




Abstract
Alternative RNA splicing is now known to be pervasive throughout the genome and a target of human disease. We evaluated if targeting intronic splicing regulatory sequences with antisense oligonucleotides could be used to correct aberrant exon skipping. As a model, we targeted the intronic silencing sequence (ISS) elements flanking the alternatively spliced α-exon of the endogenous fibroblast growth factor receptor 1 (FGFR1) gene, which is aberrantly skipped in human glioblastoma. Antisense morpholino oligonucleotides targeting either upstream or downstream ISS elements increased α-exon inclusion from 10% up to 70% in vivo. The effect was dose dependent, sequence specific and reproducible in several human cell lines, but did not necessarily correlate with blocking of protein association in vitro. Simultaneous targeting of the ISS elements had no additive effect, suggesting that splicing regulation occurred through a shared mechanism. Broad applicability of this approach was demonstrated by similar targeting of the ISS elements of the human hnRNPA1 gene. The correction of FGFR1 gene splicing to >90% α-exon inclusion in glioblastoma cells had no discernable effect on cell growth in culture, but was associated with an increase in unstimulated caspase-3 and -7 activity. The ability to manipulate endogenously expressed mRNA variants allows exploration of their functional relevance under normal and diseased physiological states.
INTRODUCTION
Alternative RNA splicing is a widespread process that allows for the production of multiple mRNAs from a single gene. Recent estimates suggest that between 40 and 74% of human genes undergo some form of alternative RNA splicing (1,2). For this reason, the process has been proposed as an important mechanism for the creation of functional and genetic diversity in many higher organisms (3,4). At a posttranscriptional level, alternative RNA splicing allows for the expression of distinct protein or RNA isoforms within different tissues or cell types, at specific developmental stages, or simply as a response to changing cellular needs (5,6). Alternative recognition of exons during splicing requires the presence of additional positive and negative regulatory elements that are distinct from the constitutive splicing signals (7,8). These elements are typically located within or immediately flanking the alternatively spliced exon and are presumed to function by enhancing or inhibiting spliceosome formation.
A widely accepted model of splicing activation involves the binding of serine/arginine (SR) proteins to an exonic splicing enhancer to stabilize interactions that occur at neighboring splice sites (7,9). The absence of specific SR proteins or the disruption of their interactions, sometimes mediated through exonic splicing silencers, is one pathway that causes exon skipping. Less well-studied regulatory sequences are the intronic splicing enhancer and intronic splicing silencer (ISS) sequences. In many cases, these intronic regulatory elements are found adjacent to branch point or splice sites and even within the polypyrimidine tract of the 3′ splice site. Binding of regulatory factors to these sequences is thought to directly modulate splice site recognition by blocking the formation of the spliceosome (7). In addition to directly enhancing or blocking splice site recognition, some intronic elements exert their regulatory effects at considerable distances. How many genes use this strategy and precisely how spliceosome assembly is modulated, in these cases are not well defined.
In recent years, the revelation of the pervasiveness of alternative RNA splicing has led to an increased appreciation that this process may be a clinically significant target of many human diseases. Approximately 10% of the known disease-causing mutations have been attributed to some defect in constitutive splicing (10). This percentage is most likely an underestimate, however, because mutation analysis rarely extends beyond an examination of constitutive splice site sequence, and examining RNA for aberrant splicing is often impractical (11). Several diseases, including various cancers, have been associated with the expression of aberrantly spliced RNA (12). In some cases, genetic mutation has even served to identify important cis-regulatory sequences. Aberrant splicing in the pre-mRNA for the fibroblast growth factor receptor 1 (FGFR1), a transmembrane tyrosine kinase receptor, has been found in association with human disease but in the absence of splice site mutation. Alternative splicing of FGFR1 modifies both the extracellular and the intracellular domains of this receptor; the resulting variants manifest receptors with different ligand specificities, binding affinities and kinase activities (13).
One variant generated by the skipping of FGFR1 exon 3, also termed the α-exon, produces a receptor that lacks an amino-terminal extracellular immunoglobin-like domain. This splice variant, known as FGFR1β, is associated with increased ligand affinity and altered subcellular localization (14–17). Aberrant splicing of the α-exon has been associated with pancreatic cancer (18), breast cancer (19) and glioblastoma (20). The precise mechanism by which FGFR1β is enhanced in tumors is unclear. The α-exon is flanked by ISS sequences ISS1 and ISS2 (Fig. 1A); these elements appear to regulate α-exon exclusion in glioblastoma cells (21). The absence of mutations in these elements suggested that the aberrant splicing observed in tumors resulted from abnormal expression of trans-regulators. Polypyrimidine tract-binding (PTB) protein, a known inhibitor of exon recognition, has been shown to be overexpressed in glial cell tumors and to bind the ISS1 element (22). Manipulating PTB levels in glioblastoma cell lines correlated with changes in α-exon splicing, but did not completely account for the skipping phenotype (23). Therefore, in addition to PTB, other splicing inhibitors must contribute to regulating α-exon skipping (Fig. 1B).
The complexity associated with the regulation of alternative RNA splicing impedes understanding of how this process can be targeted in human disease and further complicates the development of methods to correct aberrant splicing. To date, approaches involving, trans-RNA splicing (24), tethering of splicing factors (25), pharmacologic agents (26–28) and targeted antisense RNA oligonucleotides (29,30), have been successfully applied to correct aberrant splicing. The latter approach has been broadly applied to correct aberrant splicing of the β-globin, CFTR, dystrophin, tau and bcl-x pre-mRNAs. Antisense RNA oligonucleotides targeted through sequence-specific hybridization to splice sites or adjacent sequences block inappropriate exon selection by inhibiting the binding of trans-acting factors (29,31). Unfortunately, these antisense approaches currently function only to correct mutation-induced changes in splicing and is unclear whether this approach can be extended to targeting other sequences responsible for the regulation of alternative RNA splicing.
In our current study, we demonstrated that antisense morpholino oligonucleotides (MOs) targeted to distant intronic elements can modulate RNA splicing. We addressed whether the hybridization of such MOs to the ISS elements of endogenous FGFR1 RNA to block the binding of trans-acting factors could be used to correct aberrant splicing in glioblastoma cell line SNB-19 (Fig. 1C). We observed that MOs enhanced α-exon inclusion in the SNB-19 cells as well as in several other cancer cell lines. Thus, manipulating the expression of alternative spliced variants under physiologic conditions by targeting intronic elements can serve as a means of modulating alternative splicing of aberrant transcripts and can provide an opportunity to develop new therapeutic agents for use in humans.
RESULTS
Antisense MOs block in vitro protein binding of the ISS1 element in SNB-19 nuclear extracts
In SNB-19 cells, two ISS elements, ISS1 and ISS2, function to inhibit FGFR1 α-exon recognition. These two elements, identified by the deletion and mutation analysis of a transiently transfected FGFR1 mini-gene reporter, are functionally redundant and independently account for 65–75% of the exon-exclusion phenotype (21). As the mini-gene reporter does not accurately duplicate the FGFR1 gene structure, which has extremely large 5′ introns, we sought to confirm the relevance of the ISS1 and ISS2 sequences in endogenous FGFR1 RNA splicing. Previous studies correlated the in vitro binding of PTB to the ISS1 element with enhanced PTB expression in glioblastomas and α-exon skipping (22).
