosomes 1 and 5 are shown in Figure 4 E. Cmn2 corresponds to a broad interval on Chr 10, potentially representing more than one QTL. We are currently constructing congenic lines harboring the CAST-derived QTLs on the neuron-specific modifier B6.CAST- Mnm C background. Once full N10 congenic strains are established for these QTLs, their potential effects on DCM as well as the onset and progression of motor neuron disease can be assessed individually. The cumulative survival rates of cardiac modified nmd mice in the N2 and N3 segregating background generations are significantly greater than that of Mnm C -nmd mice as shown in Figure 5 A ( P <0.0001). Likewise, cardiac modified nmd mice carrying at least one CAST allele for each of the first three Cmn loci showed significantly greater mean lifespan than those of Mnm C - nmd mice (228.2±37.5 days versus 68.6±1.8 days, * P <0.0001) (Fig. 5 B). Overall, these results suggest that, even at the N4 generation, wherein the genome is expected to be over 90% B6-like, the CAST-derived QTLs on Chrs 9, 10 and 16, individually or in-combination, are still able to modify the onset and severity of DCM.
Morphological and functional assessment of DCM modification
To investigate the nature of the cardiac improvement in N4F1 generation of Cmn-nmd mice, plasma creatine kinase (CK)-MB (brain/heart CK) levels were examined. As shown in Figure 6 A, nmd mice between 4 and 8 weeks of age carrying the neuron-specific transgene TgNI showed the greatest average plasma CK-MB when compared with non-transgenic nmd as shown previously ( 5 ). Although cardiac modified nmd mice had appreciably higher average plasma CK-MB levels when compared with controls (367±66.7 versus 174.4±10.6, *** P =0.0086), they were significantly lower than TgNI- nmd (826.6±380, * P =0.0002) or nmd mice without the modifiers (620.8±63.5, ** P =0.0013), suggesting that relatively fewer cardiomyocytes were undergoing degeneration. Because the neuron-specific TgNI construct rescues nmd paralysis, the observed increase in activity and cardiac demand hastens DCM, resulting in marked elevation in plasma CK-MB. With relatively fewer cardiomyocytes being lost, the cardiac morphology and the ventricular free-wall thickness in the hearts of N3–N5 generation Cmn-nmd mice were significantly rescued (* P <0.0001) when compared with nmd mice without the modifier loci (Fig. 6 B). The mean ventricular free-wall thickness and total heart weights of Cmn-nmd mice are not significantly different from those of control and reflect compensatory cardiac adaptation and remodeling (Fig. 6 C). This difference is further illustrated by increased left ventricular (LV) weight in Cmn-nmd versus B6- nmd hearts (* P =0.0024). In contrast, both Cmn-nmd and B6- nmd mice showed significantly heavier mean atrial weights when compared with normal sib-control (* P =0.028), suggesting that the CAST-derived modifiers have no effect on atrial changes. Alternatively, subtle residual valvular insufficiency in Cmn-nmd hearts may promote compensatory atrial hypertrophy, which is more pronounced in B6- nmd mice ( 5 ). The mean right ventricular free-wall weights, and interventricular septum weights of Cmn-nmd hearts, as in B6- nmd hearts, are markedly reduced when compared with sib-control hearts (** P <0.014) (Fig. 6 D). Assessment of cardiac function using ECG corroborated the observed atrial changes of either TgNI or Cmn-nmd mice with significantly greater P-wave amplitudes when compared with control (* P <0.002, ** P <0.009) (Fig. 6 E). Both B6- nmd and Cmn-nmd showed greater mean LV volumes at diastole than control mice, consequently resulting in greater cardiac output (Fig. 6 F).
