




Abstract
Duchenne muscular dystrophy (DMD) is a fatal X-linked muscle-wasting disease caused by mutations of the gene encoding the cytoskeletal protein dystrophin. Therapeutic options for DMD are limited because the pathogenetic mechanism by which dystrophin deficiency produces the clinical phenotype remains obscure. Recent reports of abnormal α-adrenergic vasoregulation in the exercising muscles of DMD patients and in the mdx mouse, an animal model of DMD, prompted us to hypothesize that the dystrophin-deficient smooth muscle contributes to the vascular and dystrophic phenotypes of DMD. To test this, we generated transgenic mdx mice that express dystrophin only in smooth muscle (SMTg/mdx). We found that α-adrenergic vasoconstriction was markedly attenuated in the contracting hindlimbs of C57BL/10 wild-type mice, an effect that was mediated by nitric oxide (NO) and was severely impaired in the mdx mice. SMTg/mdx mice showed an intermediate phenotype, with partial restoration of the NO-dependent modulation of α-adrenergic vasoconstriction in active muscle. In addition, the elevated serum creatine kinase levels observed in mdx mice were significantly reduced in SMTg/mdx mice. This is the first report of a functional role of dystrophin in vascular smooth muscle.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a severe muscle-wasting disease caused by mutations in the dystrophin gene that result in the absence of dystrophin in the sarcolemma of muscle fibers ( 1 ). DMD is the most common muscular dystrophy, an X-linked recessive disorder that affects one in 3500 live males at birth ( 2 ). A milder form of DMD, known as Becker muscular dystrophy, is also caused by mutations in the dystrophin gene, resulting in low-level expression of a truncated protein ( 3 , 4 ). Currently, there is no effective therapy for the dystrophinopathies because the mechanism by which dystrophin deficiency produces the clinical phenotype of progressive muscle weakness and respiratory and cardiac failure is poorly understood.
Dystrophin is localized to the sarcolemma in association with a highly organized complex of transmembrane and cytosolic proteins that forms a structural link between the extracellular matrix and the intracellular actin cytoskeleton ( 1 , 5–7 ). The loss of dystrophin destabilizes this complex, which is thought to increase the susceptibility of muscle cells to contraction-induced damage ( 8 ). Despite a uniform genetic defect, the characteristic histological phenotype observed in dystrophin-deficient muscle is one of small, random groups of necrotic muscle cells surrounded by normal cells ( 9 , 10 ). We and other researchers have shown that a similar pattern of focal necrosis can be induced by artificial infarction in healthy muscle, suggesting that vascular insufficiency may contribute to the dystrophic pathology ( 11–13 ). We have also previously observed strong nerve growth factor receptor (NGFR) immunoreactivity on the tunica adventitia of blood vessels in muscle specimens from DMD patients, but not in controls ( 14 ). This suggests an aberration of the neurovascular system and/or blood vessels in DMD, because NGFR plays essential roles in the survival and maintenance of sympathetic and sensory neurons ( 15 ).
In fact, recent reports have indicated that sympathetic neurovascular control is abnormal in dystrophin-deficient muscle ( 16–18 ). This defective regulation is observed during exercise, when sympathetic vasoconstriction normally is blunted in the active muscles by locally produced dilator substances, including nitric oxide (NO) ( 16 , 17 , 19–21 ). In the exercising muscles of DMD patients and mdx mice, a naturally occuring animal model of DMD ( 22 ), constrictor responses to activation of either sympathetic nerves or vascular α-adrenergic receptors were abnormally preserved ( 16–18 ). This was attributed to a deficiency of neuronal NO synthase (nNOS) that normally associates with the dystrophin complex at the sarcolemma, but is secondarily reduced in the absence of dystrophin ( 23 , 24 ). On the basis of these studies, dystrophin-deficiency in skeletal muscle was identified as the principal cause of the impairments in NO signaling and vasoregulation.
However, another possibility is that the impaired vasoregulation is due to dystrophin-deficiency in vascular smooth muscle cells (VSMCs). Dystrophin is normally expressed in the tunica media of blood vessels, especially in the muscular arteries, but it is absent in vessels from mdx mice ( 25 , 26 ). We now report that selective smooth muscle expression of full-length human dystrophin (h-dys), which is easily distinguished from endogenous mouse dystrophin, partially restores the NO-dependent attenuation of α-adrenergic vasoconstriction in the exercising muscles of mdx mice. Moreover, these transgenic mdx mice have significantly reduced serum creatine kinase (CK) levels compared with those of the non-transgenic mdx mice. These data suggest that dystrophin in VSMCs may play an important role in the local autocrine regulation of α-adrenergic constriction, and that the loss of this regulatory mechanism may exacerbate muscle fiber necrosis.
RESULTS
Generation of SMTg/mdx transgenic mice
To determine if dystrophin in VSMCs contributes to the α-adrenergic regulation of blood flow, we generated transgenic mdx mice that express full-length h-dys only in smooth muscle, but not in skeletal muscle. A smooth muscle-specific dystrophin (SMTg) construct (Fig. 1 A) was generated under the control of human smooth muscle α-actin promoter (EA4.7 promoter). It comprises of exon 1, intron 1 and the first 14 bp of exon 2 in addition to the 5′-flanking sequence of the smooth muscle α-actin gene. The intron 1 region plays a pivotal role for inducing a high level of smooth muscle-specific transcription ( 27 ). The construct was microinjected into 800 fertilized eggs of C57BL/6 mice, which were transferred into female recipient mice. We obtained 41 mice of which seven harbored the transgene. These transgenic mice were bred to dystrophin-deficient mdx mice with the C57BL/10 genetic background. Three of the seven lines reproduced, resulting in three independent SMTg/mdx lines (10, 23, 26) that exhibited normal dystrophin expression only in smooth muscles. All of the SMTg/mdx mice were back-crossed to mdx mice, and over N5 generations were analyzed. The transgenic mice were of normal appearance and development, similar to the mdx mice.
