




Abstract
X-linked cyclin-dependent kinase-like 5 (CDKL5 or STK9) has recently been implicated in atypical Rett and X-linked West syndromes, severe neurological disorders associated with mental retardation, loss of communication and motor skills and infantile spasms and seizures in predominantly females. Besides CDKL5, these disease phenotypes are also linked to mutations in the MECP2 and ARX genes. Here, we have expressed and characterized CDKL5 and its mutant forms. CDKL5 is a 118 kDa protein that is widely distributed in all tissues, with highest levels in brain, thymus and testes. Whole mount embryo staining reveals CDKL5 to be ubiquitous. Within cells, CDKL5 is localized primarily in the nucleus. Removal of the C-terminal domain increases CDKL5 expression, enhances autophosphorylation activity and causes perinuclear localization, indicating that the C-terminus regulates CDKL5 function. Although we detect MeCP2 but not ARX binding to CDKL5, our results suggest that neither of these proteins are direct substrates of the CDKL5 kinase. Finally, the CDKL5 mutations associated with the disease phenotype cause loss of kinase activity as assessed by autophosphorylation. These results suggest that inactivation of the CDKL5 kinase can lead to severe neurodevelopmental disorders.
INTRODUCTION
Protein kinases regulate a wide range of cellular processes from cell proliferation and differentiation to apoptosis. Aberrations in kinase activity and expression, whether due to mutation or mis-regulation, have increasingly been linked to the pathogenesis of diseases and genetic developmental disorders. Recently, mutations in c yclin- d ependant k inase- l ike 5 (CDKL5), previously known as serine/threonine kinase 9 (STK9), have been connected to X-linked neurological developmental disorders involving severe mental retardation and seizures appearing in the first postnatal months ( 1 – 5 ). The first described mutations of CDKL5 were X/autosomal translocations in two unrelated girls who, in addition to seizures and retardation, had hypsarrythmia and infantile spasms ( 1 ). These symptoms were collectively diagnosed as X-linked West syndrome, a disorder previously attributed to the mutation of a homeobox transcription factor, aristaless (ARX). More recently, point mutations in CDKL5 have been reported in patients with atypical variants of Rett syndrome (RTT), infantile spasms and severe neurodevelopmental retardation. Classical RTT is a neurodevelopmental disorder in predominantly young females and causes retardation with loss in motor and vocal skills and many additional problems such as microencephaly, ataxia, growth retardation and scoliosis. Atypical variants of RTT include early onset of epileptic seizures and infantile spasms. Ninety five percent of classical RTT and 40–50% of atypical cases have been attributed to mutations in the gene coding for methyl CpG binding protein, MECP2 ( 6 ). As indicated earlier, mutations in CDKL5 are responsible for some of the RTT atypical cases.
CDKL5 was cloned via an exon trapping method utilized to screen candidate genes in Xp22, a region where a number of genetic disorders have been found ( 7 ). Sequence analysis suggests that CDKL5 is a member of a proline-directed kinase subfamily that has homology to both MAP and cell-cycle-dependent kinases known as the cyclin-dependent kinase-like (CDKL) kinases. Other members of this family include p56KKIAMRE (CDKL1) and p42KKIALRE (CDKL2) and NKIAMRE (CDKL3). All CDKLs have a TXY sequence that aligns and corresponds to the TXY activation motifs of classic MAP kinases such as ERK2.
Because mutations in either the CDKL5 or MECP2 genes lead to similar genetic disorders, the relationship between them was recently investigated. Two isoforms of MeCP2 have been identified, the brain-rich isoform MeCP2E1, and the less abundant but previously characterized isoform, MeCP2E2 ( 8 , 9 ). CDKL5 had overlapping expression patterns with MeCP2 in the brain and was associated in vivo with MeCP2 ( 10 ). In addition, the authors reported that the non-brain isoform of MeCP2, now termed MeCP2E2, is a substrate of CDKL5 in an in vitro kinase assay, raising the possibility that MeCP2E2 is the direct target of CDKL5 action.
In this study, we further characterize CDKL5 and demonstrate that the C-terminal domain of CDKL5 regulates cellular localization, kinase activity and stability. We also conducted kinase assays with both MeCP2E2 and ARX and found no evidence that they are direct substrates of CDKL5. Finally, we constructed mutant variants of CDKL5 corresponding to those found in diseased patients and examined their function. Our results suggest that loss of CDKL5 kinase activity can lead to neurological disorders such as the West syndrome and the RTT.
RESULTS
Previous northern analyses have established that CDKL5 transcripts are expressed in a variety of human and mouse tissues including brain, placenta, lung, testes, heart, pancreas, liver and kidney. To confirm and extend these observations, we screened EST databases for tissue-specific expressed sequence tags. These analyses also identified EST transcripts from prostate, bone marrow and eye, and the species expressing CDKL5 ranged from human, bovine, rat and mouse to frog (Supplementary Material, Table S1). Interestingly, a bioinformatics search of insect genomes did not identify any apparent CDKL5 orthologues (data not shown). In order to identify cell lines that express CDKL5, we utilized several CDKL5 probes from both the N and C-termini of human CDKL5 and analyzed lysates by reverse transcription–polymerase chain reaction (RT–PCR). As shown in Figure 1 , we identified CDKL5 transcripts in a variety of human and rat cell lines. These include human androgen-dependent and androgen-independent prostate tumor cells (PC3, Du145, LnCAP), human embryonic kidney cells (293), rat neural cell lines [H19-7 (hippocampal), H19-5 (glial), RN33B (Raphe), PC12 (pheochromocytoma)] and human lung cells [Human bronchial epithelial (HBE), human airway epithelial (HAE), human tracheal epithelial (HTE)]. In general, only one transcript was observed, although two bands were detected by RT–PCR in PC12 cells, possibly due to a splice variant of CDKL5. These results suggest that CDKL5 is a kinase that is highly conserved and widely expressed in a variety of eukaryotic species and cell types.
In order to confirm the expression of CDKL5 in tissues and cells, we developed an antibody against a 17-mer peptide from the C-terminus of CDKL5. As shown in Figure 2 A, the CDKL5-C antibody specifically recognizes FLAG-CDKL5 that had been transfected into COS cells. Using this antibody, we screened lysates from a variety of rat tissues for CDKL5 protein expression by western blotting and normalized expression to total protein levels. By this measure, CDKL5 is expressed at highest levels in testes, brain and thymus, moderate levels in heart, lung, spleen and prostate and lower but detectable levels in liver, pancreas and skeletal muscle (Fig. 2 B). Interestingly, two bands were observed in many lysates including those from brain and most neural cell lines. The second smaller band may reflect a splice variant, as two transcripts were detected in PC12 cells, or may result from protein degradation either within cells or during sample preparation. No CDKL5 was observed in the rat kidney tissue lysates despite previous detection of CDKL5 transcripts in kidney ( 7 ). This lack of protein detection may be due to low CDKL5 expression or sample degradation. Finally, western blotting analysis of neural cell lines (H19-7, H19-5, RN3B, PC12) that express CDKL5 transcripts revealed CDKL5 protein as well (Fig. 2 C). These results confirm that the CDKL5 protein has a broad tissue distribution and is also present in cells derived from these tissues.