Gross and microscopic evaluation of the Cmn-nmd hearts revealed a wide range of morphological alterations but showed marked reduction in chamber dilation and nearly normal cardiac wall thickness (Fig. 7 A and C). However, most of these Cmn-nmd mice eventually develop CHF due to other causes, such as obstructive cardiomyopathy (due to organizing thrombi, Fig. 7 C, black arrow), restrictive cardiomyopathy (due to compensatory cardiomyocyte hypertrophy, Fig. 7 G), ischemic cardiomyopathy (due to myocardial infarction, Fig. 6 C, red arrows) or deficits in cardiac conduction (Fig. 7 I), resulting in sudden death. Closer examination of Cmn-nmd hearts revealed that putative cardiac modifiers may reduce the occurrence of replacement fibrosis as shown in Figure 7 B and in Figure 7 E. Furthermore, functional evaluation of heart rates, blood pressure, ECGs and echocardiograms in these mice indicated normal systolic function, fully compensated hearts and remarkably normal cardio-respiratory function, in contrast to B6- nmd mice (Fig. 7 H–O) (data not shown). The diverse morphological changes in the hearts of long-lived Cmn-nmd mice reflect the effects of segregating CAST-derived cardiac modifiers. In the extremes, the Cmn-nmd heart in Figure 7 F appear morphologically normal, but functionally the strength of its conduction proved weak (Fig. 7 J) when compared with the Cmn-nmd ECG in Figure 7 K or the normal B6 ECG in Figure 7 H. The echocardiogram (M-mode) in Figure 7 N depicts cardiac functional integrity in the presence of a mild, but noticeably increase in LVEDD and LVESD when compared with Cmn-nmd heart in Figure 7 O or B6-control in Figure 7 L. Likewise, histopathology of long-lived Cmn-nmd ventricles in Figure 7 G revealed the presence of hypertrophic cardiomyocytes which is consistent with that observed grossly in Figure 7 C. The amplitude of (QRS) cardiac conduction in Figure 7 K (∼300 mV) is only half that of the B6‐control in Figure 7 L (∼600 mV) even with apparent hypertrophic cardiac remodeling. The functional consequences of these changes are not clearly apparent in the normal echocardiogram in Figure 7 O when compared with Figure 7 L.
Early clinical signs of disease in nmd mice with or without cardiac modification included sudden death, respiratory distress, anorexia/dysphagia resulting in weight loss, anasarca (generalized subcutaneous edema) and cessation of normal activities. Given the wide range of maximum lifespan, dead or euthanized-moribund cardiac modified nmd mice were subjected to a full necropsy examination; not only to establish the cause of death but also to validate clinical diagnostic findings with grossly observed pathological changes. Although gross and microscopic alterations of the heart were observed, the cause of death was not exclusively cardiovascular in origin. Presumptively, because of their underlying neuromuscular degeneration, Cmn-nmd mice like the transgenic rescued TgMI- nmd showed increased vulnerability to mega-esophagus and gastrointestinal motility disorders (data not shown). Hearts from Cmn-nmd mice with overt clinical signs revealed a range of morphological changes including, but not exclusively, the presence of degenerating, apoptotic and necrotic, cardiomyocytes and replacement-interstitial fibrosis without inflammation. Furthermore, we did not observe pathognomonic pleural effusion previously associated with cardio-respiratory failure or secondary valvular insufficiency in nmd mice ( 5 ). In the extreme situation, however, signs of compensatory hypertrophic DCM were occasionally observed, suggesting a role for cardiac modifiers in promoting cardiovascular adaptation and remodeling in order to maintain tissue perfusion.
DISCUSSION
We have shown that muscle-specific expression of an Ighmbp2 transgene in nmd mice prevents cardiomyocyte loss and restores normal cardiac morphology and function. This tissue-specific rescue of cardiomyopathy confirms the essential role of IGHMBP2 in the maintenance and survival of not only neurons but also cardiac and skeletal myocytes. Regardless of the severity and progression of the motor neuron disease, these transgenic TgMI- nmd mice failed to develop DCM that would have resulted in early lethality. Rather, their lifespan was markedly extended, albeit with severely restricted mobility, and were more prone to sibling competition and complications stemming from their underlying progressive paralysis. This experiment further suggests that the early lethality of nmd mice is primarily due to the development of DCM, not the progressive motor neuron disease (Fig. 2 C and D). Autosomal recessive mutations in the human IGHMBP2 gene cause a phenotypically similar motor neuron disease, spinal muscular atrophy with respiratory distress (SMARD1) ( 4 ). Like the mutant nmd mouse, SMARD1 patients develop a progressive neurogenic muscular atrophy as infants. However, unlike SMARD1 patients, the absence of diaphragmatic paralysis and the prevalence of DCM in nmd mice suggest that these muscle tissues may be differentially spared and/or readily modified by background strain polymorphisms. Whether this is a species-specific manifestation or a consequence of residual Ighmbp2 expression (20–25% of normal) from the hypomorphic nmd mutation ( 2 ) remains to be determined.