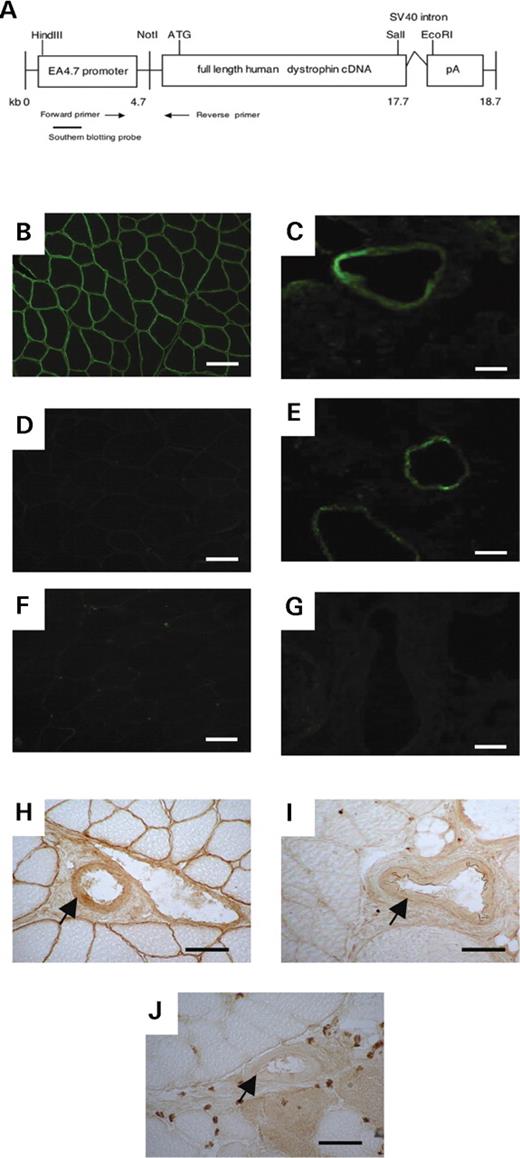
Generation of SMTg/mdx mice. ( A ) The construct used to generate transgenic mice. The full-length h-dys cDNA was expressed under control of EA 4.7 promoter. PCR primers and Southern blotting probe were used for the detection of the transgenic mouse lines. ( B – G ) Immunofluorescent staining for dystrophin in TA muscle using the 60 kDa antibody shows that dystrophin expression at the sarcolemma was detected only in C57BL/10 mice (B), but not in SMTg/mdx (D) or mdx mice (F). Dystrophin expression in lung tissue was detected in C57BL/10 (C) and SMTg/mdx (E) mice, but not in mdx (G) mice. ( H – J ) Dystrophin expression in the smooth muscle of blood vessels (arrows) found in skeletal muscle was observed by immunohistochemistry using the DYS2 antibody in both C57BL/10 (H) and SMTg/mdx mice (I), but not in mdx (J) mice. Scale bar: 50 µm.
Expression pattern of dystrophin in SMTg/mdx mice
We have previously reported that dystrophin expression can be easily detected in VSMCs of lung in normal control mice by immunofluorescent staining, whereas it is lacking in mdx mice ( 25 ). Therefore, to confirm dystrophin expression, we performed immunofluorescence labeling of tissue samples of skeletal muscle and lung (Fig. 1 ) using a polyclonal sheep anti-dystrophin antibody (60 kDa; kindly provided by Dr E.P. Hoffman, Children's National Medical Center, Washington, DC, USA) and a monoclonal mouse anti-dystrophin antibody (DYS2). As expected, dystrophin expression was detected in the skeletal muscle sarcolemma and in VSMCs of skeletal muscle and lung in C57BL/10 mice, but not in mdx mice. In the three lines of SMTg/mdx mice, dystrophin expression was detected only in VSMCs of skeletal muscle and lung (Fig. 1 B–J). We also analyzed the expression of other components of the dystrophin-glycoprotein complex in smooth muscle using immunofluorescent staining for β-sarcoglycan and β-dystroglycan, and found no differences among the C57BL/10, SMTg/mdx and mdx mice (data not shown).
Immunoblotting analysis confirmed that dystrophin was expressed in skeletal muscle of C57BL/10 mice, but not of mdx or SMTg/mdx mice (Fig. 2 A). Dystrophin was also expressed in aorta from C57BL/10 mice and from each of the SMTg/mdx lines, although the level of expression was lower than in C57BL/10 mice (Fig. 2 B). Similar results were obtained in the lung (data not shown). We estimated the expression pattern of the h-dys transgene in various tissues of one line of SMTg/mdx mice by semi-quantitative real-time reverse transcription-polymerase chain reaction (RT–PCR) using a probe and primers specific for h-dys. Expression of h-dys was high in the lung, aorta, small intestine and heart, but minimal in skeletal muscle (Fig. 2 C). This expression pattern coincides with the distribution of reporter genes under the control of the EA4.7 promoter ( 28 ). Taken together, these data indicate that the transgenic construct successfully expresses dystrophin in the VSMCs of SMTg/mdx mice with high specificity for smooth muscle, but that the dystrophin expression levels of SMTg/mdx lines were lower than those of C57BL/10 mice.
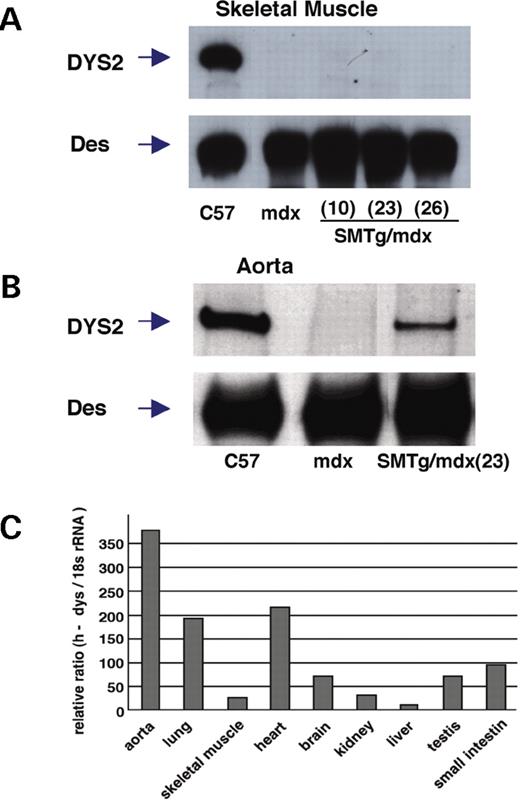
Dystrophin expression in SMTg/mdx mice. ( A – B ) Western blotting analysis was performed on protein extracts from TA muscles and aorta using a C-terminal specific dystrophin antibody (DYS2). The arrows indicate the 427 kDa dystrophin (DYS2) and the 50–55 kDa desmin (Des). Dystrophin expression was detectable only in TA muscle of C57BL/10 mice (A) and in aorta of C57BL/10 and SMTg/mdx mice, but not in mdx mice (B). Des was used as a loading control. ( C ) Semi-quantitative real-time RT-PCR shows that dystrophin mRNA expression was high in tissues containing abundant smooth muscle, but low in skeletal muscle.