To address whether PTB association with the ISS1 element could be blocked in vitro, we designed three overlapping 25-mer antisense MOs that hybridized to the ISS1 sequence (Fig. 2A). The ability of each of these MOs to block protein association was tested with an ultraviolet (UV) cross-linking assay. Radiolabeled ISS1 RNA was incubated with increasing concentrations of oligonucleotide in the presence of SNB-19 nuclear extracts. In the absence of MO, several proteins bound the ISS1 sequence, including the 57 kDa PTB (Fig. 2B, lane N). Two of the three ISS1 MOs, ISS1B and ISS1C, blocked protein binding (most notably of PTB) to the ISS1 element (Fig. 2B). Treatment with a random RNA MO had no effect on protein binding, even at the highest concentration tested (Fig. 2B, lane R). Incubation with sense MO specific to ISS1A, ISS1B and ISS1C also had no effect on UV cross-linking (data not shown), suggesting that unlike antisense MOs, sense MOs do not effectively compete for protein binding. In addition, all three MOs induced the binding of an ∼50 kDa protein of unknown identity to the ISS1 element (Fig. 2B). This protein may be recognizing the double-stranded RNA structure or may only be able to bind to the ISS1 element after the MOs disrupt the binding of other trans-acting factors.
ISS1 antisense MOs modulate splicing in vivo in SNB-19 cells
We delivered MOs into SNB-19 cells by using the scrape-loading method, which entails the transient disruption of the cell membrane to allow passive diffusion into the cytoplasm (32). Using a fluorescently labeled random MO, we examined the efficiency of oligonucleotide uptake after scrape-loading. Oligonucleotide concentration and uptake were directly correlated: uptake was nearly 80% at the highest concentration (2 µM) (Table 1). Furthermore, scrape-loading had no noticeable effect on cell viability regardless of the concentration of random MO used.
To assess the effect of MO treatment on FGFR1 α-exon splicing, we treated SNB-19 cells using the same delivery method, concentrations and time point, followed by extraction of total RNA for analysis by low-cycle reverse transcriptase (RT)–polymerase chain reaction (PCR). Without MO treatment, the rate of α-exon inclusion varied between 1 and 15% (Figs 2–6, data not shown). The rate of α-exon inclusion in cells treated with random MO did not differ from the rate in untreated cells, even at the highest concentration of random MO (Fig. 2C). Compared with random MO, treatment with each of the ISS1 MOs enhanced α-exon inclusion in a dose dependent manner from ∼2- to ∼3-fold (Fig. 2C). Although the ISS1A MO did not affect in vitro binding of proteins, this oligonucleotide caused the greatest increase in the rate of α-exon inclusion (54% inclusion level following treatment with 2 µM MO). Overall, these data clearly support the idea that the ISS1 element plays a functional role in endogenous FGFR1 RNA splicing. Furthermore, the values obtained for α-exon inclusion likely under represent the effect of oligo treatment when differences in transfection efficiency and existing mRNA pools are considered. However, results obtained using the ISS1A MO suggest that the in vivo binding of trans-acting factors may not be accurately recreated in vitro.
Antisense MOs block in vitro protein binding of the ISS2 element and modulate in vivo RNA splicing
We also synthesized two antisense MOs that targeted the downstream intronic element ISS2 (Fig. 3A). UV cross-linking assays revealed that these oligonucleotides were equally effective in blocking the in vitro association of most proteins in SNB-19 nuclear extracts (Fig. 3B), which suggested either redundant binding sequences or nonspecific association. When we combined both ISS2 MOs, binding of all proteins was completely blocked (data not shown).
Next, we examined the effect of these two ISS2 MOs on FGFR1 splicing in vivo using the same conditions and protocol as for the three ISS1 MOs. Both ISS2 MOs increased the rate of α-exon inclusion in a dose-dependent manner (Fig. 3C). With higher concentrations of the ISS2B MO, α-exon inclusion increased to 70% at 10 µM (Fig. 3D). This finding indicated that with sufficient oligonucleotide and time, inducing a complete switch in RNA splicing may be possible. Overall, the ISS2 MOs were more effective than the ISS1 MOs at changing the rate of α-exon inclusion, suggesting that the ISS2 element is a more important regulator of in vivo splicing or that the ISS2 MOs are more effective competitors of binding.
Finally, to confirm that the changes in the level of α-exon splicing produced functional mRNA, we examined protein expression by western-blot analysis using an FGFR1-specific antibody (Fig. 3E). Treatment of SNB-19 with 5 µM ISS2B MO induced the appearance of a 145 kDa immunoreactive band, consistent with a previous reports of the full-length FGFR1 (16). Analysis also revealed the presence of previously described 80 and 55 kDa bands, which represent truncated receptor isoforms (33).
Modulation of α-exon splicing is specific to the targeting of the ISS elements in SNB-19 cells
To demonstrate that the increased rate of α-exon inclusion was specific to the targeting of the ISS1 and ISS2 elements in SNB-19 cells, we created two additional control MOs—intronic antisense and ISS1A sense (Fig. 4A)—and noted their effect on α-exon splicing. Direct comparison of all MOs at a concentration of 2 µM confirmed that the ISS1A, ISS2A and ISS2B antisense MOs were more effective than the ISS1B and ISS1C antisense MOs in enhancing α-exon inclusion and demonstrated that neither of the additional control MOs appreciably changed the rate of α-exon inclusion compared with that observed using random MO (Fig. 4B). We concluded that the increased level of α-exon inclusion resulted from the sequence-specific hybridization of antisense ISS oligonucleotides. Splicing of the closely related FGFR2 gene was not affected by treatment with ISS1A or ISS2B MO (data not shown), further validating the specificity of targeting intronic elements.
In previous studies, it has been reported that antisense MO targeted to a cryptic 5′ splice site of an endogenous mutant β-globin gene resulted in a corrected splicing rate of ∼70%, 8–15 days following syringe loading of 45 µM MO in culture human mononuclear cells (34). Therefore, as an additional control, we evaluated the efficacy of targeting the 5′ splice site of FGFR1 exon 5 with a targeted antisense MO (Fig. 4A). Blocking the downstream splice site resulted in an overall exon 5 skipping rate of 28±6.1%, without altering the level of α-exon exclusion (Fig. 4B, right lane). This further supports the specificity of targeting and suggests that that targeting FGFR1 ISS elements is at least as effective as targeting splice sites.
Changes in α-exon splicing occur in other cell lines
We focused on characterizing the mechanisms involved in α-exon exclusion in the SNB-19 glioblastoma cell line, but because of the role in cell growth and differentiation, this protein is also expressed in a wide variety of cell types. Again, through the use of targeted MOs, we directly assessed the relevance of the ISS elements in α-exon splicing in human cervical carcinoma (HeLa), prostate carcinoma (PC3), thyroid carcinoma (TPC-1) and pancreatic adenocarcinoma (Panc 28) cell lines. HeLa cells were included because they have been widely used to study the mechanism of splicing, whereas the other three cell lines were included because FGFR1 protein expression has been implicated in progression of these cancers (18,35,36).