The role of IGHMBP2 in cardiac and skeletal muscles
Gross morphological and histopathological examinations of mutant nmd hearts shortly after birth revealed no discernable abnormalities. However, this does not necessarily reflect the absence of pathology at the molecular or functional levels in these hearts. Indeed, if this is the case, our original hypothesis that IGHMBP2 is not critical for normal cardiac morphogenesis may have to be revised. From birth until 7 days post-natal, the major cardiac myosin heavy chain (MHC) isoform in the mouse is beta-MHC (manifested by slower heart rates). Thereafter, complete transition to the adult alpha-MHC isoform (manifested by faster heart rates) occurs ( 19 ). Although this transition appears to occur normally in nmd mice based on extrapolation of functional and biochemical analyses in newly weaned 3-week-old mice, temporally regulated cardiac morphogenesis and functional markers will need to be examined in much younger (prenatal and newborn) nmd mice. Preliminary echocardiographic examination of newborn P0 nmd mice revealed a remarkable difference in heart rates in mutants when compared with sib-controls. The mutant nmd mice showed almost 30% greater heart rates relative to sib-controls, suggesting signs of precocious cardiac functional development. Thus, besides normal levels of IGHMBP2 being essential for cardiomyocyte maturation during this stage of rapid growth (3–6 weeks of age), it may also be critical for appropriate cardiomyocyte differentiation during early cardiac morphogenesis. The essential functions of IGHMBP2 in cardiac muscle, therefore, could be defined by proper temporal expression of muscle isoforms, adequate remodeling and compensatory responses of the heart to changing load and demand. Given that IGHMBP2 is putatively involved in transcriptional activation or splicing, not only will altered expression of pro-survival and/or pro-apoptotic genes result in a cascade of signaling events culminating in apoptosis, but it may also promote changes in cellular development that could ultimately affect function ( 11 , 20 , 21 ). To date, descriptions of DCM in SMARD1 patients are lacking, however, five SMARD1 infants were described with cardiac arrhythmia ( 3 , 4 , 22 , 23 ) and treatments to prolong their lives may reveal a role for IGHMBP2 in human heart disease. Alternatively, the presence of background modifier genes that significantly alter the onset and duration of DCM in nmd mice suggests that similar genetic modifiers may exist in humans to suppress or enhance the manifestation of DCM in SMARD1 patients.
Cardiac-specific genetic modifiers prolong the lifespan of nmd mice
Although the cardiac modified Cmn C -nmd mice display a remarkable amelioration of DCM, they still succumb to CHF and premature death. In contrast, the transgenic TgMI- nmd mice show complete cardiac rescue, suggesting that the protective alleles of the cardiac modifier loci can only partially suppress the cardiomyocyte degeneration. At the N4 and N5 backcross generations of B6.CAST- Cmn C ; Mnm C ; Ighmbp2 nmd-2J mice, the mean lifespan and cumulative survival of these mice is significantly greater than that of nmd mice without the CAST-derived cardiac-specific modifier loci (Fig. 5 ). Effective treatments for cardiomyopathic diseases, such as familial, idiopathic and/or hypertrophic DCM, beyond heart transplant are currently not available ( 24 ). Our previous transgenic rescue of the neuromuscular degeneration phenotype ( 5 ) and now the cardiomyocyte degeneration in nmd mice offer the promise for an effective gene therapy. Apparent functional rescue of the nmd phenotype was achieved by transgenic expression of IGHMBP2 in motor neurons and in cardiac and skeletal myocytes. Moreover, the identification of genetic modifier loci delaying the onset and progression of cardiomyopathy in nmd mice suggest that molecular pathways exist that can be targeted to alter the pathogenesis of CHF.