Selective expression of dystrophin in VSMCs partially normalizes α-adrenergic vasoregulation in the hindlimbs of the SMTg/mdx mice
To evaluate the effect of dystrophin expression in VSMCs on vascular function in vivo , we assessed the α-adrenergic vasoregulation in the hindlimbs of SMTg/mdx mice as previously described ( 16 , 17 ). Compared with C57BL/10 mice, mean arterial blood pressures, femoral blood flow velocities (FBFV) and femoral vascular conductances (FVC) at rest and during contraction were similar in each group of mice, with the exception of higher resting FBFV and FVC in SMTg/mdx (line 23) and in mdx mice (Table 1 ). Norepinephrine (NE) injected into the arterial supply of the resting hindlimbs caused prominent vasoconstriction in all of the mice, maximally decreasing FBFV by 31–46% and FVC by 38–47% (Figs 3 and 4 A). The amount of NE injected to cause these vasoconstrictor responses did not differ among C57BL/10 (6.7±0.8 ng), SMTg/mdx (7.7±1.1, 8.6±0.9 and 8.1±0.5 ng for lines 10, 23 and 26, respectively) and mdx mice (7.1±0.6 ng). When injected into the contracting hindlimbs, peak NE vasoconstriction was markedly attenuated by 73±2% in the C57BL/10 mice, but by only 15±2% in the mdx mice (Figs 3 A, C and 4 A). All three lines of transgenic mice displayed an intermediate phenotype, such that peak NE-mediated vasoconstriction was reduced by 32±7%, 42±9% and 33±3% in SMTg/mdx lines 10, 23 and 26, respectively (Fig. 4 A). Similar results were obtained when the NE-mediated decreases in FBFV and FVC were expressed as integrated areas to reflect the total, rather than just the peak, vasoconstrictor responses (Fig. 4 B and C). Collectively, these data indicate that dystrophin in VSMCs plays an important role in the coordinated vascular response to competing constrictor and dilator signals.
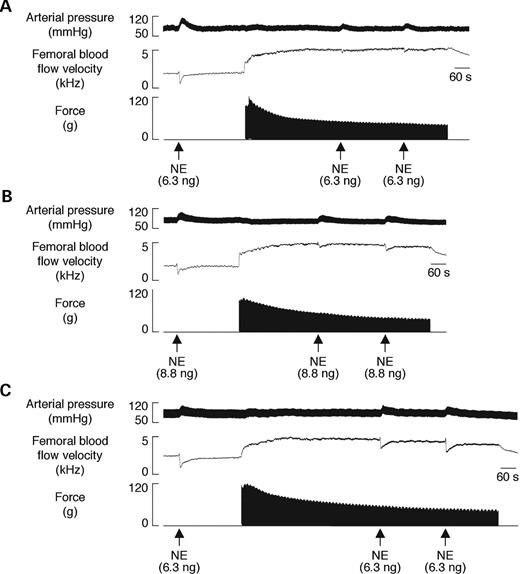
Vasoconstrictor responses to intra-arterial injection of the α-adrenergic agonist NE in resting and contracting hindlimbs. ( A – C ) Segments of original records from experiments in a C57BL/10 (A), SMTg/mdx (line 23) (B) and mdx (C) mouse showing the increases in arterial blood pressure and decreases in FBFV in response to NE. The robust vasoconstrictor responses to NE that were observed in the resting hindlimbs of all three mice were attenuated in the contracting hindlimbs of the C57BL/10 mouse and to a lesser degree in the SMTg/mdx mouse, but not in the mdx mouse.
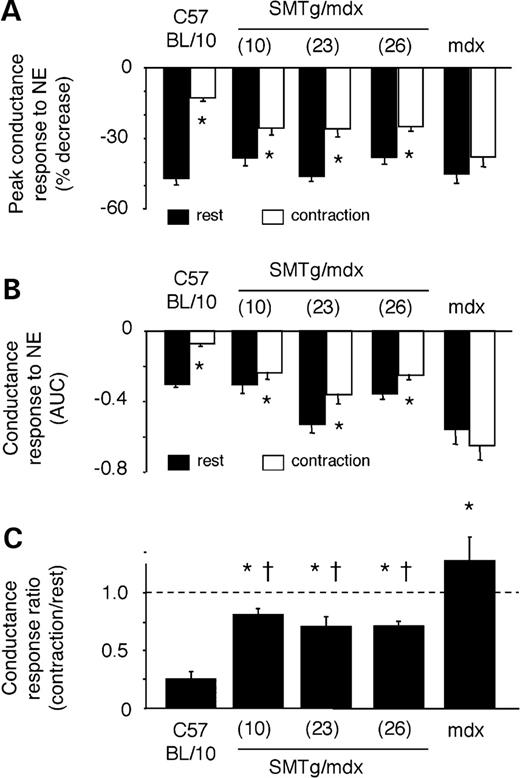
Comparison of NE-mediated vasoconstrictor responses in resting and contracting hindlimbs ( A and B ). Decreases in FVC evoked by NE in resting (black bars) and contracting (white bars) hindlimbs expressed as peak changes (A) or as integrated changes (B). * P <0.05 versus rest; AUC, area under the curve. ( C ) The degree of attenuation of the NE-mediated vasoconstrictor responses (expressed as the ratio of the integrated decreases in vascular conductance evoked by NE in the contracting versus resting hindlimbs) was greatest in the C57BL/10 mice, least in the mdx mice and intermediate in the SMTg/mdx mice. * P <0.05 versus C57BL/10; †P <0.05 versus mdx; C57BL/10, n =9; SMTg/mdx (line 10), n =6; SMTg/mdx (line 23), n =11; SMTg/mdx (line 26), n =11; mdx, n =10.
Hemodynamics at rest and during unilateral hindlimb contraction
| C57BL/10 | SMTg/mdx (line 10) | SMTg/mdx (line 23) | SMTg/mdx (line 26) | mdx | |
|---|---|---|---|---|---|
| n | 9 | 6 | 11 | 11 | 10 |
| MAP, mmHg | |||||
| Rest | 77±1 | 78±2 | 73±1 | 76±1 | 76±1 |
| Contraction | 75±1 | 75±1 | 73±1 | 72±1* | 76±1 |
| FBFV, kHz | |||||
| Rest | 1.7±0.1 | 2.2±0.2 | 3.0±0.2** | 2.4±0.2 | 3.1±0.2** |
| Contraction | 3.7±0.4* | 2.7±0.3* | 4.4±0.4* | 2.9±0.3* | 4.6±0.2* |
| FVC, kHz/mmHg | |||||
| Rest | 0.022±0.002 | 0.028±0.003 | 0.041±0.003** | 0.031±0.003 | 0.041±0.003** |
| Contraction | 0.050±0.005* | 0.036±0.004* | 0.060±0.005* | 0.040±0.004* | 0.060±0.003* |
| Peak hindlimb force, g | 113±7 | 121±7 | 105±7 | 108±4 | 98±7 |
| C57BL/10 | SMTg/mdx (line 10) | SMTg/mdx (line 23) | SMTg/mdx (line 26) | mdx | |
|---|---|---|---|---|---|
| n | 9 | 6 | 11 | 11 | 10 |
| MAP, mmHg | |||||
| Rest | 77±1 | 78±2 | 73±1 | 76±1 | 76±1 |
| Contraction | 75±1 | 75±1 | 73±1 | 72±1* | 76±1 |
| FBFV, kHz | |||||
| Rest | 1.7±0.1 | 2.2±0.2 | 3.0±0.2** | 2.4±0.2 | 3.1±0.2** |
| Contraction | 3.7±0.4* | 2.7±0.3* | 4.4±0.4* | 2.9±0.3* | 4.6±0.2* |
| FVC, kHz/mmHg | |||||
| Rest | 0.022±0.002 | 0.028±0.003 | 0.041±0.003** | 0.031±0.003 | 0.041±0.003** |
| Contraction | 0.050±0.005* | 0.036±0.004* | 0.060±0.005* | 0.040±0.004* | 0.060±0.003* |
| Peak hindlimb force, g | 113±7 | 121±7 | 105±7 | 108±4 | 98±7 |
Values are mean ± SE. * P <0.05 versus rest; ** P <0.05 versus C57BL/10.