We found that for all four cell lines α-exon skipping was the predominant pathway in the absence of MO treatment (data not shown). Compared with the treatment with random MO, ISS1 or ISS2 MOs increased the α-exon inclusion (Fig. 5), confirming the generalization of this approach. Although the four cell lines had approximately the same level of α-exon inclusion (10–15%), their response to specific MO treatment differed from that of the SNB-19 cells and from each other. HeLa cells were the least responsive to the treatment and TPC-1 cells were the most responsive. ISS2 targeting appeared more efficient than ISS1 targeting in the TPC-1 and Panc 28 cells but not in the HeLa or PC3 cells. The level of α-exon inclusion in TPC-1 cells treated with the ISS1C, ISS2A or ISS2B MO and in Panc 28 cells treated with the ISS2B MO mimicked the results obtained in SNB-19 cells treated with the same oligonucleotides (compare Figs 4 and 5). The reason for the differences in response to MO treatment is unclear, but the results suggested that a cell-specific regulatory mechanism is present (e.g. unique splicing factors, cis-elements or a difference in the relative ratio of constitutive splicing factors) or that cell-specific differences in oligonucleotide uptake exist.
Combination of ISS1 and ISS2 antisense MOs suggests a mechanism for splicing regulation
Using an FGFR1 mini-gene reporter, we previously demonstrated that simultaneous deletion of the ISS1 and ISS2 elements resulted in nearly 100% α-exon inclusion (21). This finding suggested that simultaneous treatment with multiple MOs could lead to greater levels of α-exon inclusion than that observed using single MOs. In this study, we used two strategies to test this possibility: simultaneously targeting both elements (1A+2A or 1A+2B) or targeting a single element with multiple element-specific MOs (1A+1C or 2A+2B). Regardless of our strategy, MOs combinations had no additive or synergistic effects on the rate of α-exon inclusion; in fact, the rates were indistinguishable from those obtained with the individual MOs (Fig. 6A; compare 1A+R with 1A+1C, and 2A+R with 1A+2A, 1A+2B and 2A+2B). This finding suggested that the ISS1 and ISS2 elements regulated α-exon skipping through a shared mechanism.
One mechanism that might account for our observations has already been described for the self-regulation of alternative hnRNP A1 RNA splicing (37). Mammalian hnRNP A1 pre-mRNA contains an alternatively spliced exon (7B) that is negatively regulated by a series of conserved intronic elements. In the mouse gene, two of these elements flanking the exon 7B, CE1 and CE4, contain high-affinity protein-binding sites for the hnRNP A1 protein. It has been proposed that through the interaction of the RNA-bound A1 protein, the exon is ‘looped out’ and hidden from the splicing machinery while simultaneously bringing close the more distant splice sites (Fig. 6B, left). Preventing A1 protein association at either CE element would be predicted to disrupt formation of the loop structure (Fig. 6B, right).
To test this prediction, we designed three specific antisense MOs to target the predicted human CE1 and CE4 elements defined through sequence conservation (37). For the 107 nucleotide CE1 element, we chose to specifically target the CE1a and CE1d subregions, which have been demonstrated to be the critical regulatory A1 binding sites upstream of exon 7B (37,38). The small size of the CE4 (24 nt), which appears to have a two strong A1-binding sites (39), allowed targeting using a single MO. We treated HeLa cells with random, CE1a, CE1d or CE4 MOs using the same protocol as described for the FGFR1 gene. Treatment with 0.5 or 2 µM random or targeted CE MO had no effect on the level of exon 7B inclusion (data not shown). Given the reduced targeting efficiency observed in HeLa cells for the FGFR1 ISS elements (Fig. 5) and the relative abundance of A1, this finding is not entirely unexpected. Treatment with 5 µM CE4 MO was able to consistently increase exon 7B inclusion levels (Fig. 6C, data not shown). To our surprise treatment with 5 µM CE1a or CE1d MO alone had no effect on exon 7B inclusion levels (Fig. 6C). A similar finding was observed when CE1a and CE1d were used in combination (Fig. 6C) and at levels as high as 10 µM (data not shown). It is unclear if this finding reflects a failure of targeting, a redundancy of regulatory sequences, or a difference in the regulation of human hnRNP A1. However, the simultaneous targeting of either CE1a or CE1d in combination with CE4 MO reproducibly enhanced exon 7B inclusion to a greater extent than treatment with CE4 alone (Fig. 6C). These results confirm a functional role for the CE1a, CE1d and CE4 elements in regulating endogenous human hnRNP A1 splicing, and further suggest that exon 7B exclusion occurs through cooperative rather than a shared mechanism. Therefore, it appears that regulation of FGFR1 α-exon occurs through a different mechanism than that used for human hnRNP A1 exon 7B.
Effect of changing FGFR1 splicing on glioblastoma cell growth
FGFR1 α-exon is aberrantly spliced concomitant with the progression of glial cell malignancy (20). It is unclear if aberrant FGFR1 splicing serves a direct functional role in glial cell malignancy or is merely a coincidental marker of tumor progression. The ability to correct the aberrant RNA splicing of the endogenous FGFR1 gene provides an approach to examine the relevance of this event on tumor cell growth. We observed no differences in glioblastoma cell growth using standard treatment conditions capable inducing a 50–70% level of α-exon inclusion after 72 h (data not shown). This finding is not entirely unexpected given that one functional role attributed to the inclusion of the α-exon is a 5-fold reduction in ligand affinity (14). Culture conditions employing 10% serum provide potentially saturating concentrations of the FGF ligand, and MO treatment clearly does not completely abolish production of FGFR1β. We believe that the inability to completely shift α-exon inclusion derives from a combination of factors, including uptake of MO and turnover of existing pools of FGFR1β mRNA. Extending the exposure time to MO by including a second scrape-loading was found to further enhance the level of α-exon inclusion. A nearly complete change in α-exon splicing was observed by 96 h when the human U251 glioblastoma cell line was treated with a combination of ISS1A and ISS2B (5 µM each), and a second scrape-loading was performed at 72 h (Fig. 7A). As the presence of serum was found to be required for cell attachment following scrape-loading, we were unable to perform extended growth studies using previously described serum-free conditions (40). Instead, short-term growth studies were performed examining the response to a reduction in serum (from 10 to 1%) following a second scrape-loading. Conditions that resulted in a >90% inclusion of the α-exon had no effect on cell viability, but did induce a significant elevation in the total combined level of activated caspase-3 and -7 (Fig. 7B). Therefore, while short-term differences in growth rate were not observed, the elevation in the proapoptotic enzymes, caspase-3 and -7, suggests that long-term glioblastoma cell growth may be impaired.