We then examined CDKL5 protein expression in the whole embryo by immunostaining with the anti-CDKL5 antibody. Analysis of mouse embryos at days E7.5 and E9.5 revealed a broad CDKL5 distribution at both embryonic stages. As indicated in Figure 3 A and C, there were no distinctive areas of intense staining, indicating that CDKL5 is ubiquitous throughout the embryo. Incubation with a blocking peptide corresponding to the antibody epitope suppressed immunoreactivity (Fig. 3 B), suggesting that the immunostaining is specific and correlates well with the previously described western analysis of the tissues.
The methyl CpG binding protein, MeCP2, has been found to localize to the nucleus ( 11 ). As CDKL5 has been reported to associate with MeCP2E2 ( 10 ), we determined the cellular localization of CDKL5. We analyzed both transfected FLAG-CDKL5 and endogenous CDKL5 in HEK293 cells. As shown in Figure 4 A and B, both transfected and endogenous CDKL5 are localized primarily in the nucleus although some cytoplasmic expression was also observed. These results suggest that CDKL5 and MeCP2 co-localize in cells, strengthening the evidence for their association and the likelihood that they functionally interact.
The sequence of CDKL5 reveals a kinase domain at the N-terminus and a large C-terminal region that spans two-thirds of the protein. To examine the function of this C-terminal region, we constructed a truncation mutant, CDKL5ΔC, that contains solely the kinase domain. This mutant is 352 residues and is similar in size to p42 ERK2. Transfection of this construct in cells revealed that CDKL5ΔC has increased expression relative to full-length CDKL5 that had been transfected at a comparable level (Fig. 5 A). Analysis of a co-transfected GFP expression vector showed comparable transfection efficiencies for both the full-length CDKL5 and mutant CDKL5ΔC (data not shown). This data indicates that the C-terminal region may be involved in the stability of CDKL5.
We also examined the truncation mutant's kinase activity via an in vitro autophosphorylation assay. To control non-specific phosphorylation, kinase-dead mutants (CDKL5 K42R and CDKL5ΔC K42R) were constructed and also tested. The kinase-dead constructs have a mutation in the ATP binding pocket of the kinase that removes their ability to bind ATP and transfer the phosphoryl group to the substrate. The results show that CDKL5ΔC has a significantly higher autophosphorylation activity than full-length CDKL5 (Fig. 5 B). To determine whether this difference was due to increased kinase activity resulting from lack of the C-terminus or was merely a reflection of higher truncated protein expression, we normalized the autophosphorylation to protein levels (Fig. 5 C). The results indicate that CDKL5 lacking the C-terminus has higher kinase activity. It should be noted that the full-length kinase-dead CDKL5 K42R has apparent autophosphorylation activity that is due to an associated kinase binding to the C-terminal domain. Thus, the C-terminal region apparently acts as a negative regulatory domain that can also destabilize the kinase.
These results are supported by analysis of the TEY motif in the activation loop of CDKL5. The TXY motif must be phosphorylated at both the threonine and tyrosine residues for maximal activation of MAP kinases ( 12 ), and the degree of phosphorylation can be assessed using immunoblotting with anti-phosphoMAP kinase antibodies. Analysis of TEY phosphorylation in the full-length versus C-terminal truncated CDKL5 enzymes shows higher phosphorylation of CDKL5ΔC relative to full-length CDKL5 (Fig. 5 D an E). This difference is comparable with that observed for CDKL5ΔC versus CDKL5 kinase activity (discussed earlier). No TEY phosphorylation is observed in kinase-dead mutants (Fig. 5 D and E), and mutation of the T and/or Y residues of the TEY motif causes loss of autophosphorylation (data not shown). These results indicate that the C-terminus of CDKL5 is a negative regulator of TEY phosphorylation and are consistent with the possibility that TEY phosphorylation is required for CDKL5 activity.
The influence of the C-terminal domain on cellular localization and protein association was also investigated. Immunocytochemistry shows that the truncated CDKL5ΔC localizes in the perinucleus and the cytoplasm, distinct from the predominantly nuclear localization of the wild-type CDKL5 (Fig. 5 F). To confirm that MeCP2E2 requires the C-terminal domain of CDKL5 for association with the kinase ( 10 ), we co-transfected cells with expression vectors for Myc-MeCP2E2 and either full-length FLAG-CDKL5 or truncated FLAG-CDKL5ΔC and immunoprecipitated cell lysates with antibody to Myc. Analysis of the immunoprecipitates by immunoblotting with anti-FLAG antibody revealed some co-precipitation of the full-length but not the truncated CDKL5 (Fig. 6 A). These results indicate that the C-terminal region of CDKL5 is required for proper nuclear subcellular localization, and the truncated CDKL5ΔC does not associate with the nuclear protein MeCP2E2.
As the reported in vitro phosphorylation of MeCP2E2 by CDKL5 was relatively weak ( 10 ), we examined whether the more active CDKL5ΔC would be more effective at phosphorylating MeCP2E2 than the full-length enzyme. Thus, both CDKL5 and CDKL5ΔC were incubated with MeCP2E2 in an in vitro kinase assay (Fig. 6 B and C). HEK293T cells were transfected with either CDKL5 or MeCP2E2, lysed and immunoprecipitated with anti-FLAG and anti-Myc, respectively, and the immunoprecipitates bound to agarose beads. Both CDKL5 and MeCP2E2 bound beads were either eluted or subjected directly to an in vitro kinase assay. The results reveal that, although there is a weak phosphorylation band at 70 kDa that corresponds to the size of MeCP2E2, the same band is also detected when MeCP2E2 is incubated with the inactive CDKL5. This effect is more prominent in lanes containing MeCP2E2 bound to beads, as the eluted MeCP2E2 is present at a lower concentration. The observed phosphorylation of MeCP2E2 results from a co-precipitating, non-specific kinase activity. Moreover, the truncated CDKL5 shows no further increase in MeCP2E2 phosphorylation than the full-length enzyme despite the fact that it is more active. Thus, the results clearly suggest that MeCP2E2 is not a direct substrate of CDKL5.
The first mutations in CDKL5 were reported in patients with X-linked West syndrome which is also caused by mutation in ARX ( 1 ). We, therefore, determined whether there was an association between CDKL5 and ARX by co-immunoprecipitation. Initially, 293T cells were transfected with tagged CDKL5 and ARX constructs. Immunoprecipitation of CDKL5 and subsequent immunoblotting for co-immunoprecipitated ARX did not reveal any association between the two proteins (Fig. 7 A). Although no stable interaction was detected, it is still possible that ARX is a substrate of CDKL5. We, therefore, measured ARX phosphorylation by CDKL5 both in vitro and in vivo . The 293T cells were transfected with FLAG-CDKL5 and ARX constructs, and isolated by immunoprecipitation. Both CDKL5 and CDKL5ΔC (either bound to beads or eluted) were incubated with ARX in an in vitro kinase assay using the same conditions as described earlier for MeCP2E2. As shown in Figure 7 B and C, no specific phosphorylation of ARX by CDKL5 was detected. Similarly, in vivo phosphorylation measured by 32 P-orthophosphate labeling of cells transfected with ARX and CDKL5 revealed that there was no additional phosphorylation of ARX in the presence of CDKL5 versus ARX alone (Fig. 7 D). Taken together, these results suggest that CDKL5 does not phosphorylate ARX. Hence, although CDKL5 and ARX may have similar mutant phenotypes, the two proteins do not appear to interact directly.