MATERIALS AND METHODS
Generation and characterization of transgenic nmd mice
Mice were bred and maintained under standard conditions in the Research Animal Facility at The Jackson Laboratory with Institutional Animal Care and Use Committee approval. For construction of the Ttn-Ighmbp2 transgene, a 5.87 kb mouse genomic fragment corresponding to 76 880 200–76 874 334 bp (chromosome 2 NCBI build-m33) was amplified from a 129-strain BAC CITB-305G19 encompassing the Ttn 5′ region ( 25 ) using primers TTN64F (5′-CTGTCATTCAATCTTCCTGACAGT A CTTTG-3′) and TTN63R (5′-GGTGCTTGAGTAGT AC TCTTTTCAGGCAC-3′). A T-A substitution (underlined-TTN64F) creates a Sca 1 restriction site, and a CA-AC substitution (underlined-TTN63R) alters the Ttn start codon (ATG to AGT). This fragment was subcloned and sequenced in pCR4-TOPO (Invitrogen) and transferred to the pBSXtra-Ighmbp2 cDNA and SV40 poly(A) vector by Eco R1 digestion as described ( 5 ). Two transgenic founders (45 and 108) of C57BL/6J-Tg( Ttn-Ighmbp2 )/Cx mice were identified from tail genomic DNA using TTN88F (5′-CAACTGCATAAGCACCAGGAG-3′) and Ighmbp2R17 (5′-GAGGTGAAGCTGTTGCTAGG-3′) generating a 565 bp PCR product. B6-TgMI was bred to B6.BKS- Ighmbp2 nmd-2J / + mice. Transgenic-+/ nmd F1 offspring were further backcrossed to generate transgenic nmd / nmd (TgMI- nmd ) mice. The Ighmbp2 nmd-2J mutation was genotyped as described ( 5 ). All nmd mice and age-matched controls were weighed weekly prior to the onset of clinical signs up to 22 weeks.
Plasma CK
Blood was obtained from the peri-orbital sinus using heparinized microhematocrit tubes after the application of Tetracaine topical anesthetic; plasma total CK and cardiac-derived CK-MB levels were assayed as described ( 5 ).
Cardiac function and histopathology
Measurements of systolic blood pressure and pulse rate were performed as described ( 5 ). Standard ECG and echocardiogram was performed in anesthetized mice (1–1.5% isofluorane in O 2 at 0.6 l/min) as described ( 5 ). Following ECG recording, hearts were isolated and fixed for at least 24 h, processed and stained for histopathology as described ( 5 ).
Quantitative trait locus analysis
Two backcross generations (N2) with an established parental congenic, B6.CAST- Mnm C Ighmbp2 nmd-2J /+ as recipient and CAST/EiJ as parental donor strain were created. Of 850 total progeny, ∼20% (166) were homozygous nmd/nmd and were aged until the development of DCM. The CAST-derived Mnm C modifier (Chr 13) was fixed in the cross to avoid confounding effects of paralysis on maximum lifespan. Genotype analyses for Mnm C and nmd loci were performed as described ( 5 ). Tail DNA from 166 nmd/nmd N2 progeny was PCR-amplified with SSLP markers (Invitrogen) as described ( 5 ). A total of 134 markers spaced at 10–20 cM, polymorphic between B6 and CAST, were typed on each of the 19 autosomes. Genotype-to-maximum lifespan correlations were performed using 40 short-lived and 40 long-lived male and female N2 nmd/nmd mice using Map Manager. Independent confirmation was carried out on all 82 nmd males and 84 nmd females using the PSEUDOMARKER set of QTL programs developed by Sen and Churchill ( 26 ). For further details and information: http://www.jax.org/staff/churchill/labsite/ . Pairwise genome scans were conducted to search for interacting QTL pairs correlated with short or prolonged lifespan. A separate genome scan analysis was performed using sex as both an additive and interacting covariate.
RT–PCR analysis
Total heart RNA was prepared from newborn (P0), P21 and P28 nmd/nmd and age-matched littermates using the Trizol method (Invitrogen) according to the manufacturer's instructions. For RT–PCR, 1 µg of total RNA from nmd mutant and control tissues were reversed transcribed using random hexamers and oligo dT as described ( 2 ). Primers within exon 1 of the Ttn gene (TTN50F 5′-TTCATGTCGGAGATGGTTGG-3′) and within the Ighmbp2 cDNA ( Ighmbp2 -R17) were used to generate a 600 bp transgene-specific RT–PCR product.
Statistical analysis
Values were expressed as the mean±SE and analyzed by paired Student's t- test or ANOVA using Statview 5.0.1 (SAS Institute Inc.). A value of P ≤0.05 was considered statistically significant.