DISCUSSION
Our findings demonstrate the feasibility of targeting intronic regulatory elements with antisense RNA oligonucleotides to specifically modulate alternative RNA splicing. The therapeutic potential of antisense oligonucleotide treatment has been clearly demonstrated at the level of gene transcription through the down-regulation of unwanted or deleterious transcripts (41) and at the level of RNA processing through the correction of aberrant splicing events (29–31). These approaches focused primarily on preventing the expression of a specific mRNA or mRNA isoform. An additional study also described the induction of apoptosis in cancer cells by using antisense oligonucleotides to modify alternative RNA splicing and force the production of Bcl-x protein (42). In all cases, modulation of RNA splicing was achieved by targeting oligonucleotides to sequences required for appropriate spliceosome assembly, most frequently splice sites and exonic enhancers (29–31). Although this technique is a useful approach for preventing the recognition of unwanted exons, in many disorders, most notably cancer, it is the aberrant exclusion of exons that is associated with the disease. For this reason, and in consideration of the increasing role attributed to alternative RNA splicing in the regulation of both normal and aberrant cellular processes, we sought to determine whether this approach could be applied to targeting intronic regulators of splicing.
In this study, we generated MOs to hybridize specifically to intronic splicing regulatory elements that regulate human FGFR1 α-exon exclusion (21). Our decision to use MOs was based on a previous study demonstration of their high-targeting specificity and superior nuclease resistance (43). Other oligonucleotide formulations have been used to alter RNA splicing, but we found MOs to be more effective than the commonly used methyl-modified phosphothioate oligonucleotides at regulating α-exon splicing (data not shown).
As MOs are uncharged, we delivered them into cells by using the scrape-loading method. This approach routinely achieved high-uptake of MO (Table 1), but we were unsuccessful at combining liposome-mediated transfection of previously characterized splicing reporters before or after scrape-loading, perhaps because of the fragile state of cells after either procedure. Therefore, we could not directly compare the effect of oligonucleotide-mediated enhancement of α-exon inclusion with that of splicing reporters containing ISS deletions. Transfection of these reporter constructs containing ISS1 or ISS2 element deletions results in an α-exon inclusion rate of 65–75% (21). In our current study, when SNB-19 cells were treated with 2 µM ISS1 or ISS2 MO, the level of endogenous α-exon inclusion ranged from 35 to 60% (Fig. 5). Although it is tempting to compare these numbers directly, the presence of pre-existing endogenous FGFR1 mRNA and the 80% transfection efficiency of the MOs lead to an underestimated rate of α-exon inclusion, and make it inappropriate to do so. Nonetheless, our findings were compelling. Clearly, treatment with targeted MOs specifically enhanced endogenous α-exon inclusion. This finding not only confirmed the functional relevance of the FGFR1 ISS elements first identified using chimeric mini-gene reporters (21), but also generally validated a widely used approach for characterizing regulatory sequences.
Although treating SNB-19 cells with antisense MOs enhanced the rate of α-exon inclusion, these splicing changes did not completely correlate with the rate of in vitro binding of proteins to the ISS1 element. Previous studies demonstrated the specific binding of PTB to the ISS1 element, the dependence of the binding on the presence of the UGC repeat sequences and a functional role for PTB binding in an α-exon exclusion (22,23). Therefore, we would have predicted that the three ISS1 MOs affect PTB binding despite the location of the UCUU binding consensus (44). The ability of the ISS1A MO to modulate splicing in vivo without affecting PTB binding is intriguing. First, it implies a role for additional regulators of α-exon splicing. Indeed, even though the modification of endogenous PTB level was able to enhance or repress α-exon inclusion (23), these changes did not begin to approach the differences we observed in our study with MO treatment. Second, and perhaps of more general importance, is the apparent paradox in the ability of a regulatory sequence to bind protein in vitro and in vivo. Compared with the ISS1A MO, the ISS1B and ISS1C MOs dramatically inhibit in vitro protein binding; however, this difference did not translate into differences in in vivo splicing (Fig. 2). Perhaps, other factors (e.g. nuclear localization, RNA secondary structure, or nucleation of proteins through cooperative binding) are important in exon recognition. Clearly, at least for the ISS1 element, in vivo mechanisms could not be completely reproduced in vitro, which raise the general question of how representative data derived from in vitro protein association are of in vivo alternative splicing mechanisms.
The regulatory elements we targeted in the FGFR1 gene were derived from detailed mapping of these elements in the human gene. Although relatively few alternative splicing pathways involving intronic splicing regulators have been fully characterized, we wanted to examine the potential to apply this approach broadly to other human genes. Our targeting of elements in the human hnRNP A1 gene not only addressed this question, but also provided some insight into the splicing regulatory mechanisms of these two genes. Alternative recognition of the mouse hnRNP A1 exon 7B has been extensively studied: exclusion of exon 7B depends on the presence of high-affinity A1-binding sites flanking the exon, and these regulatory sites are highly conserved between the human and the mouse, implying conservation of function as well (37). Our results targeting the human hnRNP A1 CE1 and CE4 elements confirmed the importance of these sequences in regulation of splicing, but raise questions regarding the precise mechanism. Three generalized mechanisms have been proposed for the negative regulation of exon recognition by ISS sequences: direct blocking of constitutive splicing factor association, enucleation of factors to create a ‘zone of silencing’ and exon ‘looping’ (7,37,45). Studies of the mouse hnRNP A1 gene led to the development of a looping/bridging model (46). In this model, A1 protein binds to high-affinity sites, CE1a, CE1d and CE4, within the introns flanking the repressed exon, and protein dimerization ‘loops out’ the exon while simultaneously bringing close the flanking exons (Fig. 6B). This model predicts that disrupting A1 association at either site would have a comparable effect on exon recognition. Although our studies targeting the predicted human CE elements do not support a looping model, we cannot rule out the possibility that inefficient targeting of the CE elements or the presence of additional A1 binding sites, led to the cooperative effects observed.
PTB has also been proposed to use the looping model for regulation of exon N1 of c-src and FGFR2 genes (45,47). The distal location of the ISS1 and ISS2 elements from the α-exon splice sites and the simultaneous blocking of both elements did not substantially enhance overall α-exon inclusion and were consistent with the looping model. Although PTB was specifically associated with the ISS1 element, our repeated UV cross-linking attempts and immunoprecipitations failed to identify PTB binding to the ISS2 element; similar experiments were unable to identify binding of A1 protein to either element (data not shown). Furthermore, the ISS1A MO, which clearly enhanced in vivo inclusion of α-exon, did not alter the in vitro binding pattern of associated proteins. Therefore, repression of α-exon inclusion is consistent with a mechanism involving exon looping, but the specific trans-regulators remain unknown.
We used the FGFR1 and hnRNPA1 genes to demonstrate how the targeting of splicing regulatory elements with antisense RNA MOs could serve as a useful approach to enhance the inclusion of specific exons. This approach also provides a mechanism to directly assess the biological impact of changing α-exon splicing. Previous studies have clearly demonstrated a strong correlation between α-exon exclusion and gliomagenesis (20). However, for human U251 glioblastoma cells, we found no effect of restoring production of the FGFR1α isoform on cell growth in culture. Unfortunately, the effect of exogenous and autocrine FGF ligands on receptor activity makes interpretation of this finding difficult. In other cell types, the FGFR1 isoforms derived from alternative splicing of the α-exon have been proposed to play a differential role in cell sensitivity to cytotoxin exposure (18,48). Treatment of pancreatic adenocarcinoma cells (PANC-1) overexpressing FGFR1α with 5-fluorouracil induced a 2-fold increase in cell death compared with cells overexpressing FGFR1β (18). In those studies, cell death was dependent on caspase-3 activation suggesting that a similar association may exist in glioblastoma cells. One caveat of these studies is that they have been performed using ectopic overexpression of FGFR1 isoforms rather than altering endogenous RNA splicing. Whether changing production of endogenous FGFR1α enhances sensitivity to cytotoxin exposure in glioblastoma or pancreatic adenocarcinoma cells remains to be addressed.