Hemodynamics at rest and during unilateral hindlimb contraction
| C57BL/10 | SMTg/mdx (line 10) | SMTg/mdx (line 23) | SMTg/mdx (line 26) | mdx | |
|---|---|---|---|---|---|
| n | 9 | 6 | 11 | 11 | 10 |
| MAP, mmHg | |||||
| Rest | 77±1 | 78±2 | 73±1 | 76±1 | 76±1 |
| Contraction | 75±1 | 75±1 | 73±1 | 72±1* | 76±1 |
| FBFV, kHz | |||||
| Rest | 1.7±0.1 | 2.2±0.2 | 3.0±0.2** | 2.4±0.2 | 3.1±0.2** |
| Contraction | 3.7±0.4* | 2.7±0.3* | 4.4±0.4* | 2.9±0.3* | 4.6±0.2* |
| FVC, kHz/mmHg | |||||
| Rest | 0.022±0.002 | 0.028±0.003 | 0.041±0.003** | 0.031±0.003 | 0.041±0.003** |
| Contraction | 0.050±0.005* | 0.036±0.004* | 0.060±0.005* | 0.040±0.004* | 0.060±0.003* |
| Peak hindlimb force, g | 113±7 | 121±7 | 105±7 | 108±4 | 98±7 |
| C57BL/10 | SMTg/mdx (line 10) | SMTg/mdx (line 23) | SMTg/mdx (line 26) | mdx | |
|---|---|---|---|---|---|
| n | 9 | 6 | 11 | 11 | 10 |
| MAP, mmHg | |||||
| Rest | 77±1 | 78±2 | 73±1 | 76±1 | 76±1 |
| Contraction | 75±1 | 75±1 | 73±1 | 72±1* | 76±1 |
| FBFV, kHz | |||||
| Rest | 1.7±0.1 | 2.2±0.2 | 3.0±0.2** | 2.4±0.2 | 3.1±0.2** |
| Contraction | 3.7±0.4* | 2.7±0.3* | 4.4±0.4* | 2.9±0.3* | 4.6±0.2* |
| FVC, kHz/mmHg | |||||
| Rest | 0.022±0.002 | 0.028±0.003 | 0.041±0.003** | 0.031±0.003 | 0.041±0.003** |
| Contraction | 0.050±0.005* | 0.036±0.004* | 0.060±0.005* | 0.040±0.004* | 0.060±0.003* |
| Peak hindlimb force, g | 113±7 | 121±7 | 105±7 | 108±4 | 98±7 |
Values are mean ± SE. * P <0.05 versus rest; ** P <0.05 versus C57BL/10.
NOS inhibition enhances α-adrenergic vasoconstriction in the contracting hindlimbs of SMTg/mdx mice
Because NO deficiency has been implicated in the impaired vasoregulation observed in mdx mice in previous studies ( 16 , 17 ), we wanted to determine if dystrophin expression in VSMCs improved NO signaling in the SMTg/mdx mice. The NOS inhibitor N-nitro-L-arginine methyl ester (L-NAME) increased the mean arterial pressure (MAP) and decreased FBFV and FVC similar to C57BL/10, SMTg/mdx and mdx mice (Table 2 ). L-NAME had no effect on NE-mediated vasoconstriction in the resting hindlimb (Fig. 5 A and B). In contrast, L-NAME significantly enhanced the vasoconstrictor responses to NE in the contracting hindlimbs of the C57BL/10 mice, whereas it caused no further enhancement of these responses in the contracting hindlimbs of the mdx mice (Fig. 5 A and C). Intriguingly, L-NAME also enhanced the NE-mediated vasoconstriction in the contracting hindlimbs of the SMTg/mdx mice, although to a lesser extent than it did in the C57BL/10 mice (Fig. 5 A and C). Western blot analysis for nNOS, the predominant NOS isoform found in smooth muscle ( 29 , 30 ), showed similar expression in the aortas of C57BL/10 and SMTg/mdx mice, but reduced expression in the mdx mice (Fig. 5 D). These results suggest that dystrophin restoration in smooth muscle upregulates nNOS expression that may contribute to the NO-dependent attenuation of NE-mediated vasoconstriction.
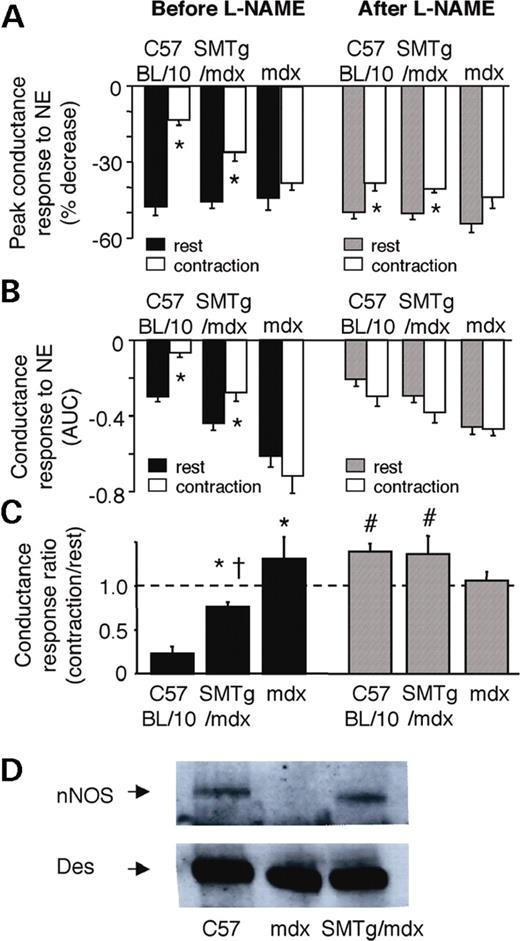
Effects of NOS inhibition on NE-mediated vasoconstriction and nNOS expression in SMTg/mdx mice. ( A and B ) Decreases in FVC evoked by NE in resting and contracting hindlimbs before and after treatment with L-NAME. Responses are expressed as peak changes (A) or as integrated changes (B). * P <0.05 versus rest; AUC, area under the curve. ( C ) Compared with the baseline responses (black bars), treatment with the NOS inhibitor L-NAME (hatched bars) significantly enhanced the vasoconstrictor responses to NE in the contracting hindlimbs of the C57BL/10 and SMTg/mdx mice, but not in the mdx mice. * P <0.05 versus C57BL/10; †P <0.05 versus mdx; #P <0.05 versus before L-NAME; C57BL/10, n =7; SMTg/mdx, n =11; mdx, n =8. ( D ) Western blot analysis showing that nNOS is expressed in aorta of C57BL/10 and SMTg/mdx mice (line 23), but not in mdx mice. Des was used as a loading control.