There have been a number of genetic studies demonstrating that the CDKL5 gene is mutated in patients diagnosed with X-linked West syndrome and atypical RTT. To characterize the functional consequence of the various naturally occurring mutations, we analyzed their kinase activity. The first study to show CDKL5 mutations, revealed a translocation event in one of the patients that would have resulted in a transcript coding for 271 residues, and, in turn, a prematurely truncated kinase domain ( 1 ). We constructed this truncated mutant as well as two mutants from atypical RTT patients ( 2 ) with point mutations in the kinase domain at residues C152F and R175S (Fig. 8 A). These point mutants were constructed without the negative regulatory C-terminus and the mutants were assessed by in vitro autophosphorylation kinase assay. The most truncated CDKL5-271 mutant, although expressed, appears to be significantly more unstable than the point mutants (see model subsequently). The results show that none of the naturally occurring CDKL5 mutants are active kinases as assessed by autophosphorylation activity (Fig. 8 B).
To further characterize the nature of these mutations, we have generated a putative structure of CDKL5 using SwissModel ( 13 ). The CDKL5 kinase domain structure was modeled using ERK2 as a template (Fig. 9 A). The overlay reveals that the backbone of the predicted CDKL5 kinase domain and that of ERK2 are nearly superimposable. The sites of the CDKL5 mutations from the West syndrome and the RTT patients that have been characterized here are shown in Figure 9 B. The deletion of residues 271–300 removes two alpha helical loops that stabilize the C-terminal lobe of the kinase domain. The cysteine at position 152 of CDKL5 precedes the invariant DFG motif that is present in all kinases and is critical for phosphoryl transfer ( 14 ). Mutation of the cysteine to a phenylalanine residue causes insertion of a bulky group that may interfere with the function of the neighboring DFG sequence, (Fig. 9 C) causing loss of kinase activity in the C152F mutant of CDKL5. Similarly, the arginine at position 175 may also play a critical role in CDKL5 kinase activation. This arginine, conserved in ERKs and other dually phosphorylated TXY activated kinases, forms a salt bridge with the phosphotyrosine of the TXY motif (Fig. 9 D) and is necessary for stabilizing the active conformation of MAP kinases ( 15 ). Mutation of this arginine to a serine removes the ability to form salt bridges leading to kinase inactivation of the R175S mutant. Thus, the model of CDKL5 provides possible explanations for the observed loss of kinase activity in these CDKL5 mutants from patients with West syndrome and RTT.
DISCUSSION
The results presented here demonstrate that CDKL5 is ubiquitously expressed in a wide variety of cell lines and tissues and is expressed early in development. The nuclear localization of CDKL5 suggests a possible role in gene regulation. The C-terminal domain of CDKL5 regulates its function by affecting the stability, kinase activity and nuclear localization of CDKL5. Contrary to a previous report ( 10 ), our analyses suggest that MeCP2E2 is not a direct substrate of CDKL5. Similarly, we find no evidence that ARX interacts with or is phosphorylated by CDKL5. Finally, examination of the naturally occurring CDKL5 mutants from patients with West and Rett neurological disorders reveals a loss of kinase activity suggesting that active CDKL5 is required for proper brain development.
The observation that the CDKL5 mutations associated with the disease state cause loss of kinase activity is consistent with the nature of the mutations. In one case, the mutation results from a deletion extending into the kinase domain, disrupting the three-dimensional structure in the lower lobe of the enzyme corresponding to subdomains X–XI. Interestingly, truncations of the C-terminus that leave an intact kinase domain, the size of ERK2, do not suppress kinase activity and in fact enhance it by removing negative regulatory sequences. The other two mutants are point mutations within the conserved kinase domain that alter residues that are in invariant regions among kinases. Surprisingly, the only substrate of CDKL5 that we have identified to date is CDKL5 itself; the enzyme favors cis -phosphorylation but not trans -phosphorylation of either full-length or truncated CDKL5 or even peptides derived from CDKL5 (data not shown). Despite its robust autophosphorylation activity, neither full-length nor truncated CDKL5 significantly phosphorylates a number of common substrates for proline-directed kinases including myelin basic protein, cdk and MAPK peptide substrates, histones and a variety of transcription factors including Fos and Myc (data not shown). Thus, CDKL5 appears to be a highly specific member of the CDKL family.
CDKL5 belongs to a unique group of kinases that share homology with both cyclin-dependent protein kinases and mitogen-activated protein kinases and are all proline-directed kinases. It has been postulated that the TXY sequences of CDKL kinases may be involved in kinase activation similar to MAP kinases where the TXY motif becomes dually phosphorylated either by autophosphorylation or an upstream kinase. Recently, dual phosphorylation of the TDY motif was found to be necessary for activation of a distantly related CDKL kinase, intestinal cell kinase ( 16 ). We have similarly determined that the TEY motif is also highly phosphorylated in the more active truncated CDKL5 kinase, and autophosphorylation is completely eliminated by mutation of the TEY motif (data not shown). Interestingly, modeling the CDKL5 structure against ERK2 shows that the general structure as well as the activation loops of both kinases are well aligned, consistent with a possible role for the TEY motif in the activation not only of ERKs but also of CDKL5. Thus, TXY activation is not exclusive to MAP kinases and may be a possible mechanism for CDKL5 activation.
Although the sequences are not very similar, CDKL5, with its large C-terminal region, is reminiscent of ERK5, a member of the big MAP kinase family. ERK5 has an extended C-terminal domain that is proline rich, binds to proteins with SH3 domains and contains an identified nuclear localization signal. Similarly, the C-terminus of CDKL5 is proline rich with many PXXP sequences and is required for nuclear localization. In fact, CDKL5 binds SH3 domains from Src and Grb2 (data not shown) suggesting that it also has scaffold functions. Removal of the C-terminus in ERK5 results in increased autophosphorylation as well as increased activity toward exogenous substrates ( 17 ). Like ERK5, the C-terminus of CDKL5 negatively regulates kinase activity and appears to influence stability. ERK5 phosphorylates Mef2c, is recruited with this protein to transcription complexes via the C-terminal domain and this domain functions in transcriptional regulation ( 17 ). Although we find no evidence for direct phosphorylation of or association with the homeobox transcription factor ARX, it is possible that CDKL5 phosphorylates other mediators or targets of ARX in the same signaling pathway. Similarly, although we also detect MeCP2E2 binding to the kinase domain of CDKL5, our data suggest that the previously reported phosphorylation of MeCP2E2 by CDKL5 ( 10 ) results from non-specific kinases in the immunoprecipitate. Although it is possible that MeCP2E1, the brain-rich isoform, is a substrate for CDKL5, as it has a conserved phosphorylation site missing in MeCP2E2 ( 8 , 9 ), this is unlikely because the putative serine phosphorylation site is lacking a proline residue at the +1 position and therefore cannot be phosphorylated by proline-directed kinases such as CDKs. The observation that MeCP2E2 is not a direct substrate, however, does not rule out the possibility that CDKL5 regulates MeCP2 function. CDKL5 and MeCP2 have been convincingly shown to associate together, have overlapping expression patterns in the brain, and, when mutated, cause similar phenotypes. Thus, MeCP2 may act to recruit CDKL5 to a DNA binding complex that contains a functional substrate of the kinase.