ACKNOWLEDGEMENTS
We are grateful to Drs Jane Barker, Beverly Paigen and Mary Ann Handel for critical review of the manuscript. This work was supported by a grant from NIH RO1AR49043 to G.A.C.; T.P.M. was supported by fellowships from NIH T32 RR07068 and The American Heart Association. Scientific services at TJL supported partly by NCI-CA34196 (NIH Cancer Center Grant).
Conflict of Interest statement.The authors have no conflicts of interest to declare.
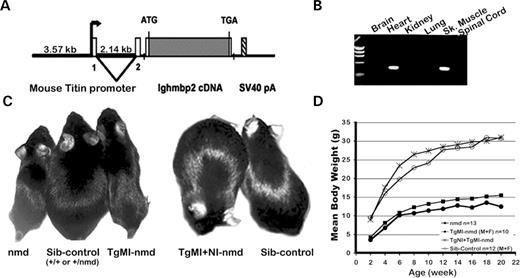
Figure 1. ( A ) TgMI transgenic construct with the mouse titin promoter (containing 3.57 kb of upstream sequences, the complete non-coding exon 1 and intron 1 and 30 bp partial exon 2 sequence with mutated Ttn ATG to AGT) upstream of a wild-type mouse Ighmbp2 cDNA and an SV40 polyadenylation signal. ( B ) RT–PCR assay for transgene expression in multiple tissues using transgene-specific primers (exon 1 of Ttn promoter and Ighmbp2 cDNA reverse primer). ( C ) Live-photograph of a (L to R) B6- nmd mouse along with an unaffected and transgenic TgMI- nmd littermate at 6 weeks of age. Note that the TgMI transgene does not rescue the nmd paralysis phenotype, as the TgMI- Ighmbp2 cDNA is not expressed in spinal cord (B). In contrast, 6-month-old double transgenic TgMI+TgNI- nmd mouse shows complete rescue of both the cardiomyopathy and the neurogenic atrophy pictured next to its littermate. ( D ) Growth curve for the groups of mice shown in (C).
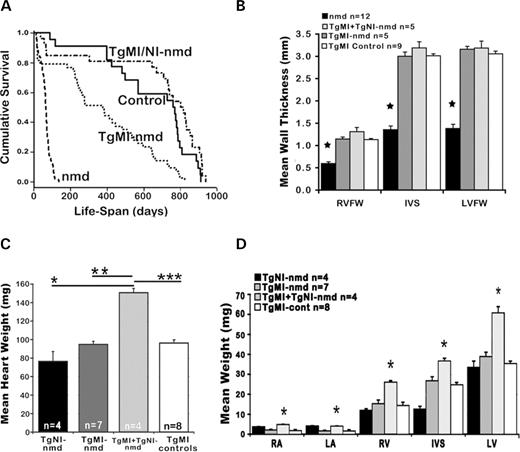
Figure 2. ( A ) Cumulative survival curve for nmd ( n =135), TgMI- nmd ( n =43), double transgenic TgMI+TgNI- nmd ( n =26) and sib-controls ( n =22). ( B ) Histo-morphometric evaluation of ventricular wall thickness. (RVFW, right ventricular free wall; IVS, interventricular septum; LVFW, left ventricular free wall) ( C ) Mean heart weights of adult transgenic and non-transgenic nmd mice at 3–8 months of age and their age-matched transgenic (+/+ or +/ nmd ) sib-controls. ( D ) Mean wet weights of individual cardiac chamber walls (RA, right atrium; LA, left atrium; RV, right ventricle).