An important task in the future will be to evaluate the expression of alternative spliced transcripts to decipher the mechanisms that govern both normal and aberrant alternative splicing. This task will likely be daunting because of the size of many genes and the complexity of alternative RNA splicing. We believe that in addition to providing a potential mechanism for correcting aberrant RNA splicing, the use of antisense oligonucleotides will be helpful in studying the splicing of full-length pre-mRNA transcripts under physiologic conditions. Clearly, many alternative splicing events are regulated through the use of intronic regulatory sequences; undoubtedly, countless others remain undiscovered. A recent comparative survey of human and mouse genes found long conserved intronic sequence flanking 77% of alternatively spliced exons compared with only 17% of constitutively spliced exons (49), further supporting a key role for intronic sequence in the regulation of alternative splicing. For genes with alternative splicing patterns conserved across species, the targeting of homologous intronic sequences may allow the identification of important regulatory elements. Our successful targeting of the human hnRNP A1 RNA sequence homologous to the mouse CE elements certainly highlights this possibility. With the ever-increasing amount of available sequence information and new algorithms for comparative analysis, the possibility now exists to apply this approach broadly to mapping and functional analysis of elements, assessment of the in vivo binding affinity of regulatory proteins and, ultimately, examination of the biological consequences of specific mRNA isoform expression.
MATERIALS AND METHODS
Cell culture and MO treatment
We cultured human SNB-19 and U251 glioblastoma cell lines in Dulbecco's Modified Eagle's Medium (low glucose) and propagated human cervical carcinoma (HeLa), prostate adenocarcinoma (PC3), papillary thyroid carcinoma (TPC1), and pancreatic adenocarcinoma (Panc 28) cells in Dulbecco's Modified Eagle's Medium (high glucose). Both media were supplemented with 10% fetal bovine serum. MOs were introduced by the scrape-loading method (32). Cells were seeded in a 100 mm dish at a density of 3×106 cells (SNB-19 cells) or 2×106 cells (HeLa, PC3, TPC1 and Panc 28), 24 h before treatment. At the time of treatment, we washed the cells once with phosphate-buffered saline and then added fresh medium containing the appropriate MO concentration. Cells were then gently removed from the plate surface using a cell scraper (Sarstedt, Newton, NC), resuspended by pipetting and finally transferred to a new plate. For double scrape-loading experiments, U251 cells were seeded in a 100 mm dish at a density of 2×106 cells and treated as described earlier. After 72 h, a second scrape-loading was performed in the presence of 1% fetal bovine serum and 5000 cells/well were transferred to 96-well plates for the assay of viability and caspase activity. The remaining cells were transferred to 6-well plates for the analysis of RNA splicing.
Preparation of nuclear extracts and UV crosslinking analysis
Preparation of nuclear extracts and UV crosslinking were performed as previously described (22). The plasmid construct used to generate the ISS1 transcript was also previously described (22). To obtain the ISS2 transcript, we amplified the entire element by PCR and subcloned it into a pGEM-T easy plasmid vector (Promega, Madison, WI, USA). RNA probes were transcribed in vitro with T7 bacteriophage RNA polymerase (Ambion, Austin, TX, USA) in the presence of [32P]UTP and gel purified. We performed UV cross-linking reactions by incubating [32P]UTP RNA transcripts with MO in 10 µl of reaction mixture containing 1% polyethylene glycol, 0.0625 mM ATP, 25 mM creatine phosphate, 1 mM MgCl2, 20% Dignam buffer D (50) and 30% of nuclear extract at 30°C for 10 min. RNA was digested with 10 µg of RNAse A and 0.65 µg of RNAse T1 at 30°C for 20 min. The cross-linked proteins were visualized by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and by autoradiography.
RNA isolation and splicing analysis
Total RNA was obtained with an Rneasy kit (Qiagen, Valencia, CA, USA) from cells 72 h after MO treatment. We performed RT reactions on 1–3 µg of total RNA in 20 µl reactions containing 50 mM Tris–HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 0.2 mM dithiothreitol, 0.25 mM dNTP, 0.5 µM reverse primer and 40 U of RNase inhibitor (Promega) for 1.5 h at 37°C. All PCRs were performed using 10 µl of the RT mixture in a 50 µl reaction containing 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 1.5 mM MgCl2, 0.25 mM dNTP, 0.3 µM each of forward and reverse primer, 0.03 µM32P-labeled reverse primer and 2.5 U of Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA). After 3 min of denaturation, cycling conditions were as follows: 94°C for 30 s, 62°C for 30 s and 72°C for 30 s, a total of 18 cycles.
We detected endogenous FGFR1 RNA splicing with the use of FP172 primers (5′-GGA AGT GCC TCC TCT TCT G-3′) and FP173 primer (5′-TTA TGA TGC TCC AGG TGG CA-3′). PCR products from the hnRNPA1 gene were amplified with HumA1-3F primer (5′-GAT GGC TAT AAT GGA TTT GG-3′), HumA1-3R primer (5′-ACC TTG GTT TCG TGG TTT TG-3′). We analyzed PCR products, as previously described, using 10% polyacrylamide gel electrophoresis and quantification using a Model GS 363 Molecular Imager System (Bio-Rad, Hercules, CA, USA). RT–PCR conditions were within the linear amplification range for RNA isolated from the tested cells.
Western blot analysis
We examined FGFR1 receptor isoforms in SNB-19 cells treated for 96 h with random MO or ISS2A MO. Total cellular proteins were extracted using a protein extraction solution (Pro-Prep, Bioplex Inc., Houston, TX, USA). Total protein was quantified with the Bio-Rad protein assay; 1 mg of total protein was immunoprecipitated overnight using 2 µg of anti-FGFR1 antibody (sc-7945) (Santa Cruz Antibodies, Santa Cruz, CA, USA) and Protein G Sepharose Beads (Amersham Bioscience, Piscataway, NJ, USA). Beads were washed three times in 150 mM NaCl, 50 mM Tris (pH 7.5) and 0.05% NP-40 (N-6507, Sigma), and the remaining bound protein was eluted by boiling in sodium dodecyl sulfate loading buffer. Western blot analysis was performed as previously described (16) and using a 1 : 1000 dilution of the anti-FGFR1 antibody.
Flow cytometry
We assessed transfection efficiency 72 h after scrape-loading using a fluorescence-labeled random MO (Gene-Tools, Inc., Philomath, OR, USA). SNB-19 cells were harvested by trypsinization, washed and resuspended in 1 ml of cell culture medium containing propidium iodide. The percentages of fluorescent cells and viable cells were analyzed with a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Cell viability and apoptosis
Cell viability and measurement of caspase-3 and -7 activity were measured using CellTiter-Blue Cell Viability Assay and Apo-ONE Homogeneous Caspase-3/7 Assay (Promega). The assays were multiplexed in the same culture well as recommended by the manufacturer, with fluorescence measured 4 h after the addition of resazurin (viability assay) and again 12 h after the subsequent addition of Z-DEVD-R110 (caspase-3/7 assay).