It is interesting to note that, as the number of clinical studies implicating CDKL5 in genetic disorders increases, early onset seizures are a common symptom. In the X-linked West syndrome cases, the disorders caused by CDKL5 mutations all showed early onset seizure symptoms, whereas in ARX cases, 50% of the patients have seizures. Similarly, CDKL5 mutations are found in atypical RTT, a variant that exhibits seizures. Additional evidence for a link between defective CDKL5 function and seizures includes a study of Danish adolescents with X-linked retinoschisis ( 18 ). One male patient with X-linked retinoschisis had seizures that are not associated with retinoschisis. The coding region of the retinoschisis gene (XLRS1) overlaps that of CDKL5, and mapping revealed a sizable deletion that involved part of CDKL5's coding region. These studies strongly suggest that inactivation of the CDKL5 kinase may be in part responsible for the occurrence of seizure symptoms. In some instances, the other gene implicated in RTT, MECP2, has also been associated with early onset and severe seizures. Thus, it is likely that there are multiple targets of the CDKL5 kinase that regulate different neurological activities including seizures. Ongoing studies should elucidate the role of CDKL5 in the pathological mechanisms of these diseases.
MATERIALS AND METHODS
Reagents
Triethylamine, glycine, peroxidase-conjugated goat anti-rat IgG, peroxidase-conjugated goat anti-rabbit IgG, peroxidase-conjugated goat anti-mouse IgG, HPLC grade phosphoric acid, monoclonal anti-Myc 9E10 antibody, monoclonal M2 anti-FLAG antibody and M2 anti-FLAG agarose beads were purchased from Sigma. Dulbecco's modified Eagle's medium, Opti-mem, Ham's F12 medium, fetal bovine serum (FBS), trypsin, penicillin, streptomycin and the eukaryotic TA cloning kit were purchased from Invitrogen. Protein A-Sepharose was purchased from RepliGen Corp. Protein G-Sepharose, Cyanogen bromide-activated Sepharose, was purchased from Amersham Pharmacia Biotech. Imject EDC kit with KLH, SuperSignal Pico enhanced chemiluminescent substrates and dialysis products (Snakeskin, Slide-a-lyzer) were purchased from Pierce. Normal Goat Serum (NGS) was purchased from Vector Labs. Fluorescent Cy3 secondary antibody was purchased from Jackson Labs. Anti-Myc antibody Profound Kit was purchased from Pierce. Anti-phosphoTEY ERK antibody was purchased from Cell Signalling. RNA isolation (RNAqueous-4PCR) and RT–PCR (Retroscript) kits were purchased from Ambion Inc. Quikchange site-directed mutagenesis kit was purchased from Stratagene. Advantage-HF 2 and Advantage-GC PCR kits were purchased from BD Biosciences Clontech. Enhanced chemiluminescence reagents and [γ- 32 P]-ATP (6000 Ci/mol) were purchased from Perkin–Elmer. Primers were obtained from Integrated DNA Technologies. Kodak X-OMAT autoradiographic film was purchased from Fisher.
Plasmid construction and preparation
CDKL5 cDNA was constructed from two DNA fragments generated previously ( 7 ). A FLAG antigenic tag, DYKDDDDK, was inserted immediately following the start methionine of Stk9 using PCR and verified by sequencing. The FLAG-CDKL5 was cloned into the mammalian expression vector, pCR3.1 (Invitrogen). Site-directed mutagenesis was performed to generate the kinase inactive K42R and other mutants using Quikchange (Stratagene). The MeCP2E2 expression plasmid was a gift from B. Minassian. The ARX expression plasmid was a gift from W. Dobyns. Plasmid DNAs were prepared by CsCl–ethidium bromide gradient centrifugation or by purification through columns according to the manufacturer's protocol (Qiagen).
Cell culture
COS cells (American Type Culture Collection) and HEK293 cells (gift from Anning Lin, University of Chicago) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and antibiotics (50 units/ml penicillin and 50 ug/ml streptomycin). PC12 cells, a gift from Angus MacNicol (University of Arkansas), were cultured in DMEM supplemented with 10% FBS, 5% horse serum and antibiotics. RN33B cells ( 19 ) were cultured in DMEM/Ham's F12 medium supplemented with 10% FBS, antibiotics and 250 µg of G418. H19-7, H19-5 and WH19-4 cell lines ( 20 ) were cultured on poly-lysine coated plates in DMEM supplemented with 10% FBS, antibiotics and 250 µg of G418. COS, HEK293 and PC12 cells were maintained at 37°C in a 95% air, 5% CO 2 environment. RN33B, H19-7, H19-5 and WH19-4 cells were maintained at 33°C in a 95% air, 5% CO 2 environment.
Reverse transcription–polymerase chain reaction
Total RNA from various cell lines was isolated by the RNAqueouse-4PCR kit according to the manufacturer's protocol (Ambion). Reverse transcripts were made from the RNA using the RETROscript kit (Ambion). Transcripts were subjected to PCR with primers covering regions 1004–1532, 1474–1998, 1968–2542 of human CDKL5 or 1000–1300 of mouse CDKL5. The RT–PCR program was five cycles at 94°C, 5 s; 72°C, 4 min; five cycles at 94°C, 5 s; 70°C, 4 min and 25 cycles at 94°C, 5 s; 68°C, 4 min.
Tissue isolation for western analysis
A 6-week-old male Sprague Dawley rat (Harlan, Indianapolis, IN, USA) was sacrificed via CO 2 and various tissues obtained from it. The tissues were frozen by liquid nitrogen and ground into powder. The ground tissue was added to RIPA lysis buffer containing 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 50 m m Tris–HCl, pH 7.5, 40 m m β-glycerophosphate, 100 m m NaCl, 50 m m NaF, 2 m m EDTA, 1 m m NaVO4 and 1:50 protease inhibitor cocktail III.
Whole mount immunochemistry
Embryos E7.5 and E9.5 were isolated and fixed with 4% paraformaldehyde and 2% Triton X-100. The embryos were incubated with CDKL5 antibody overnight and followed by Cy3 secondary antibody. The embryos were visualized on a dissection microscope with a fluorescent light source. The images were captured by a digital low light imaging camera (Roper Scientific).
Transient transfection and preparation of cell extracts
Cells were seeded on 100 mm plates and incubated overnight. The medium was changed to serum-free Opti-Mem (Invitrogen), and cells were transfected with 10–13 µg of plasmid DNA and 40 µl of Transit LT-1 as specified by the manufacturer (Pan Vera Corp, WI, USA). Four hours post-transfection, the Opti-Mem medium was changed to growth medium. The cells were harvested 24 h post-transfection, washed with ice-cold PBS and lysed with either a 1% Triton X-100 based lysis buffer (TLB) containing 1% Triton X-100, 150 m m NaCl, 50 m m Tris–HCl pH 7.5, 50 m m NaF, 40 m m β-glycerophosphate, 2 m m EDTA, 1 m m NaVO 3 , 1 m m phenylmethylsulfonyl fluoride, 1 µg/ml leupeptide, 1 µg/ml aprotinin and 20 m m ρ-nitrophenyl phosphate or RIPA lysis buffer. Cell extracts were cleared by centrifugation, and the supernatant subjected to Bradford analysis for protein concentration.