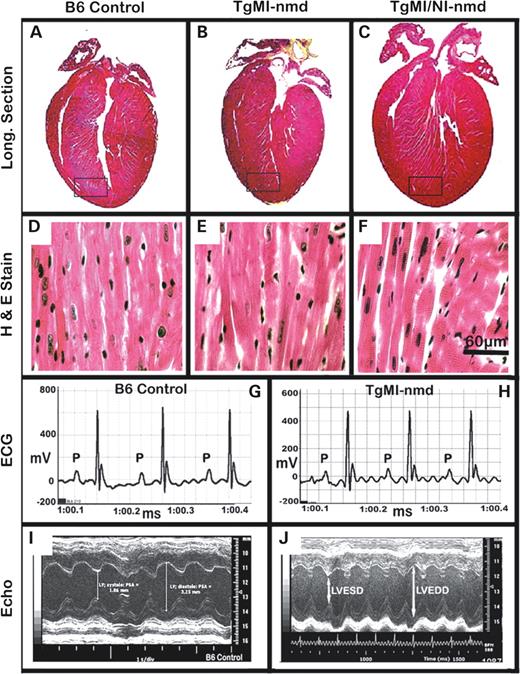
Figure 3. Gross morphology, histology, electrocardiographic and echocardiographic presentations of B6-control versus TgMI transgenic rescued nmd hearts at 8–16 weeks of age. ( A–C ) H&E-stained longitudinal sections show normal morphologic appearance of the heart. Magnification of the rectangular areas near the cardiac apex shows cardiomyocytes staining uniformly in B6-control heart ( D ) similar to transgenic ( E ) and double transgenic ( F ) hearts, indicating a lack of pathological changes. These observations are consistent with a normal ECG pattern in B6-control ( G ) analogous with the transgenic rescue ( H ), both indicative of healthy cardiac conduction. Normal echocardiograms, manifested by relatively robust LVEDD and LVESD in B6-control ( I ) heart similar to the transgenic ( J ) heart, further attest to the cardiac rescue.
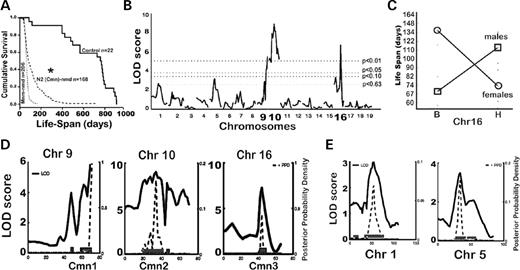
Figure 4. ( A ) Cumulative survival rates of B6.CAST- Mnm C -nmd mice with the neuron-specific modifier when compared with the *N2 backcross nmd population used for QTL analysis and unaffected ( +/+ or +/nmd ) sib-controls. ( B ) Genome-wide screen of N2 backcross nmd mice and QTL analysis of maximum lifespan revealed significant linkage of CAST-derived modifier loci to chromosomes 9, 10 and 16. The horizontal lines from bottom-up represent significance thresholds 0.63, 0.10, 0.05 and 0.01, respectively. ( C ) Gender effect of the Chr 16 CAST-modifier in males is significantly associated with increased lifespan, in contrast to females with a B6 allele beneficial to survival. ( D ) Interval map of each lifespan QTL with logarithm of odds (LOD) scores (solid line) and 95% confidence intervals. ( E ) Interval map of two suggestive B6-derived QTLs on Chrs 1 and 5.
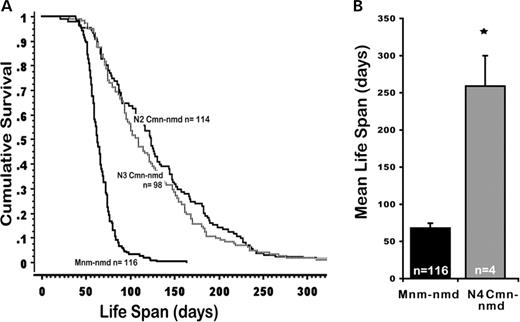
Figure 5. ( A ) Cumulative survival curves of B6- Mnm C nmd mice without CAST-derived cardiac modifiers and Cmn-nmd mice segregating modifier QTLs at the N2–N3 generations. (B) Mutant nmd mice heterozygous for Cmn1, Cmn2 and Cmn3 loci at the N4 backcross generation lived significantly longer than B6- Mnm C -nmd mice (* P <0.0001).
