




Abstract
β-Adrenergic receptor-mediated cAMP or protein kinase A (PKA)-dependent modulation of cardiac potassium currents controls ventricular action potential duration (APD) at faster heart rates. HERG (KCNH2) gene mutations are associated with congenital long-QT syndrome (LQT2) and affect IKr activity, a key determinant in ventricular repolarization. Physical activity or emotional stress often triggers lethal arrhythmias in LQT2 patients. β-Adrenergic stimulation of HERG channel activity is amplified and prolonged in vitro by the adaptor protein 14-3-3ϵ. In LQT2 families, we identified three novel heterozygous HERG mutations (G965X, R1014PfsX39, V1038AfsX21) in the C-terminus that led to protein truncation and loss of a PKA phosphorylation site required for binding of 14-3-3ϵ. When expressed in CHO cells, the mutants produced functional HERG channels with normal kinetic properties. We now provide evidence that HERG channel regulation by 14-3-3ϵ is of physiological significance in humans. Upon co-expression with 14-3-3ϵ, mutant channels still bound 14-3-3ϵ but did not respond with a hyperpolarizing shift in voltage dependence as seen in wild-type channels. Co-expression experiments of wild-type and mutant channels revealed dominant-negative behavior of all three HERG mutations. Simulations of the effects of sympathetic stimulation of HERG channel activity on the whole-cell action potential suggested a role in rate-dependent control of APD and an impaired ability of mutant cardiac myocytes to respond to a triggered event or an ectopic beat. In summary, the attenuated functional effects of 14-3-3ϵ on C-terminally truncated HERG channels demonstrate the physiological importance of coupling β-adrenergic stimulation and HERG channel activity.
INTRODUCTION
Regulation of contraction rate and force of the heart is an essential property of the cardiovascular system and crucial for the adaptation of blood flow in response to environmental factors. It is, in particular, the stimulation of the sympathetic nervous system (SNS) that in response to exercise or emotional stress can lead to an immediate and dramatic increase in heart rate, which is paralleled by a concomitant decrease in action potential duration (APD) of cardiac myocytes and shortening of the QT interval of the surface electrocardiogram. These effects are mediated by activation of β-adrenergic receptors that regulate the activity of specific cardiac ion channels via increases in intracellular cAMP concentrations.
There is growing evidence for a cAMP-dependent regulation of the activities of both slow and rapid outward potassium (K+) currents IKs and IKr (1–6). Since both channel activities directly affect repolarization currents, such a regulation is capable of adapting cellular excitability at a millisecond time scale. The role of the two K+ channels in these processes is of particular pathophysiological interest because mutations in the genes encoding their molecular correlates, KCNQ1/KCNE1 (IKs) and KCNH2 (IKr), have been linked to inherited long-QT syndrome (LQTS) (7–9). LQTS is characterized by a prolonged QT interval on the surface electrocardiogram and increased susceptibility to ventricular tachyarrhythmias that may lead to recurrent syncope and sudden cardiac death (10,11). In ∼30% of the cases, LQTS is associated with mutations in the human ether-à-go-go-related gene KCNH2 (LQT2, also called HERG). In the majority of LQT2 mutation carriers, cardiac events were triggered by conditions involving increased adrenergic tone, such as, for example, emotional stress (43%) or exercise (13%) (12). To understand the mechanistic basis of arrhythmogenesis in LQT2, it is therefore crucial to identify the molecular links between β-adrenergic stimulation and HERG channel regulation.
The KCNH2 gene encodes the α-subunit of the voltage-gated K+ channel HERG. Co-assembly of four HERG subunits results in a functional K+ channel mediating the delayed rectifier IKr current (13,14). Each HERG subunit has an intracellular N- and C-terminus, a core region consisting of six transmembrane segments, and a pore loop with a selectivity filter. Various KCNH2 mutations have been identified that lead to a reduction or complete attenuation of IKr. These mutations may result in haploinsufficiency or in dominant-negative suppression of IKr. Mutations in the N-terminal Per-Arnt-Sim (PAS) domain may profoundly alter deactivation kinetics of HERG-mediated currents (15). Sequences in the large intracellular C-terminus of HERG were shown to be important for biophysical properties (16), subunit assembly (17,18) and intracellular trafficking (19–21). However, LQT2 mutations associated with alterations in HERG channel regulation have not been reported so far, although recent findings suggest β-adrenergic modulation of HERG channel activity via cAMP binding to the cyclic nucleotide-binding domain (1,22), or via phosphorylation of particular serine (S) residues by protein kinase A (PKA) (2,4).