MO sequences
MOs were synthesized by Gene-Tools Inc. Specific antisense sequences were ISS1A (5′-GAA AGA GGC AAA GTT AGG AAG CAG C-3′), ISS1B (5′-GAA AGA GGC AAA GTT AGG AAG CAG C-3′), ISS1C (5′-AAA AGT CAA AGA AGA AAG AGG CAA-3′), ISS2A (5′-GCG GCA GGC AGG GAG CAA TGT TAG T-3′), ISS2B (5′-GGC AGC AGT TTC TGA AGC AGA GTG-3′), random (5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′), intronic control (5′-TCT TTG CCA AGA TTG CCA CTT GCC A-3′), 5′ splice site control (5′-TGC ACA CAC ACG TAC CTT GTA GCC T-3′), CE1a (5′-CTC TAC TAA CCT ATT CTA AAG ATC C-3′), CE1d (5′-GTT TGA CAT GCA CCA GAA TTT TAG T-3′) and CE4 (5′-AAG CTC TAA AAG GCT AAT CTA GCT G-3′). The sense sequences were ISS1A (5′- GCT GCT TCC TAA CTT TGC CTC TTT C-3′), ISS1B (5′-ATC CGC TTA ATG CTG CTA CAG CTG C-3′), ISS1C (5′-TTG CCT CTT TCT TCC TTT GAC TTT T-3′).
ACKNOWLEDGEMENTS
The authors thank Elizabeth L. Hess, Hannah Cheung and Miles Wilkinson for useful discussions and critical reading of this manuscript. The project was funded by the NIH grant (CA67946) and the Texas ATP Grant (003657-0147-1999) to G.J.C. and W.J. Additional support for I.G.B. was provided by a Schissler Foundation Fellowship Award and NIH Minority Supplement.
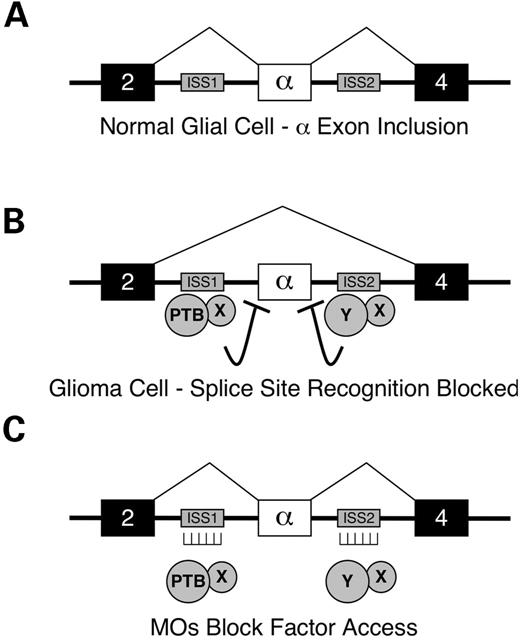
Figure 1. Model for antisense RNA MO-mediated correction of FGFR1 α-exon splicing in human glioblastoma cells. (A) In normal glial cells, exon 3 (the α-exon) of the FGFR1 gene is included during RNA splicing. Exons are represented by large boxes, introns by thick lines, RNA splicing pathways by thin lines. (B) The trans-acting factor PTB is overexpressed in glioma cells and along with another unknown inhibitor(s) (X, Y), and mediates α-exon exclusion through binding of flanking ISS elements (small grey boxes). (C) Antisense RNA MOs hybridized to the ISS elements block the binding of trans-acting factors, thereby allowing α-exon inclusion.
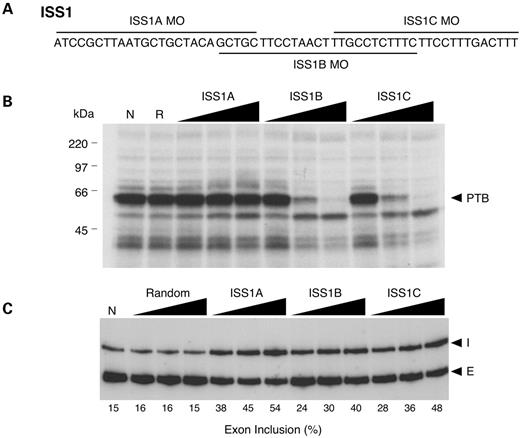
Figure 2. Targeting the ISS1 element with antisense MOs in SNB-19 cells. (A) Sequence of the ISS1 element and the targeting positions of the three overlapping MOs used. (B) UV cross-linking assay was used to assess the ability of each MO to block association of SNB-19 cell nuclear extract proteins with the ISS1 element. In the absence of MO treatment (N), PTB (∼57 kDa) is the predominant protein associated with ISS1. Random (R), ISS1A, ISS1B and ISS1C MOs were added at a transcript-to-MO ratio of 10 : 1, 1 : 1 and 1 : 10. (C) RT–PCR analysis of endogenous FGFR1 α-exon splicing in vivo. SNB-19 cells were scrape-loaded in the absence (N) or presence of increasing concentrations (0.5, 1 and 2 µM) of random, ISS1A, ISS1B or ISS1C MO. I, α-exon inclusion; E, α-exon exclusion.
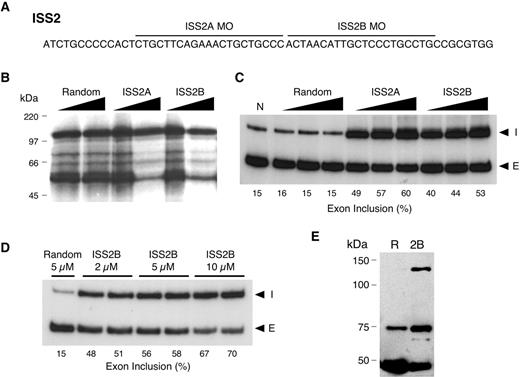
Figure 3. Targeting the ISS2 element with antisense MOs in SNB-19 cells. (A) Sequence of the ISS2 element and the targeting positions of the two MOs used. (B) UV cross-linking assay was used to assess the ability of each MO to block association of SNB-19 cell nuclear extract proteins with the ISS2 element. Random, ISS2A and ISS2B MOs were added at transcript-to-MO ratios of 1 : 1 and 1 : 5. (C) RT–PCR analysis of endogenous FGFR1 α-exon splicing in vivo. SNB-19 cells were scrape-loaded in the absence (N) or presence of increasing concentrations (0.5, 1 and 2 µM) of random, ISS2A or ISS2B MO. (I) α-exon inclusion; (E) α-exon exclusion. (D) Same as (C) except for MO concentrations. (E) Western blot analysis using an FGFR1-specific antibody after treatment of SNB-19 cells with 5 µM of random (R) or ISS2B (2B) MO. The appearance of an ∼145 kDa immunoreactive band is consistent with the expected size of the full-length receptor. The ∼55 and ∼80 kDa represent truncated FGFR1 isoforms.