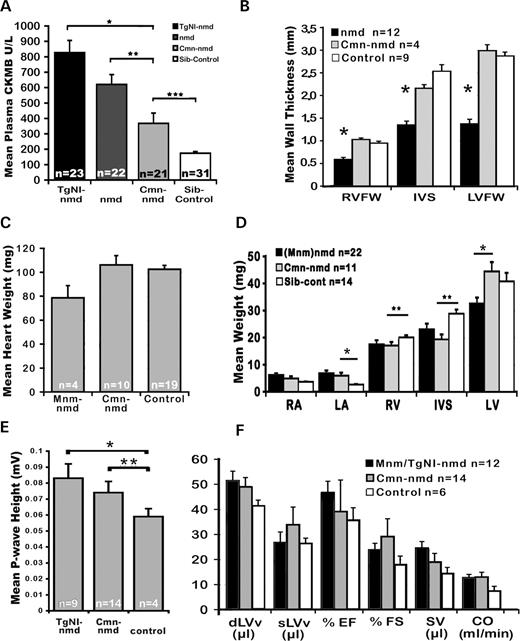
Figure 6. ( A ) Plasma CK-MB levels of 4- to 8-week-old TgNI- nmd or B6- nmd and adult (6–8 months) N4F1 Cmn-nmd and age-matched littermates. ( B ) Mean ventricular free-wall thickness of 5- to 8-week-old nmd , N3–N5 generation Cmn-nmd and unaffected littermates. ( C ) No significant differences are observed in the total heart wet weights of B6.CAST- Mnm - nmd , N3–N6 Cmn-nmd or controls. However, wet weights of individual cardiac chamber walls ( D ) do reveal significant differences. ( E) Significant elevations of mean P-wave ECG amplitudes are evident in mutant nmd hearts carrying either the neuron-specific TgNI-transgene or Cmn cardiac modifiers when compared with unaffected littermates, * P =0.002 and ** P =0.009. ( F ) Derived cardiac functional parameters in B6- nmd mice and N3F1–N4F1 generation carriers of cardiac modifiers versus sib-controls (dLVv, left ventricular volume at diastole; sLVv, left ventricular volume at systole; EF, ejection fraction; FS, fractional shortening; SV, stroke volume; CO, cardiac output).
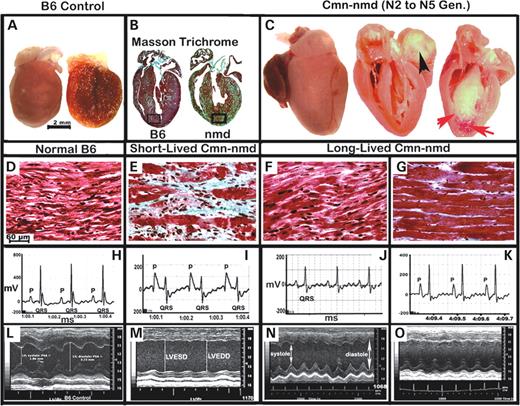
Figure 7. ( A and C ) Gross morphologic presentation of B6-control and cardiac modified B6.CAST- Cmn, nmd hearts. (B) Masson Trichrome stained longitudinal sections of B6 and Cmn-nmd hearts. Rectangular areas near the apex of the hearts were magnified to show details as depicted ( D–G ). Unlike the short-lived Cmn-nmd heart (E ) , hearts from long-lived Cmn-nmd mice (F and G ) show dramatically less fibrosis. Cardiac function was evaluated using both ECG ( H–K ) and ultrasound examinations ( L–O ). Consistent with the P-wave enlargement and shortened QRS-complex (I ) , the presence of atrial enlargement, fibrosis and markedly dilated ventricles on gross examination (B and C) is the appearance of functionally deficient cardiac contraction (M ) , depicted by LVEDD and LVESD.
Chromosomal location and LOD scores for nmd lifespan QTLs
| Name | Chr | Peak (cM) | 95% CI (cM) | High allele | Peak marker | LOD | % variance |
|---|---|---|---|---|---|---|---|
| Cmn1 | 9 | 70 | 60–70 | CAST | D9Mit17 | 5.56 | 8.2 |
| Cmn2 | 10 | 34 | 28–44 | CAST | D10Mit42 | 7.95 | 12.1 |
| Cmn3 | 16 | 44 | 42–48 | CAST | D16Mit64 | 4.28 | 6.2 |
| Name | Chr | Peak (cM) | 95% CI (cM) | High allele | Peak marker | LOD | % variance |
|---|---|---|---|---|---|---|---|
| Cmn1 | 9 | 70 | 60–70 | CAST | D9Mit17 | 5.56 | 8.2 |
| Cmn2 | 10 | 34 | 28–44 | CAST | D10Mit42 | 7.95 | 12.1 |
| Cmn3 | 16 | 44 | 42–48 | CAST | D16Mit64 | 4.28 | 6.2 |
Cmn , Cardiac modifier of nmd ; Chr, chromosome; CI, confidence interval; LOD, logarithm of the odds ratio.