Phosphorylation of S283 in the N-terminus and S1137 in the C-terminus stimulated binding of 14-3-3ϵ (2). 14-3-3ϵ is a member of a protein family that participates in a wide range of biological processes, mostly through binding to serine-phosphorylated sequence motifs in diverse partners [for review see (23)]. In the case of HERG channels, 14-3-3ϵ binding stabilizes a phosphorylated state and enhances HERG activity by shifting channel activation toward hyperpolarizing membrane potentials (2).
In our present study, we combined human genetic, functional and action potential simulation techniques to provide evidence that HERG channel regulation by 14-3-3ϵ is of physiological significance in humans. In LQT2 patients, we detected three novel mutations (G965X, R1014PfsX39, V1038AfsX21) causing a C-terminal truncation of HERG subunits and eliminating the C-terminal PKA-phosphorylation site at serine 1137. When expressed in vitro, the three truncated HERG subunits still interacted with 14-3-3ϵ. However, in contrast to wild-type channels, the activation threshold for activation of the mutant HERG channels was not shifted to hyperpolarized membrane potentials upon co-expression with 14-3-3ϵ. Co-expression experiments of wild-type and mutant channels revealed dominant-negative behavior of all three mutations. Simulation of the effects of SNS-mediated stimulation of HERG channel activity on the whole cell action potential suggested an important role in the suppression of premature ectopic beats. We conclude that the altered or absent functional effect of 14-3-3ϵ on mutant HERG channels represents a novel pathophysiological mechanism that potentially underlies adrenergically mediated arrhythmias.
RESULTS
Clinical findings and genetic analysis
We identified three novel C-terminal KCNH2 gene mutations in two unrelated LQTS families and in a single patient: one family with 3036-3048del (resulting in the amino acid change R1014PfsX39; N=12 carriers, Fig. 1), one family with 3107-3111dup (V1038AfsX21; N=4 carriers), and one proband with 2893G>T (G965X). The LQT2 gene was completely sequenced, but, except for the mutations reported here, no other disease-associated alterations were identified in the index patients. The three identified mutations were not present in a large number of normal control samples (>200 chromosomes). Furthermore, sequencing and/or co-segregation analyses were performed to exclude other mutations in the LQT1, LQT3, LQT5 and LQT6 genes at these loci. The LQT4 locus was not investigated by sequencing because the specific and rare phenotype consisting of QT interval prolongation, sinus bradycardia and atrial fibrillation (24) was not present in our subpopulation of patients.
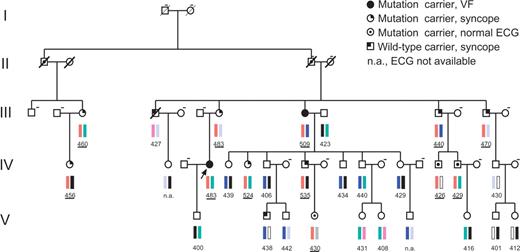
Pedigree of a large multi-generation family (generation I–V) with congenital LQTS and a C-terminal HERG mutation (R1014PfsX39). Haplotypes at the LQT2 locus (D7S483–D7S636) are shown; red bars indicate the chromosomal haplotype marker associated with the disease; other colored bars indicate markers unrelated to the disease. Males are shown as squares and females as circles. Values below symbols indicate QTc values (from lead II) in ms1/2 and were underlined when referring to mutation carriers. Dashed line on the right top of a symbol means ‘DNA not available for analysis’. The disease haplotype was found in nine individuals with a QTc prolongation (>440 ms1/2). In contrast to other individuals with borderline QTc values, they all suffered from syncope. The disease haplotype was also found in three individuals with normal QTc values (IV-16: 426 ms1/2, IV-17: 429 ms1/2, V-4: 430 ms1/2). In these cases, the mutation was identified retrospectively. Altogether, the disease haplotype was identified in 12 individuals, but the disease penetrance was incomplete (75%), because only nine patients had LQTS symptoms.