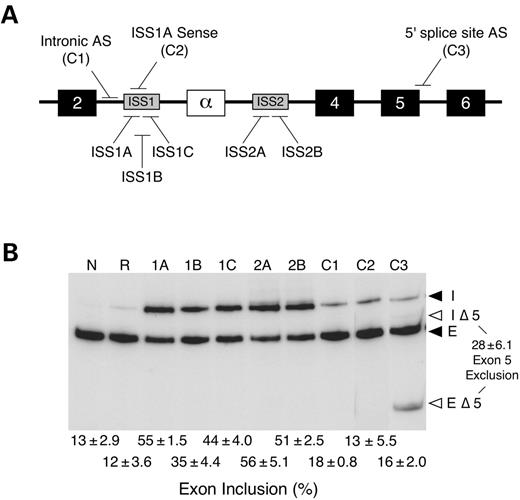
Figure 4. Enhanced α-exon inclusion requires specific antisense MO targeting in SNB-19 cells. (A) Relative positions of the ISS antisense MOs, control MOs: intronic antisense (C1), ISS1A sense (C2) and 5′ splice site antisense (C3). The C1 MO targets the sequence ∼100 nt upstream of ISS1. (B) RT–PCR analysis of endogenous FGFR1 α-exon splicing in vivo. SNB-19 cells were scrape-loaded in the absence (N) or presence of random (R), ISS1 (1A–1C), ISS2 (2A, 2B) or control (C1–C3) MOs. The level of α-exon inclusion is presented below the gel. The level of exon 5 exclusion is indicated to the right of the gel. All values represent the mean±standard deviation of three independent experiments. I, α-exon inclusion; IΔ5, α-exon inclusion product lacking exon 5; E, α-exon exclusion; EΔ5, α-exon exclusion product lacking exon 5.
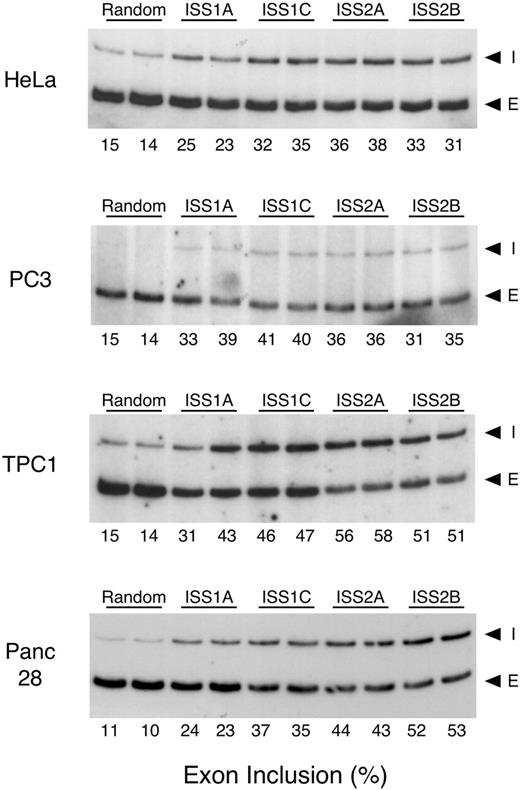
Figure 5. Effect of ISS antisense MO treatment in other human cell lines. RT–PCR analysis of endogenous FGFR1 α-exon splicing was performed on cervical carcinoma (HeLa), prostate carcinoma (PC3), thyroid carcinoma (TPC-1), and pancreatic adenocarcinoma (Panc 28) cells, after treatment with the 2 µM MOs. I, α-exon inclusion; E, α-exon exclusion.
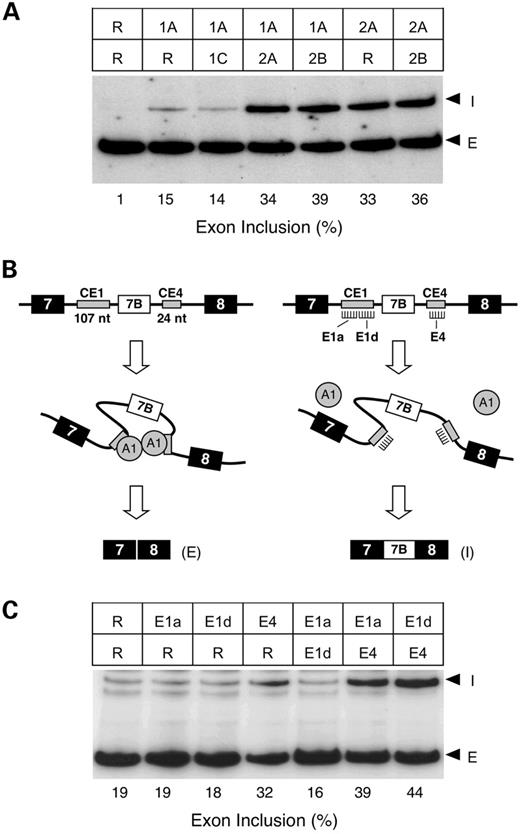
Figure 6. Antisense MO treatment supports an exon ‘looping’ model. (A) RT–PCR analysis of endogenous FGFR1 α-exon splicing in vivo. SNB-19 cells were scraped in the presence of various combinations of 0.5 µM random (R), ISS1 and ISS2 MOs. The random only contains a final concentration of 1 µM MO. (I) α-exon inclusion; (E) α-exon exclusion. (B) The exon ‘looping out’ model proposed for regulation of mouse hnRNP A1 exon 7B exclusion, which involves the binding of A1 protein to intronic ISS elements, CE1 and CE4 and the dimerization of A1 protein. Left: no MO treatment. Right: treatment with CE4 MO. (C) RT–PCR analysis of hnRNP A1 exon 7B splicing in RNA isolated from human cervical carcinoma (HeLa) cells treated in vivo with various combinations of 5 µM random (R), CE1a antisense (E1a), CE1d antisense (E1a) and CE4 antisense (E4) MOs. The random only lane contains a final concentration of 10 µM MO. I, exon 7B inclusion; E, exon 7B exclusion.
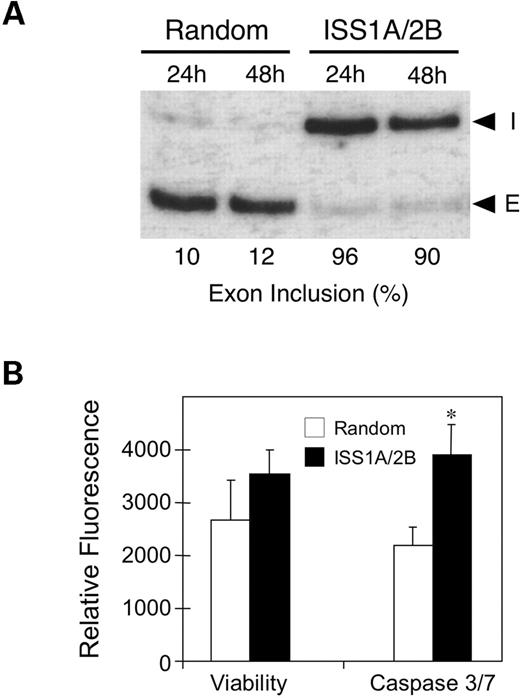
Figure 7. Extended treatment with antisense MO completely corrects FGFR1 α-exon splicing without affecting cell viability. U251 glioblastoma cells were treated with either 10 µM random MO or a combination of 5 µM of each ISS1A and ISS2B. After 72 h, a second treatment was performed in reduced serum. (A) RT–PCR analysis of endogenous FGFR1 α-exon splicing at 24 and 48 h following the second scrape-loading. (I) α-exon inclusion; (E) α-exon exclusion. (B) Cell viability and combined caspase-3/7 activity was performed on parallel cultures 24 h following the second scrape-loading. Data provided are the average relative fluorescent determined for two independent experiments performed in triplicate ± the standard deviation. The asterisk (*) indicates a P-value of <0.0001 determined by Student's t-test.