Anti-CDKL5 antibody
Anti-CDKL5 rabbit antiserum (Cocalico Biologicals) was raised against a peptide corresponding to the last 17 amino acids of the C-terminal domain of CDKL5. The CDKL5 peptide was synthesized by the University of Chicago Cancer Research Center Peptide Facility and was conjugated to keyhole limpet hemocyanin protein via EDC coupling (Pierce). The anti-CDKL5 antiserum was purified by affinity chromatography using the CDKL5 peptide coupled to cyanogen bromide activated sepharose. Antibodies were eluted by 100 m m glycine, pH 2.5, followed by 100 m m triethylamine, pH 11.5. The eluents were neutralized with 1 m Tris–HCl, pH 8.0 and dialyzed against PBS, pH 7.5. A protein A-sepharose column was used to isolate the IgG fraction. The IgG fraction was eluted using 100 m m glycine, pH 3.0, neutralized by 1 m Tris–HCl and dialyzed against PBS, pH 7.5.
Immunocytochemistry
Cells were grown on 12 mm glass coverslips for 24 h, fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100 in PBS for 5 min. Following fixation, the cells were incubated with 2% NGS in PBS for blocking for 30 min. Primary antibody was added and incubated overnight. The cells were washed with PBS and incubated with FITC-labeled secondary antibody for 1 h. The cells were washed and visualized 200× on a Zeiss Axioplan fluorescence microscope supplied with FITC long filters and the images captured by a digital low light imaging camera (Roper Scientific) at the University of Chicago Cancer Center Digital Light Microscopy Facility. The images were deconvoluted by OPENLAB software (Improvision, Ltd).
Western analysis
Cell extracts (50–100 µg of protein per lane) were resolved on a 7.5% acrylamide separating gel by SDS–PAGE. Proteins were transferred to a nitrocellulose membrane. Membrane blocking, washing, antibody incubation and detection by enhanced chemiluminescence were preformed as described previously ( 21 ).
Immunoprecipitation and in vitro kinase assay
Five hundred micrograms of ectopically expressed FLAG-tagged CDKL5 was incubated with 5 µg of M2 bound agarose for 4 h. The beads were washed three times with TLB and twice with kinase buffer (25 m m HEPES, pH 7.4, 10 m m MgCl 2 , 10 m m MnCl 2 , 10 m m dithiothreitol, 0.2 m m sodium vanadate and 10 m m nitro-phenyl-phosphate). FLAG peptide was used to elute CDKL5 in some samples according to manufacturer's instructions (SIGMA). The resultant beads were resuspended in 30 µl of kinase buffer with addition of 100 µ m ATP, 5 µCi of [γ- 32 P]-ATP (NEN) and substrate. The kinase assays were carried out 15 min at 30°C and terminated by addition of SDS–PAGE protein loading buffer.
Substrate preparation
Vectors expressing MeCP2E2 or ARX were transfected into HEK293T cells, and the cells lysed with TLB. The resulting lysates were incubated with either anti-MeCP2 or anti-ARX antibody cross-linked to agarose beads for 4 h. The beads were washed three times with TLB and either eluted by acid elution (Pierce) or used directly in kinase assays.
In vivo labeling of ARX
ARX and CDKL5 were transfected into 293T cells. Twenty four hours post-transfection, cells were incubated with 2 mCi of 32 P-orthophosphoric acid/ml of phosphate-free DMEM supplemented with 10% dialyzed FBS for 4 h. Cells were washed with 20 m m HEPES pH 7.4 and 150 m m NaCl and lysed with TLB. Cell extracts were immunoprecipitated with anti-HA antibody (3F10, Roche) for 4 h. Immunoprecipitates were separated by 10% SDS–PAGE, transferred onto nitrocellulose membrane and exposed to film by autoradiography.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
This work has been supported by NIH grant NS033858 to M.R.R., a gift from the Cornelius Crane Trust for Eczema Research to M.R.R. and an NIH Pharmacology training grand T32 GM07151 to C.L. We thank Drs Mark Abe, Akira Imamoto, Anning Lin, Piers Nash and Mitchel Villereal for valuable discussions and assistance and Yaling Wu for assistance with structural modeling.
Conflict of Interest statement. None declared.
![Figure 1. Cellular expression of the CDKL5 mRNA transcript. Total RNA was isolated from human lung cells (HBE, HAE, HTE), human androgen-dependent and androgen-independent prostate tumor cells (PC3, Du145, LnCAP), human embryonic kidney cells (293) and rat neural cell lines [H19-7 (hippocampal), H19-5 (glial), RN33B (Raphe), PC12 (pheochromocytoma)]. RT–PCR was performed on the RNA samples using primers derived from CDKL5 cDNA. A FLAG-CDKL5 cDNA template was used as a positive control. This result is representative of three independent experiments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/24/10.1093_hmg_ddi391/1/m_ddi39101.jpeg?Expires=1716545969&Signature=crtxCOHlvnJ91aWuEWEzrDCJZpFzu-pjdkyC3U7H0lNSgsbFEjcO4oabpqMJ3rnlinV9bAOmzSLQKN1seifKG5isNpGHnSa9KYr2XSrqo8zRq0YHpLS-e7AAnf2f83q365s1mhLzn57glpA1PP81tbxv8alEZ-O8QZVMn~XcOdZobt-F7uKiAUELnPJrLXUfsemjoMgKJBEx8lZOr67ABT8aQ5WnGESpbkr0Zz47-zDB55dX84t2wILwnxaJqN~Szw-tsZ3Wxgoc~yMpNmQdOvR7HjjiOGcqqhs-zABSjzktS4GduepIDvCvNhFoN0Y0raAeoj69lk54SeGVoc-Jtw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 1. Cellular expression of the CDKL5 mRNA transcript. Total RNA was isolated from human lung cells (HBE, HAE, HTE), human androgen-dependent and androgen-independent prostate tumor cells (PC3, Du145, LnCAP), human embryonic kidney cells (293) and rat neural cell lines [H19-7 (hippocampal), H19-5 (glial), RN33B (Raphe), PC12 (pheochromocytoma)]. RT–PCR was performed on the RNA samples using primers derived from CDKL5 cDNA. A FLAG-CDKL5 cDNA template was used as a positive control. This result is representative of three independent experiments.