The heterozygous mutation R1014PfsX39 was found in a large German multi-generation family (N=36 related members) with LQTS (Fig. 1). Because of the large size of the family, we first performed a co-segregation analysis in affected patients (symptomatic patients + QTc >440 ms1/2) using microsatellite markers of all LQT gene loci. Segregation analysis was only positive for LQT2 markers (see Materials and Methods; Fig. 1). The disease haplotype was found in nine individuals with a QTc prolongation (>440 ms1/2). In contrast to other individuals with borderline QTc values, they all suffered from syncope. The 31-year-old female index patient of the family had suffered from stress-induced syncopal attacks, initially categorized as ‘epileptic seizures’, since the age of 16. At the age of 25, she had a cardiac arrest due to ventricular fibrillation, which was successfully treated. A pronounced prolongation of the QTc interval (500 ms1/2) led to the diagnosis of LQTS and was finally confirmed genetically by the presence of the KCNH2 mutation. Further evaluation of the family history revealed that one aunt had also suffered a cardiac arrest. A history of syncope was frequent in the family (N=11). Case histories revealed that in two of these family members who had neither the LQT2 mutation nor a QT interval prolongation, syncope was most likely related to orthostatic hypotension. In addition to the index patient, four other mutation carriers had a history of stress-induced syncope; others had cardiac events related to arousal due to sudden acoustic noise. Following identification of the KCNH2 mutation R1014PfsX39, all family members were analyzed retrospectively. The analysis showed that all patients with the disease haplotype (N=9) carried the heterozygous mutation. The disease haplotype was also found in three mutation carriers with apparently normal QTc values (426–430 ms1/2). Taken together, 12 individuals were carriers of the disease haplotype, but the disease penetrance in this family was incomplete (75%), because only nine mutation carriers had LQTS symptoms. Interestingly, borderline QTc values (430–440 ms1/2) were also found in asymptomatic family members. Reliable identification of LQT2 patients in this family was only possible through genetic analysis (Fig. 1). The prominent ECG phenotype of R1014PfsX39 mutation carriers was the presence of notched or biphasic T waves (not shown).
A heterozygous duplication of nucleotides 3107-3111 (V1038AfsX21) was detected in the second family. Four out of seven family members were mutation carriers. Furthermore, LQTS was diagnosed in four other members of this family, who, however, were not available for genetic analysis. The 25-year-old male proband had a syncope related to physical activity at the age of 23. On the surface ECG, the T wave was notched (V4) or biphasic (V2-V3), with a QTc of 503 ms1/2 (lead II). The 52-year-old father of the proband was also carrier of this mutation, but he was asymptomatic (QTc: 480 ms1/2).
Mutation G956X was identified in a 28-year-old woman with a history of syncope. The first episode had occurred at the age of 25 and was followed by recurrent episodes usually triggered by loud noises (alarm clock, telephone ringing). At the age of 27, polymorphic ventricular tachycardia was documented during a syncopal episode that occurred while the patient underwent a neurological examination. Subsequently, the finding of a prolonged QT interval (QTc: 498 ms1/2 in lead II) confirmed the diagnosis of LQTS. Following treatment with nadolol (1 mg/kg/day), no further cardiac events occurred during 3 years of follow-up. The family history was negative for syncope and/or unexplained cardiac arrest at this age. Family screening demonstrated that this mutation had been inherited from the asymptomatic father (no ECG available). No further family members were available for clinical evaluation and/or genetic testing.
Functional expression of mutant HERG channels in CHO cells
All three HERG mutations resulted in predicted protein truncations of varying lengths in the HERG C-terminus (Fig. 2). We investigated the consequences for channel properties and function by transient expression in CHO cells and compared the biophysical properties of wild-type and mutant HERG subunits.