In vivo uptake of fluorescent random MO in SNB19 cells
| SNB19 cells | Oligonucleotide concentration (µM) | |||
|---|---|---|---|---|
| 0 | 0.5 | 1.0 | 2.0 | |
| Positive [mean±standard deviation (%)] | 00.8±0.9 | 26.8±2.9 | 48.4±3.3 | 77.7±1.4 |
| Viable [mean±standard deviation (%)] | 93.5±2.0 | 94.2±0.8 | 95.1±0.8 | 95.0±0.3 |
| SNB19 cells | Oligonucleotide concentration (µM) | |||
|---|---|---|---|---|
| 0 | 0.5 | 1.0 | 2.0 | |
| Positive [mean±standard deviation (%)] | 00.8±0.9 | 26.8±2.9 | 48.4±3.3 | 77.7±1.4 |
| Viable [mean±standard deviation (%)] | 93.5±2.0 | 94.2±0.8 | 95.1±0.8 | 95.0±0.3 |
In vivo uptake of fluorescent random MO in SNB19 cells
| SNB19 cells | Oligonucleotide concentration (µM) | |||
|---|---|---|---|---|
| 0 | 0.5 | 1.0 | 2.0 | |
| Positive [mean±standard deviation (%)] | 00.8±0.9 | 26.8±2.9 | 48.4±3.3 | 77.7±1.4 |
| Viable [mean±standard deviation (%)] | 93.5±2.0 | 94.2±0.8 | 95.1±0.8 | 95.0±0.3 |
| SNB19 cells | Oligonucleotide concentration (µM) | |||
|---|---|---|---|---|
| 0 | 0.5 | 1.0 | 2.0 | |
| Positive [mean±standard deviation (%)] | 00.8±0.9 | 26.8±2.9 | 48.4±3.3 | 77.7±1.4 |
| Viable [mean±standard deviation (%)] | 93.5±2.0 | 94.2±0.8 | 95.1±0.8 | 95.0±0.3 |
References
Johnson, J.M., Castle, J., Garrett-Engele, P., Kan, Z., Loerch, P.M., Armour, C.D., Santos, R., Schadt, E.E., Stoughton, R. and Shoemaker, D.D. (
Maniatis, T. and Tasic, B. (
Graveley, B.R. (
Herbert, A. and Rich, A. (
Lopez, A.J. (
Black, D.L. (
Smith, C.W. and Valcarcel, J. (
Graveley, B.R. (
Stenson, P.D., Ball, E.V., Mort, M., Phillips, A.D., Shiel, J.A., Thomas, N.S., Abeysinghe, S., Krawczak, M. and Cooper, D.N. (
Stamm, S. (
Faustino, N.A. and Cooper, T.A. (
Groth, C. and Lardelli, M. (
Wang, F., Kan, M., Yan, G., Xu, J. and McKeehan, W.L. (
Stachowiak, M.K., Fang, X., Myers, J.M., Dunham, S.M., Berezney, R., Maher, P.A. and Stachowiak, E.K. (
Clarke, W.E., Berry, M., Smith, C., Kent, A. and Logan, A. (
Maciag, T. and Friesel, R.E. (
Vickers, S.M., Huang, Z.Q., MacMillan-Crow, L., Greendorfer, J.S. and Thompson, J.A. (
Luqmani, Y.A., Mortimer, C., Yiangou, C., Johnston, C.L., Bansal, G.S., Sinnett, D., Law, M. and Coombes, R.C. (
Yamaguchi, F., Saya, H., Bruner, J.M. and Morrison, R.S. (
Jin, W., Huang, E.S., Bi, W. and Cote, G.J. (
Jin, W., McCutcheon, I.E., Fuller, G.N., Huang, E.S. and Cote, G.J. (
Jin, W., Bruno, I.G., Xie, T.X., Sanger, L.J. and Cote, G.J. (
Garcia-Blanco, M.A. (
Cartegni, L. and Krainer, A.R. (
Chang, J.G., Hsieh-Li, H.M., Jong, Y.J., Wang, N.M., Tsai, C.H. and Li, H. (
Andreassi, C., Jarecki, J., Zhou, J., Coovert, D.D., Monani, U.R., Chen, X., Whitney, M., Pollok, B., Zhang, M., Androphy, E. et al. (
Slaugenhaupt, S.A., Mull, J., Leyne, M., Cuajungco, M.P., Gill, S.P., Hims, M.M., Quitero, F., Axelrod, F.B. and Gusella, J.F. (
Sazani, P. and Kole, R. (
Gebski, B.L., Mann, C.J., Fletcher, S. and Wilton, S.D. (
Villemaire, J., Dion, I., Elela, S.A. and Chabot, B. (
Partridge, M., Vincent, A., Matthews, P., Puma, J., Stein, D. and Summerton, J. (
Hanneken, A., Ying, W., Ling, N. and Baird, A. (
Suwanmanee, T., Sierakowska, H., Lacerra, G., Svasti, S., Kirby, S., Walsh, C.E., Fucharoen, S. and Kole, R. (
Cocks, H.C., Thompson, S., Turner, F.E., Logan, A., Franklyn, J.A., Watkinson, J.C. and Eggo, M.C. (
Feng, S., Wang, F., Matsubara, A., Kan, M. and McKeehan, W.L. (
Chabot, B., LeBel, C., Hutchison, S., Nasim, F.H. and Simard, M.J. (
Hutchison, S., LeBel, C., Blanchette, M. and Chabot, B. (
Blanchette, M. and Chabot, B. (
Yamada, S.M., Yamaguchi, F., Brown, R., Berger, M.S. and Morrison, R.S. (
Mercatante, D.R., Bortner, C.D., Cidlowski, J.A. and Kole, R. (
Sazani, P., Kang, S.H., Maier, M.A., Wei, C., Dillman, J., Summerton, J., Manoharan, M. and Kole, R. (
Perez, I., Lin, C.H., McAfee, J.G. and Patton, J.G. (
Wagner, E.J. and Garcia-Blanco, M.A. (
Nasim, F.U., Hutchison, S., Cordeau, M. and Chabot, B. (
Chan, R.C. and Black, D.L. (
Jiao, J., Greendorfer, J.S., Zhang, P., Zinn, K.R., Diglio, C.A. and Thompson, J.A. (
Sorek, R. and Ast, G. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}