Chromosomal location and LOD scores for nmd lifespan QTLs
| Name | Chr | Peak (cM) | 95% CI (cM) | High allele | Peak marker | LOD | % variance |
|---|---|---|---|---|---|---|---|
| Cmn1 | 9 | 70 | 60–70 | CAST | D9Mit17 | 5.56 | 8.2 |
| Cmn2 | 10 | 34 | 28–44 | CAST | D10Mit42 | 7.95 | 12.1 |
| Cmn3 | 16 | 44 | 42–48 | CAST | D16Mit64 | 4.28 | 6.2 |
| Name | Chr | Peak (cM) | 95% CI (cM) | High allele | Peak marker | LOD | % variance |
|---|---|---|---|---|---|---|---|
| Cmn1 | 9 | 70 | 60–70 | CAST | D9Mit17 | 5.56 | 8.2 |
| Cmn2 | 10 | 34 | 28–44 | CAST | D10Mit42 | 7.95 | 12.1 |
| Cmn3 | 16 | 44 | 42–48 | CAST | D16Mit64 | 4.28 | 6.2 |
Cmn , Cardiac modifier of nmd ; Chr, chromosome; CI, confidence interval; LOD, logarithm of the odds ratio.
References
Morita, H., Seidman, J. and Seidman, C.E. (
Cox, G.A., Mahaffey, C.L. and Frankel, W.N. (
Pitt, M., Houlden, H., Jacobs, J., Mok, Q., Harding, B., Reilly, M. and Surtees, R. (
Grohmann, K., Schuelke, M., Diers, A., Hoffmann, K., Lucke, B., Adams, C., Bertini, E., Leonhardt-Horti, H., Muntoni, F., Ouvrier, R. et al. (
Maddatu, T.P., Garvey, S.M., Schroeder, D.G., Hampton, T.G. and Cox, G.A. (
Ross, J., Jr. (
Pyle, W.G. and Solaro, R.J. (
Nicol, R.L., Frey, N. and Olson, E.N. (
Wallace, D.C. (
Czubryt, M.P. and Olson, E.N. (
Wencker, D., Chandra, M., Nguyen, K., Miao, W., Garantziotis, S., Factor, S.M., Shirani, J., Armstrong, R.C. and Kitsis, R.N. (
Crone, S.A., Zhao, Y.Y., Fan, L., Gu, Y., Minamisawa, S., Liu, Y., Peterson, K.L., Chen, J., Kahn, R., Condorelli, G. et al. (
Erickson, S.L., O'Shea, K.S., Ghaboosi, N., Loverro, L., Frantz, G., Bauer, M., Lu, L.H. and Moore, M.W. (
Grohmann, K., Rossoll, W., Kobsar, I., Holtmann, B., Jablonka, S., Wessig, C., Stoltenburg-Didinger, G., Fischer, U., Hubner, C., Martini, R. et al. (
Pasotti, M., Repetto, A., Tavazzi, L. and Arbustini, E. (
Darvasi, A. (
Suzuki, M., Carlson, K.M., Marchuk, D.A. and Rockman, H.A. (
Le Corvoisier, P., Park, H.Y., Carlson, K.M., Marchuk, D.A. and Rockman, H.A. (
Fijnvandraat, A.C., Lekanne Deprez, R.H. and Moorman, A.F. (
Sabbah, H.N. and Sharov, V.G. (
Russo, G.L. and Russo, M. (
Grohmann, K., Varon, R., Stolz, P., Schuelke, M., Janetzki, C., Bertini, E., Bushby, K., Muntoni, F., Ouvrier, R., Van Maldergem, L. et al. (
Rudnik-Schoneborn, S., Stolz, P., Varon, R., Grohmann, K., Schachtele, M., Ketelsen, U.P., Stavrou, D., Kurz, H., Hubner, C. and Zerres, K. (
Jain, P., Massie, B.M., Gattis, W.A., Klein, L. and Gheorghiade, M. (
Garvey, S.M., Rajan, C., Lerner, A.P., Frankel, W.N. and Cox, G.A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}