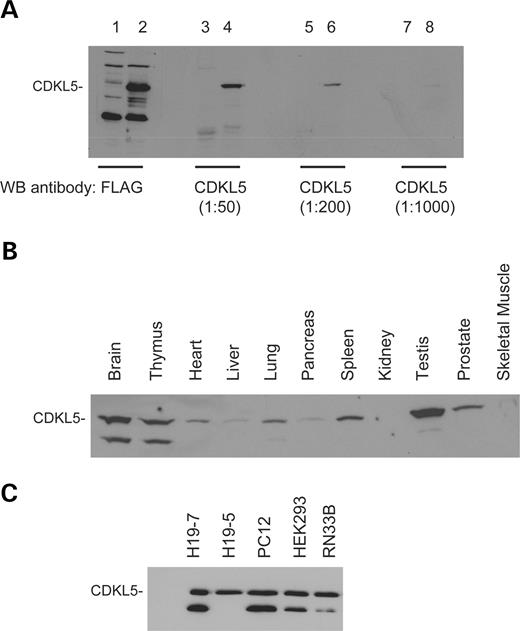
Figure 2. The CDKL5 protein is expressed in a variety of tissues and cell lines. ( A ) Specificity of the polyclonal anti-CDKL5 antibody. COS cells were transfected with either FLAG-CDKL5 (Lanes 2, 4, 6 and 8) or empty pc-DNA vector (Lanes 1, 3, 5 and 7). Lysates were prepared and resolved by SDS–PAGE. Western blotting analysis of the samples was performed with the anti-FLAG monoclonal antibody (Lanes 1 and 2) or the anti-CDKL5 polyclonal antibody (Lanes 3–8) at the indicated concentrations. ( B) Expression of CDKL5 in tissues. Extracts from adult male rat tissues were resolved by SDS–PAGE. The samples were analyzed by western blotting using the anti-CDKL5 polyclonal antibody. ( C) Expression of CDKL5 in cell lines. Lysates were prepared from H19-7, H19-5, PC12, HEK293 and RN33B cell lines and resolved by SDS–PAGE. The samples were analyzed by immunoblotting with the anti-CDKL5 polyclonal antibody. This result is representative of three independent experiments.
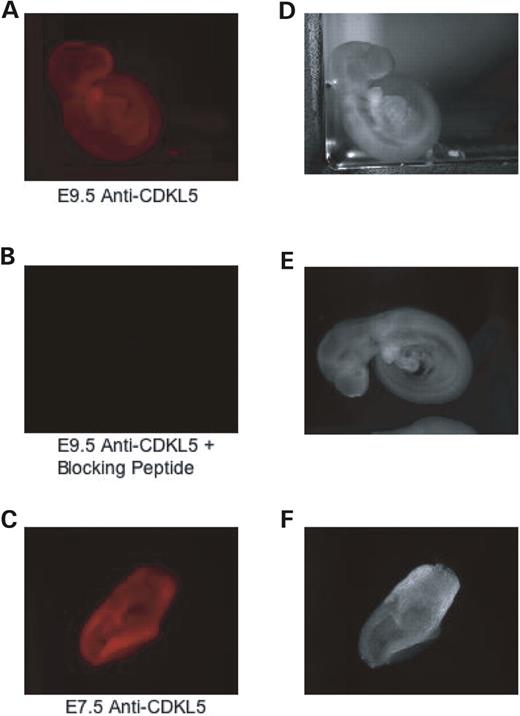
Figure 3. CDKL5 is ubiquitously expressed in mouse embryos. ( A) Immunofluorescence of endogenous CDKL5 at embryonic stage day 9.5. E9.5 embryos were fixed and visualized by immunocytochemistry with the polyclonal anti-CDKL5 antibody. ( B ) Immunostaining is specific. E9.5 embryos were fixed and visualized by immunocytochemistry with polyclonal anti-CDKL5 antibody and a blocking peptide corresponding to the anti-CDKL5 antibody epitope. ( C ) Immunofluorescence of endogenous CDKL5 at embryonic stage day 7.5. E7.5 embryos were fixed and visualized by immunocytochemistry with polyclonal anti-CDKL5 antibody. ( D–F) Light phase microscopy depicting the whole mouse embryo in samples A–C. This result is representative of two independent experiments.
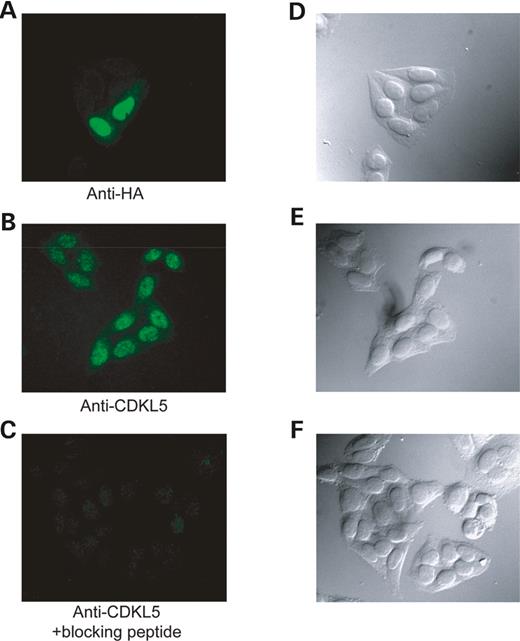
Figure 4. CDKL5 is localized primarily in the nucleus. ( A ) Localization of transiently transfected CDKL5 in HEK293 cells. HEK293 cells were transiently transfected with HA-CDKL5, fixed and visualized by immunocytochemistry with the monoclonal anti-HA antibody (3F10). ( B ) Localization of endogenous CDKL5. HEK293 cells were fixed and visualized by immunocytochemistry with the polyclonal anti-CDKL5 antibody. ( C ) Immunostaining is specific. HEK293 cells were fixed and visualized by immunocytochemistry with polyclonal anti-CDKL5 antibody and a blocking peptide corresponding to the anti-CDKL5 antibody epitope. ( D–F) Light-phase microscopy depicting the HEK293 cells in samples A–C. These results are representative of three independent experiments.
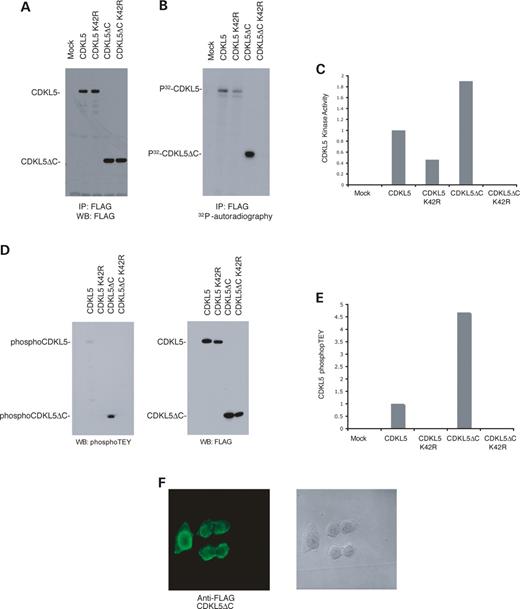
Figure 5. The CDKL5 C-terminal region is important for stability, activity and localization. ( A ) The C-terminus of CDKL5 is important for stability. Lysates from HEK293T cells transfected with FLAG-tagged CDKL5, CDKL5 K42R, CDKL5ΔC or CDKL5ΔC K42R were immunoprecipitated, resolved by SDS–PAGE and analyzed by immunoblotting with the anti-FLAG monoclonal antibody (M2). ( B ) Activity of C-terminal truncated (CDKL5ΔC) versus full-length CDKL5. Immunoprecipitated FLAG-tagged CDKL5, CDKL5ΔC and kinase-dead counterparts were subjected to an in vitro kinase assay and subsequently resolved by SDS–PAGE. Samples were then transferred onto nitrocellulose membrane and exposed to film by autoradiography. ( C ) C-terminal truncated CDKL5 has higher kinase activity than full-length CDKL5. The protein levels of the samples in (B) were quantitated by densitometry imaging, and kinase activity was measured by 32 P-phosphor imaging. The resulting graph shows the kinase activity of CDKL5 normalized to protein levels. ( D ) TEY phosphorylation in CDKL5 and CDKL5ΔC. Lysates from HEK293T cells transfected with CDKL5, CDKL5 K42R, CDKL5ΔC and CDKL5ΔC K42R were immunoprecipitated, separated by SDS–PAGE and analyzed by immunoblotting with the anti-phosphoTEY ERK antibody (left panel) or anti-FLAG antibody (right panel) to quantitate protein levels of CDKL5 and CDKL5ΔC. ( E) C-terminal truncated CDKL5 has higher TEY phosphorylation than full-length CDKL5. The protein levels and phosphoTEY levels in sample ‘E’ were quantitated by densitometry imaging. The resulting graph shows the relative amount of phosphorylated TEY in CDKL5 and CDKLΔC normalized to their respective protein levels. ( F) CDKL5ΔC fails to localize properly. HEK293T cells transfected with FLAG-tagged CDKL5ΔC were fixed and visualized by immunocytochemistry with anti-FLAG antibody. The left panel depicts fluorescence and the right panel shows light phase microscopy of the fixed cells. These results are representative of three independent experiments.