Effect on NOS inhinition on hemodynamics at rest and during unilateral hindlimb contraction
| C57BL/10 | SMTg/mdx | mdx | ||||
|---|---|---|---|---|---|---|
| −L-NAME | +L-NAME | −L-NAME | +L-NAME | −L-NAME | +L-NAME | |
| MAP, mmHg | ||||||
| Rest | 78±2 | 103±2* | 76±1 | 102±3* | 77±1 | 101±3* |
| Contraction | 75±1 | 96±3*,** | 75±1 | 90±2*,** | 75±1 | 92±2* |
| FBFV, kHz | ||||||
| Rest | 1.6±0.1 | 1.1±0.1* | 2.8±0.3*** | 1.5±0.1* | 3.2±0.2*** | 1.7±0.1*,*** |
| Contraction | 3.6±0.4** | 2.4±0.4*,** | 3.8±0.5** | 2.7±0.3*,** | 4.5±0.3** | 2.7±0.2*,** |
| FVC, kHz/mmHg | ||||||
| Rest | 0.020±0.002 | 0.011±0.001* | 0.036±0.004*** | 0.015±0.001* | 0.042±0.003*** | 0.017±0.002*,*** |
| Contraction | 0.048±0.007** | 0.025±0.004*,** | 0.051±0.006** | 0.030±0.004*,** | 0.060±0.004** | 0.030±0.004*,** |
| Peak hindlimb force, g | 108±7 | 92±5* | 99±9 | 89±7 | 110±7 | 105±8 |
| C57BL/10 | SMTg/mdx | mdx | ||||
|---|---|---|---|---|---|---|
| −L-NAME | +L-NAME | −L-NAME | +L-NAME | −L-NAME | +L-NAME | |
| MAP, mmHg | ||||||
| Rest | 78±2 | 103±2* | 76±1 | 102±3* | 77±1 | 101±3* |
| Contraction | 75±1 | 96±3*,** | 75±1 | 90±2*,** | 75±1 | 92±2* |
| FBFV, kHz | ||||||
| Rest | 1.6±0.1 | 1.1±0.1* | 2.8±0.3*** | 1.5±0.1* | 3.2±0.2*** | 1.7±0.1*,*** |
| Contraction | 3.6±0.4** | 2.4±0.4*,** | 3.8±0.5** | 2.7±0.3*,** | 4.5±0.3** | 2.7±0.2*,** |
| FVC, kHz/mmHg | ||||||
| Rest | 0.020±0.002 | 0.011±0.001* | 0.036±0.004*** | 0.015±0.001* | 0.042±0.003*** | 0.017±0.002*,*** |
| Contraction | 0.048±0.007** | 0.025±0.004*,** | 0.051±0.006** | 0.030±0.004*,** | 0.060±0.004** | 0.030±0.004*,** |
| Peak hindlimb force, g | 108±7 | 92±5* | 99±9 | 89±7 | 110±7 | 105±8 |
C57BL/10, n =7; SMTg/mdx, n =11; mdx, n =8. Values are mean ± SE. * P <0.05 versus −L-NAME; ** P <0.05 versus rest; *** P <0.05 versus C57BL/10.
Effect on NOS inhinition on hemodynamics at rest and during unilateral hindlimb contraction
| C57BL/10 | SMTg/mdx | mdx | ||||
|---|---|---|---|---|---|---|
| −L-NAME | +L-NAME | −L-NAME | +L-NAME | −L-NAME | +L-NAME | |
| MAP, mmHg | ||||||
| Rest | 78±2 | 103±2* | 76±1 | 102±3* | 77±1 | 101±3* |
| Contraction | 75±1 | 96±3*,** | 75±1 | 90±2*,** | 75±1 | 92±2* |
| FBFV, kHz | ||||||
| Rest | 1.6±0.1 | 1.1±0.1* | 2.8±0.3*** | 1.5±0.1* | 3.2±0.2*** | 1.7±0.1*,*** |
| Contraction | 3.6±0.4** | 2.4±0.4*,** | 3.8±0.5** | 2.7±0.3*,** | 4.5±0.3** | 2.7±0.2*,** |
| FVC, kHz/mmHg | ||||||
| Rest | 0.020±0.002 | 0.011±0.001* | 0.036±0.004*** | 0.015±0.001* | 0.042±0.003*** | 0.017±0.002*,*** |
| Contraction | 0.048±0.007** | 0.025±0.004*,** | 0.051±0.006** | 0.030±0.004*,** | 0.060±0.004** | 0.030±0.004*,** |
| Peak hindlimb force, g | 108±7 | 92±5* | 99±9 | 89±7 | 110±7 | 105±8 |
| C57BL/10 | SMTg/mdx | mdx | ||||
|---|---|---|---|---|---|---|
| −L-NAME | +L-NAME | −L-NAME | +L-NAME | −L-NAME | +L-NAME | |
| MAP, mmHg | ||||||
| Rest | 78±2 | 103±2* | 76±1 | 102±3* | 77±1 | 101±3* |
| Contraction | 75±1 | 96±3*,** | 75±1 | 90±2*,** | 75±1 | 92±2* |
| FBFV, kHz | ||||||
| Rest | 1.6±0.1 | 1.1±0.1* | 2.8±0.3*** | 1.5±0.1* | 3.2±0.2*** | 1.7±0.1*,*** |
| Contraction | 3.6±0.4** | 2.4±0.4*,** | 3.8±0.5** | 2.7±0.3*,** | 4.5±0.3** | 2.7±0.2*,** |
| FVC, kHz/mmHg | ||||||
| Rest | 0.020±0.002 | 0.011±0.001* | 0.036±0.004*** | 0.015±0.001* | 0.042±0.003*** | 0.017±0.002*,*** |
| Contraction | 0.048±0.007** | 0.025±0.004*,** | 0.051±0.006** | 0.030±0.004*,** | 0.060±0.004** | 0.030±0.004*,** |
| Peak hindlimb force, g | 108±7 | 92±5* | 99±9 | 89±7 | 110±7 | 105±8 |
C57BL/10, n =7; SMTg/mdx, n =11; mdx, n =8. Values are mean ± SE. * P <0.05 versus −L-NAME; ** P <0.05 versus rest; *** P <0.05 versus C57BL/10.
Restoration of dystrophin in VSMCs reduces serum CK levels in SMTg/mdx mice
To determine if dystrophin restoration in smooth muscle ameliorates the skeletal muscle phenotype, we evaluated serum CK levels and muscle histopathology in the SMTg/mdx mice (Fig. 6 ). CK levels of 1-year-old SMTg/mdx mice (1030±170, 766±207 and 1128±301 units in lines 10, 23 and 26, respectively) were higher than those of C57BL/10 mice (151±43 units), but they were dramatically reduced compared with those of age-matched mdx mice (4145±861 units) (Fig. 6 A). However, the histopathology of SMTg/mdx mice was not improved (Fig. 6 B–D). The number of centrally nucleated muscle fibers in SMTg/mdx mice was not significantly different from that in mdx mice (78±2, 86±3 and 81±2% in SMTg/mdx lines 10, 23 and 26, respectively, versus 80±4% in mdx).