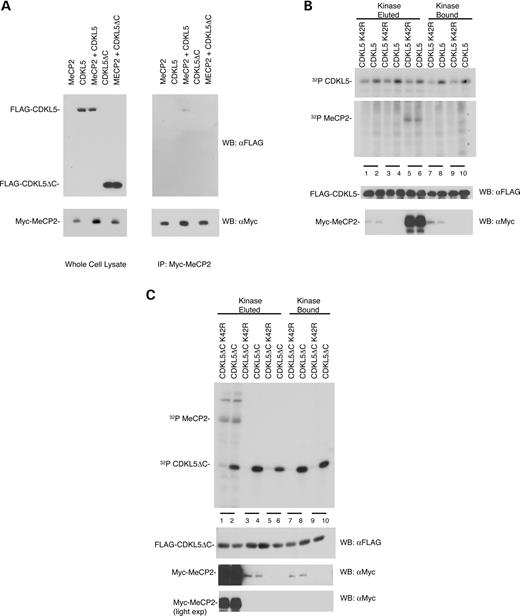
Figure 6. CDKL5 associates with but does not phosphorylate MeCP2E2 in vitro . ( A ) Full length CDKL5 but not truncated CDKL5ΔC associates with MeCP2E2. HEK293T cells transfected with either FLAG-tagged CDKL5 or CDKL5ΔC and Myc-tagged MeCP2E2 were immunoprecipitated by anti-Myc antibody. The immunoprecipates were resolved by SDS–PAGE and visualized by immunoblotting with anti-FLAG antibody. ( B ) Full length CDKL5 and CDKL5 K42R do not phosphorylate MeCP2E2. HEK293T cells transfected with either FLAG-tagged CDKL5 or CDKL5 K42R were immunoprecipitated by anti-FLAG M2 agarose beads. CDKL5 either remained bound to the beads or was eluted by FLAG peptide and then assayed for in vitro kinase activity using either eluted MeCP2E2 or MeCP2E2 bound to beads as described in Materials and Methods. Samples were resolved by SDS–PAGE and visualized by autoradiography. (Lanes 1 and 2) Eluted MeCP2E2; (Lanes 3 and 4) No substrate; (Lanes 5 and 6) Bound MeCP2E2; (Lanes 7 and 8) Eluted MeCP2E2 and (Lanes 9 and 10) No substrate. (Lower panels) Immunoblots of kinase assay samples showing CDKL5 and MeCP2E2 expression. ( C ) Truncated CDKL5 does not phosphorylate MeCP2E2. HEK293T cells transfected with either FLAG-tagged CDKL5ΔC or CDKL5ΔC K42R were immunoprecipitated by anti-FLAG M2 agarose beads. Kinase bound on beads was either eluted by FLAG peptide or used directly. The substrate MeCP2E2 was prepared similarly. The bound and eluted kinases and substrates were subjected to an in vitro kinase assay separated by SDS–PAGE and visualized by autoradiography. Lanes 1 and 2 contain MeCP2E2 bound; lanes 3 and 4 have MeCP2E2 eluted; lanes 5 and 6 have no substrate; lanes 7 and 8 have MeCP2E2 eluted and lanes 9 and 10 contain no substrate. (Lower panels) Immunoblots of kinase assay samples showing CDKL5ΔC and MeCP2E2 expression. In the figure, MeCP2 refers to the MeCP2E2 isoform. These results are representative of two independent experiments.
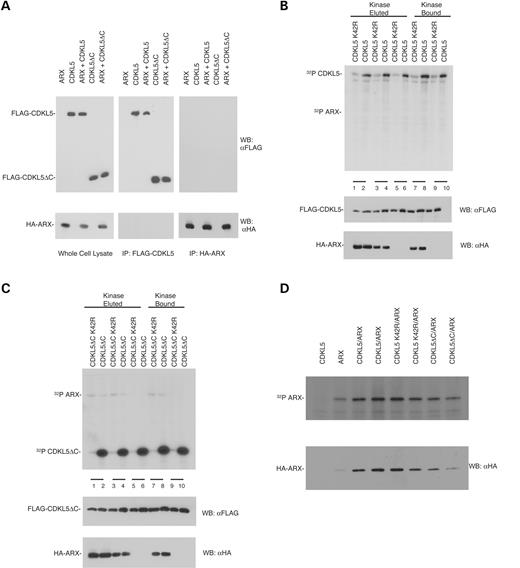
Figure 7. CDKL5 does not associate with or phosphorylate ARX in vitro . ( A ) CDKL5 does not co-immunoprecipitate with ARX. HEK293T cells, transfected with FLAG-CDKL5, HA-ARX or both were lysed and immunoprecipiated by either anti-FLAG or anti-HA antibody. The immunoprecipitates were separated by SDS–PAGE and transferred onto nitrocellulose membrane. The membrane was western blotted for the presence of the co-immunoprecipitated protein. ( B ) Full length CDKL5 and CDKL5 K42R do not phosphorylate ARX. HEK293T cells transfected with either FLAG-tagged CDKL5 or CDKL5 K42R were immunoprecipitated by anti-FLAG M2 agarose beads. CDKL5 either remained bound to the beads or was eluted by FLAG peptide and then assayed for in vitro kinase activity using either eluted ARX or ARX bound to beads as described in Materials and Methods. Samples were resolved by SDS–PAGE and visualized by autoradiography. Lanes 1 and 2 contain ARX bound; lanes 3 and 4 have ARX eluted; lanes 5 and 6 have no substrate; lanes 7 and 8 ARX eluted and lanes 9 and 10 contain no substrate. (Lower panels) Immunoblots of kinase assay samples showing CDKL5 and ARX expression. ( C ) Truncated CDKL5 does not phosphorylate ARX. HEK293T cells transfected with either FLAG-tagged CDKL5ΔC or CDKL5ΔC K42R were immunoprecipitated by anti-FLAG M2 agarose beads. Kinase bound on beads was either eluted by FLAG peptide or used directly. The substrate ARX was prepared similarly. The bound and eluted kinases and substrates were subjected to an in vitro kinase assay and separated by SDS–PAGE and visualized by autoradiography. Lanes 1 and 2 contain ARX bound; 3 and 4 have ARX eluted; 5 and 6 have no substrate; 7 and 8 ARX eluted and 9 and 10 contain no substrate. (Lower panels) Immunoblots of kinase assay samples showing CDKL5ΔC and ARX expression. These results are representative of two independent experiments. ( D) CDKL5 does not increase in vivo phosphorylation of ARX. HEK293T cells transfected with either ARX, CDKL5 or both were starved and labeled with 32 P-orthophosphate for 4 h. The resulting lysate was immunoprecipitated for ARX and separated by SDS–PAGE. The SDS–PAGE gel was transferred onto nitrocellulose membrane and visualized by autoradiography. These results are representative of at least two independent experiments.
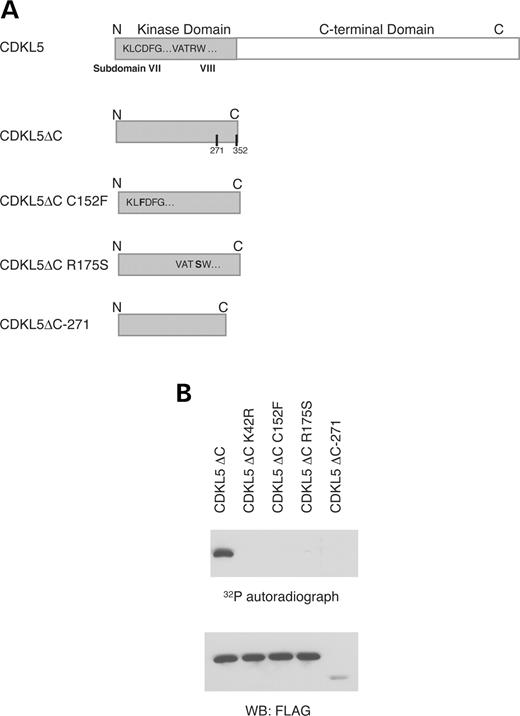
Figure 8. CDKL5 mutations associated with the disease phenotype cause a loss in activity. ( A ) Schematic depicting truncation and point mutants of CDKL5. ( B ) Truncation and point mutants of CDKL5 were expressed in HEK293T cells. The cells were lysed, immunoprecipitated by anti-FLAG antibody and subjected to an in vitro kinase assay. The immunoprecipitates were resolved by SDS–PAGE, transferred to a nitrocellulose membrane, and the membrane was visualized by autoradiography. These results are representative of at least three independent experiments.
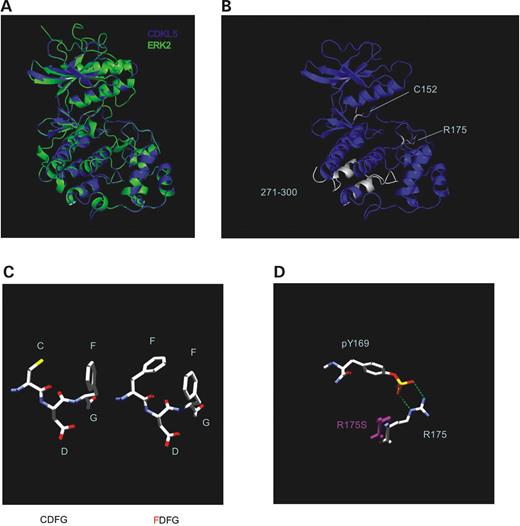
Figure 9. A Putative structure of CDKL5 kinase domain and molecular modeling of West syndrome and RTT mutations. ( A ) Superimposition of ERK2 kinase domain and putative CDKL5 kinase domain generated by SwissModel. ( B ) Sites affected by West syndrome and RTT mutations. The sites of mutations are highlighted in white. ( C ) Molecular modeling of the C152F mutation. The left structure depicts normal amino acid sequence and the right structure is a representation of cysteine substituted for phenylalanine. ( D ) The R175S mutation is unable to form a salt bridge with phospho-tyrosine 169. The R175S serine is shown in purple .
References
Kalscheuer, V.M., Tao, J., Donnelly, A., Hollway, G., Schwinger, E., Kubart, S., Menzel, C., Hoeltzenbein, M., Tommerup, N., Eyre, H. et al. (
Tao, J., Van Esch, H., Hagedorn-Greiwe, M., Hoffmann, K., Moser, B., Raynaud, M., Sperner, J., Fryns, J.P., Schwinger, E., Gecz, J. et al. (
Weaving, L.S., Christodoulou, J., Williamson, S.L., Friend, K.L., McKenzie, O.L., Archer, H., Evans, J., Clarke, A., Pelka, G.J., Tam, P.P. et al. (
Scala, E., Ariani, F., Mari, F., Caselli, R., Pescucci, C., Longo, I., Meloni, I., Giachino, D., Bruttini, M., Hayek, G. et al. (
Evans, J.C., Archer, H.L., Colley, J.P., Ravn, K., Nielsen, J.B., Kerr, A., Williams, E., Christodoulou, J., Gecz, J., Jardine, P.E. et al. (
Van den Veyver, I.B. and Zoghbi, H.Y. (
Montini, E., Andolfi, G., Caruso, A., Buchner, G., Walpole, S.M., Mariani, M., Consalez, G., Trump, D., Ballabio, A. and Franco, B. (
Kriaucionis, S. and Bird, A. (
Mnatzakanian, G.N., Lohi, H., Munteanu, I., Alfred, S.E., Yamada, T., MacLeod, P.J., Jones, J.R., Scherer, S.W., Schanen, N.C., Friez, M.J. et al. (
Mari, F., Azimonti, S., Bertani, I., Bolognese, F., Colombo, E., Caselli, R., Scala, E., Longo, I., Grosso, S., Pescucci, C. et al. (
Nan, X., Campoy, F.J. and Bird, A. (
Pearson, G., Robinson, F., Beers Gibson, T., Xu, B., Karandikar, M., Berman, K. and Cobb, M.H. (
Schwede, T., Kopp, J., Guex, N. and Peitsch, M.C. (
Hanks, S.K. and Hunter, T. (
Canagarajah, B.J., Khokhlatchev, A., Cobb, M.H. and Goldsmith, E.J. (
Fu, Z., Schroeder, M.J., Shabanowitz, J., Kaldis, P., Togawa, K., Rustgi, A.K., Hunt, D.F. and Sturgill, T.W. (
Hayashi, M. and Lee, J.D. (
Huopaniemi, L., Tyynismaa, H., Rantala, A., Rosenberg, T. and Alitalo, T. (
White, L.A., Keane, R.W. and Whittemore, S.R. (
Eves, E.M., Kwon, J., Downen, M., Tucker, M.S., Wainer, B.H. and Rosner, M.R. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}