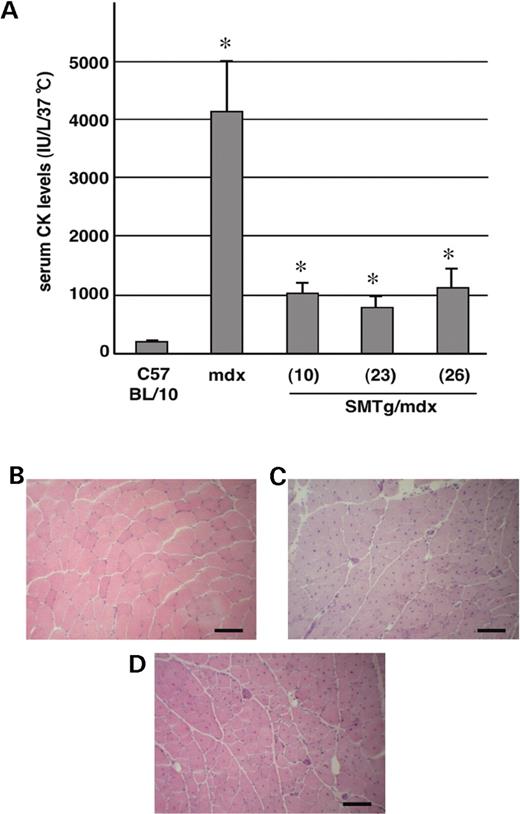
Skeletal muscle pathology in the SMTg/mdx mice. ( A ) Serum CK levels of 1-year-old SMTg/mdx mice were significantly lower than those of age-matched mdx mice ( n ≥7 each group). * P <0.01 for comparisons between mdx and each line of SMTg/mdx. ( B – D ) Sections of TA muscles from a 1-year-old C57BL/10 (B), SMTg/mdx (line 23) (C), and mdx (D) mouse stained with H&E. There were no discernible histological differences between TA muscles of the SMTg/mdx and mdx mice, both of which had a predominant number of fibers with centralized nuclei. Scale bar: 100 µm.
DISCUSSION
This is the first study to evaluate the effect of selective dystrophin expression in smooth muscle on vascular regulation and skeletal muscle dystrophy in the mdx mouse. Previous studies have shown that the normally robust NO-dependent attenuation of α-adrenergic vasoconstriction that occurs in the exercising muscle is impaired in the dystrophin-deficient mdx mouse ( 16 , 17 , 20 ). We now report that this impairment is partially reversed in three independent lines of transgenic mdx mice that express dystrophin only in smooth muscle (SMTg/mdx mice). In addition, the characteristic elevation in serum CK that occurs in mdx mice was markedly attenuated in the SMTg/mdx mice. These findings suggest that expression of dystrophin in smooth muscle improved both the vascular and dystrophic phenotypes of the mdx mouse.
Our study provides new insight into the function of dystrophin in blood vessels, particularly in the resistance vessels of the skeletal muscle circulation. We found that there were no consistent or striking differences in basal blood pressures or hindlimb blood flows among the wild-type, SMTg/mdx and mdx mice under anesthesia. Also, peak vasoconstrictor responses to intra-arterial injections of NE into the resting hindlimbs were similar among the three genotypes. Taken together, these data suggest that dystrophin in VSMCs is not essential to maintain normal vascular tone or to evoke acute increases in tone in the skeletal muscle circulation. In agreement with these in vivo results, a previous in vitro study of smooth muscle from wild-type and mdx mice showed that cytosolic Ca 2+ concentrations were similar under basal conditions, and increased similarly in response to a variety of contractile stimuli ( 31 ).
In contrast, dystrophin expression in VSMCs appeared to influence the attenuation of NE-mediated vasoconstriction that occurs in the contracting hindlimb, with the degree of attenuation being greatest in the wild-type mice, least in the mdx mice and intermediate in the SMTg/mdx mice. Previous studies have attributed this contraction-induced attenuation to a paracrine effect of vasodilatory metabolites that are produced in the active muscle cells ( 32 ). We therefore considered the possibility that dystrophin in VSMCs enhanced the dilator response to muscle-derived metabolites. However, we felt that this explanation was unlikely given the findings of our study and others that dystrophin deficiency in the mdx mouse does not appear to alter vascular reactivity per se to either constrictor (e.g. phenylephrine, KCl) or dilator (e.g. acetylcholine, nitroprusside) stimuli ( 17 , 31 , 33 ).
Instead, our results suggest that dystrophin in VSMCs facilitates an autocrine effect of NO to attenuate α-adrenergic constriction in the contracting mouse hindlimb. NOS inhibition prevented the contraction-induced attenuation of NE-mediated vasoconstriction in both the wild-type and SMTg/mdx mice. The lack of an effect of NOS inhibition that we observed in the mdx mice was previously attributed to a deficiency of nNOS in skeletal muscle ( 16 , 17 ) that normally associates with the dystrophin complex at the sarcolemma, but is secondarily reduced in the absence of dystrophin ( 23 , 24 ). Whether nNOS associates with the dystrophin complex in VSMCs is not known, but we did find that nNOS protein was present in aortic tissue of wild-type and SMTg/mdx mice, but not in that of mdx mice. Other investigators also have reported that nNOS is expressed in VSMCs, and have shown that NO generated in VSMCs can blunt constrictor responses, supporting an autocrine signaling role for NO in vascular tissue ( 29 , 30 , 34 , 35 ). Because dystrophin, nNOS and α-adrenergic receptors have been localized to the cell membrane caveolae ( 36–38 ), we speculate that dystrophin deficiency may disrupt intrinsic signaling complexes that act in concert to regulate VSMC contractile activity.
Our novel finding that a reduced autocrine effect of NO in dystrophin-deficient VSMCs contributes to vascular dysregulation in the mdx mouse hindlimb complements previous studies showing impairments in paracrine signaling by NO in this same animal model ( 16 , 17 , 20 , 33 ). In addition to the effect of dystrophin deficiency in skeletal muscle cells to reduce sarcolemmal nNOS and attenuate the effect of muscle-derived NO to blunt α-adrenergic vasoconstriction, dystrophin deficiency in vascular endothelial cells has recently been implicated in an impaired NO-dependent dilator response to increased flow (shear stress) ( 16–18 , 20 , 33 ). Collectively, the implications of these findings are that some dystrophin-deficient tissues, such as skeletal muscle, may have an increased susceptibility to develop ischaemia. Whether the skeletal muscles of DMD patients or mdx mice are subject to varying degrees of functional ischaemia during the course of normal daily activities is an important, but unanswered, question.
Another key unresolved question is whether aberrant vascular regulation contributes to the pathogenesis of the dystrophinopathies, for example, by inducing an ischaemic stress that initiates or exacerbates injury of the dystrophin-deficient skeletal muscles. A remarkable pathological feature of the muscles of DMD patients and mdx mice that is observed early in the disease is the appearance of random groups of necrotic muscle fibers surrounded by histologically normal fibers ( 9 , 10 , 39 , 40 ). Such grouped or focal necrosis can be reproduced in normal muscle by microvascular infarction, suggesting a causal role for ischaemia ( 11–13 ). This concept has been advanced in recent studies investigating the pathogenesis of cardiomyopathy characterized by focal necrosis in sarcoglycan-deficient mice, which are models of limb girdle muscular dystrophy ( 41–43 ). In these mice, coronary vessel abnormalities resembling spasms precede the development of myocardial necrosis and elevation of cardiac-specific troponin I ( 41 ). Treatment with the calcium channel blocker verapamil ameliorated both the microvascular spasms and the cardiac muscle pathology, suggesting a pathogenetic role for vascular dysfunction ( 41 , 43 ).
In our study, serum CK levels were markedly reduced in 1-year-old SMTg/mdx mice compared with the values in the mdx mice, although they remained above the values in the wild-type mice. Interestingly, the CK levels in the three genotypes corresponded with the degree of vascular dysregulation that we observed in the contracting hindlimbs of mice studied at 4 to 6 months of age. Elevated CK levels are characteristic of increased membrane permeability ( 44 ). For this reason, we also evaluated the uptake of Evans blue dye in muscle ( 45 ), but we observed no differences between SMTg/mdx and mdx mice at 6 to 7 months of age in these initial experiments (data not shown). Clearly, further experiments are warranted to assess serial changes in CK levels and Evans blue dye uptake in these transgenic mice.
We also found that the proportion of muscle fibers with centralized nuclei did not differ in 1-year-old mdx and SMTg/mdx mice, suggesting that muscle regeneration was not altered by dystrophin expression in smooth muscle. However, because of the characteristically high levels of muscle regeneration in the mdx mice, the majority of fibers already contain central nuclei by the age of 12 weeks ( 39 , 40 , 46 ). Other explanations for the lack of improvement in the histological phenotype of the SMTg/mdx mice might include the partial rather than complete normalization of vasoregulation in active muscle or lower than the normal level of dystrophin expression in VSMCs. Because we did not study transgenic mice that were not crossed onto the mdx background, we do not know if the lower level of dystrophin expression in the SMTg/mdx mice was due to protein degradation on the mdx background. However, β-sarcoglycan and β-dystroglycan were detected by immunofluorescent staining in smooth muscle of SMTg/mdx mice, suggesting that this level of dystrophin was sufficient to restore the dystrophin–glycoprotein complex ( 41 ).
The SMTg/mdx mouse may provide a useful model to elucidate the effect of dystrophin in VSMCs on vascular structure as well as function. For example, arteriogenesis is enhanced in mdx mice in chronically ischaemic hindlimb muscle and in wounded skin, but the tissue-specific role of dystrophin-deficiency in this neovascularization is unknown ( 47 ). The SMTg/mdx mouse also could be used to determine if dystrophin expression in non-vascular smooth muscle ameliorates some of the gastrointestinal disorders or respiratory dysfunction that manifest in the dystrophinopathies.
In conclusion, we have shown that the expression of dystrophin in VSMCs of the dystrophin-deficient mdx mouse partially corrects the aberrant α-adrenergic vasoconstriction in the exercising muscle and reduces serum CK, which is a classical index of active dystrophy. Dystrophin appears to be part of a local regulatory system in smooth muscle that uses NO as an autocrine modulator of α-adrenergic constriction. In the clinical setting of DMD, loss of this protective mechanism may leave the dystrophin-deficient skeletal muscle fibers vulnerable to ischaemic insults, thereby exacerbating muscle fiber necrosis ( 48 ). Our data suggest a potential benefit to restoring dystrophin in vascular smooth muscle in DMD.
MATERIALS AND METHODS
Transgene construct and production of transgenic mice
All animal experiments were approved by the committee of the Center for Animal Resources and Development, Kumamoto University, Japan.
Oligo-DNA including restriction enzyme, Apa I, Spe I, Hind III, Not I, Sal I, Eco RI and Pst I site was synthesized, and subcloned into Apa I– Pst I sites pBluescript II KS-vectors (Stratagene) that removed NotI site by T4 DNApolymerase treatment, to generate BlueII-oligo. The Hind III– Not I fragment containing human smooth muscle α-actin promoter (EA 4.7 promoter) from pBS-HSMA-EA4.7, Not I– Sal I fragment containing full-length h-dys cDNA, and Eco RI– Xho I fragment containing the SV-40 intron and poly-A signal from pBINIacZ was inserted into BlueII-oligo in order to generate pEA 4.7-Dys-An (SMTg construct). Finally, the SMTg construct that expresses h-dys under control of EA 4.7 promoter was generated in order to analyze the dystrophin function in smooth muscle.
The construct, which was excised with Spe I, was microinjected into 800 fertilized oocyte pronuclei from C57BL/6 mice. They were returned to C57BL/6 recipients. The mice that express dystrophin only in smooth muscle (SMTg/mdx) were produced by back-cross of the transgenic mice with mdx mice.
Genotyping
Transgenic founders and offspring back-crossed with mdx mice were identified by PCR and Southern blot analysis on DNA from tissue samples of the tail. For PCR, the sense primer sequence is located in EA 4.7 promoter and the antisense primer sequence is in h-dys in the construct. The following primers and PCR conditions were used: The forward primer 5′-TGCTCACGAGAACACATGATA-3′ and the reverse primer 5′-AGTGTATATCAAGGCAGCGAT-3′: first denaturation at 94°C for 5 min, followed by 40 cycles of 1 min at 94°C, 1 min at 57°C, 3 min at 72°C and 10 min last extension at 72°C, which amplify the 504 bp fragment. For Southern hybridization, genomic DNA was digested with Pst 1, and resolved on a 0.9% agarose gel. The bands were then transferred to nylon membrane (Amersham Pharmacia Biotech) and hybridized with the 32 P-labeled DNA probe of a 634 kb fragment constructed in the EA4.7 promoter. The membrane was placed on autoradiographic film (Fuji Photo Film Co., Ltd) and exposed for 24 h.
Reverse transcription-polymerase chain reaction
Semi-quantitative real-time RT–PCR was performed using the ABI Prism 7900 Sequence Detector System ® (PE Applied Biosystems). Total RNA was extracted from nine tissues including brain, lung, aorta, heart, liver, kidney, skeletal muscle, small intestine and testis of one of SMTg/mdx lines (line 23, n =1) with TRIzol reagent (Invitrogen life technologies). For cDNA synthesis, 2 µg of total RNA were reverse-transcribed using SuperScript™ First-Strand Synthesis System for RT–PCR (Invitrogen life technologies) according to the manufacturer's Specification. TaqMan ® probe and primers specific for h-dys cDNA that can detect only h-dys were designed as follows. The sense primer 5′-AGACCTTGGGCAGCTTGAAAA-3′, the antisense primer 5′-GGAGATAACCACAGCAGCAGATG-3′ and the TaqMan ® probe 5′-TTGAAGACCTTGAAGAGC-3′. cDNA samples were mixed with primers, probe and TaqMan ® Universal PCR Master Mix as described in the manufacturer's directions (PE Applied Biosystems). The PCR was conducted using the following parameters: 50°C for 2 min, 95°C for 10 min and 40 cycles at 95°C for 15 s and 60°C for 1 min. Standard curve for the target amplicon was drawn using the transgene construction. All PCR assays were performed in duplicate and results are represented by the mean values. 18S rRNA was used as housekeeping gene against which all the samples were normalized for differences in the amount of total RNA added to each cDNA reaction and for variation in the reverse transcriptase efficiency among the different cDNA reactions.
Immunoblot analysis
Protein extracts of tibialis anterior muscle (TA) and aorta from three SMTg/mdx lines ( n =2 per each line) at 1 year of age and age-matched control C57BL/10 ( n =2) and mdx mice ( n =2) were prepared from frozen tissue, homogenized in extraction buffer (2% SDS, 5% β-mercaptoethanol, 4 m m EDTA, 40 m m Tris, 0.24 m glycine, 40% glycerin, 0.03% bromphenol blue, 20 µg/ml PMSF, pH 8.5), quantified and diluted to equal concentrations with extraction buffer. Equivalent amounts of protein were separated by SDS-PAGE on 4–12% gradient gels and electrophoretically transferred to nitrocellulose membranes (BioScience). Membranes were incubated with the specific antibody (DYS2 1:100, Novocastra Laboratories, Ltd; Anti-Desmin 1 : 5000, Sigma; Anti-nNOS 1 : 5000, Zymed Laboratories), washed and then probed with secondary biotinylated anti-mouse antibody (1:10 000, Vector laboratories) or anti-rabbit antibody (1:10 000, Vector Laboratories). The antigen-antibody complex was magnified using the avidin–biotin complex (ABC) method, using the ABC kit (Vector Laboratories). Blots were developed using the ECL chemiluminescence system (Amersham).
Immunochemical staining for dystrophin and histological analysis
Immunohistochemistry for dystrophin was performed in order to analyze dystrophin expression of smooth muscle in the lungs of all the SMTg/mdx lines and age-matched controls. The primary antibody for dystrophin (60 kDa; a kind gift from Dr E.P. Hoffman) was diluted 1:300 in 1% bovine serum albumin and the secondary antibody (FITC-conjugated rabbit anti-sheep; Southern Biotechnology Associates, Inc.) was diluted 1:200. Stained sections were viewed on a microscope and captured using image analysis software (Photograb-2500; Fuji Photo Film Co., Ltd). The expression of dystrophin, β-sarcoglycan and β-dystroglycan on blood vessels in skeletal muscles was analyzed with mouse monoclonal anti-dystrophin, anti-β-sarcoglycan and anti-β-dystroglycan antibodies (DYS2, 1:25; NCL-β-DG, 1:20; NCL-β-SARC, 1:20; Novocastra Ltd) and M.O.M.™ peroxidase kit (Vector). The immunoreactivity was visualized using 3,3′-diaminobenzidine as a chromogen. Skeletal muscles were frozen in isopentane pre-cooled in liquid nitrogen and 10-µm-thick sections were cut with a cryostat. For central nuclei counts, the sections were stained with hematoxylin and eosin (H&E) in standard fashion. The percentage of the total muscle fibers containing central nuclei that were present in complete cross-sections of TAs was determined microscopically in 1-year-old SMTg/mdx ( n =2, 4 and 4 of SMTg/mdx 10, 23 and 26, respectively) and age-matched control C57BL/10 ( n =2) and mdx ( n =4) mice.
Hemodynamic measurements and hind limb contraction
All protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. C57BL/10, SMTg/mdx (lines 10, 23, 26), and mdx mice of both sexes (4–6-month-old; 23–38 g) were initially anesthetized with a mixture of tiletamine (4 mg/kg, i.p.), zolazepam (4 mg/kg, i.p.) and xylazine (20 mg/kg, i.p.). After surgical instrumentation, mice were switched to inhalation anesthesia (1% isoflurane) for the remainder of the experiment. Catheters were placed in a carotid artery for the measurement of arterial blood pressure and in a jugular vein for drug infusion. A third catheter was inserted into the right femoral artery and advanced to the abdominal aorta and was used for intra-arterial hindlimb infusion of NE. A Doppler crystal embedded in a soft silastic cuff was placed around the left femoral artery to measure changes in blood flow velocity by recording the pulsatile and mean Doppler shifts using a VF-1 pulsed Doppler flow system (Crystal Biotech). The left triceps surae muscles were connected to a force-displacement transducer (FT-10, Grass Instruments) via the calcaneal tendon. The left sciatic nerve was exposed, covered with warm mineral oil, and affixed to stimulating electrodes. To produce intermittent, tetanic contractions, the sciatic nerve was stimulated (model S88, Grass Instruments) at 2–3 times the motor threshold voltage with 100 ms trains of pulses (100 Hz, 0.2 ms duration) at a rate of 30 trains per min.
Following a 20-min stabilization period, arterial blood pressures and FBFV were sampled continuously at 100 Hz and saved as digital files (PowerLab, AD Instruments). FVC was calculated online as mean blood flow velocity divided by MAP. Hemodynamic responses to intra-arterial hindlimb infusion of NE (4–13 ng in volumes of 3–10 ml) were evaluated when the hindlimb was at rest, and then again during hindlimb contractions. These measurements were repeated in a subset of mice from each group after treatment with L-NAME (10 mg/kg, i.v.).
NE-mediated vasoconstriction was analyzed by measuring the change in FBFV or vascular conductance expressed as the maximal decrease from the pre-injection baseline (peak response) or as the integrated area below baseline (total response). Between group differences were evaluated using ANOVA followed by Tukey-Kramer post hoc tests. Within group differences (rest versus contraction, before L-NAME versus after L-NAME) were evaluated using paired t -tests (GraphPad Prism software, 4.0).
Serum CK level
The concentration of CK in serum collected by tail cutting was measured by a laboratory examination agency (SRL, Inc.) as described by the manufacturer of the assay kit (Kanto Kagaku). CK levels were determined in 1-year-old C57BL/10, SMTg/mdx and mdx mice ( n ≥7 each). Statistical significance of differences between groups was determined using Mann–Whitney test.
ACKNOWLEDGEMENTS
The authors thank Dr Ryan Pruchnic (Cook MyoSite, Inc., USA) for his assistance. This study was supported by a research grant for Nervous and Mental Disorders from the Ministry of Health, Labour and Welfare, a grant for research in Brain Science from the Ministry of Health, Labour and Welfare, a grant from the Ministry of Education, Science, Culture and Sports of Japan, and US National Institutes of Health grants HL06296 and AR051034 (to G.D.T.).
Conflict of Interest statement. The authors declare that they have no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}