Schematic representation of the primary structure of wild-type and mutant HERG subunits analyzed in this study. This figure shows key structural elements in wild-type HERG, such as transmembrane domains (S1–S6), pore loop (P), cyclic nucleotide-binding domain (cNBD), C-terminal endoplasmic reticulum retention (RGR), ‘tetramerizing coiled coil’ (TCC) sequences and serine phosphorylation sites that are critical for 14-3-3ϵ binding (S283, S1137). Gray horizontal lines indicate amino acids that are identical to HERG subunits; black horizontal lines indicate additional C-terminal amino acid sequences resulting from reading frame shifts introduced by the mutations R1014PfsX39 and V1038AfsX21. Scale bar unit: amino acids.
In agreement with previous data (2), wild-type HERG channels mediated slowly activating outward currents at test potentials positive to −40 mV. Current densities reached a maximum at about 10 mV (Fig. 3A and E; N=12) and then decreased at membrane potentials positive to 10 mV, reflecting the well-described inactivation properties of HERG channels at more depolarized potentials (25). Expression of mutant HERG subunits (HERG-G965X, N=7; HERG-R1014PfsX39, N=7; and HERG-V1038AfsX21 N=7) in CHO cells yielded functional HERG channels that mediated similar currents as wild-type HERG (Fig. 3B–D). Current density–voltage curves revealed a one-third reduction in peak current densities for HERG-G965X channels, but no significant differences could be found between wild-type and the other two mutant HERG channels (Fig. 3E). Half maximal activation (V1/2) and slope factor (z) of wild-type HERG currents were −6.28±0.62 mV for V1/2 and 6.79±0.22 mV (N=12) for z (Fig. 3F). All three mutant HERG channels exhibited comparable conductance–voltage relationships (Fig. 3F and Table 1). Activation time constants, τact, which showed a steady decrease between test potentials of −20 to 20 mV, were similar for wild-type and all three mutant HERG channels [P>0.05 (ANOVA), Fig. 3G and Table 1]. Next, we investigated HERG channel inactivation, which is known to be accelerated at more positive potentials (25). Accordingly, we observed a voltage-dependent decrease in the inactivation time constant [9.25±0.27 ms at −40 mV to 4.31±0.26 ms at +40 mV (N=8)]. Mutant channels showed a similar voltage-dependence of inactivation and were not significantly different from wild-type [τinact −40 mV/τinact+40 mV (ms)]: G965X (N=9) 10.25±0.4/4.85±0.1; R1014PfsX39 (N=12) 10.6±0.3/4.7±0.3; V1038AfsX21 (N=9) 9.9±0.4/4.4±0.2 (P>0.05 for all comparisons of HERG versus mutants). The deactivation time course of the inwardly rectifying HERG currents measured at test potentials between −140 and −50 mV was well described by an exponential fit with two time constants, τ1 and τ2. They were similar in magnitude and ratio for wild-type or mutant HERG channels [τfast/τslow at −120 mV (ms)]: HERG (N=8) 13±1/84±10; G965X (N=10) 14±2/75±9; R1014PfsX39 (N=10) 14±1/82±8; V1038AfsX21 (N=9) 12±1/70±8 (P>0.05 for all comparisons of HERG versus mutants).
![Voltage-dependence and activation kinetics of HERG and mutant HERG subunits harboring either mutation R1014PfsX39, V1038AfsX21 or G965X. Potassium currents measured by 3.5-s depolarizing test pulses from a holding potential of −80 mV to test potentials between −50 and +50 mV, followed by repolarization to −40 mV [protocol as depicted in (A, bottom)]. (A) Representative current traces from wild-type HERG-transfected cells. (B–D) Representative current traces obtained from HERG-G965X, HERG-R1014PfsX39 or HERG-V1038AfsX21-transfected cells. (E) Bell-shaped current–voltage relationships for HERG, HERG-G965X, HERG-V1038AfsX21 and HERG-R1014PfsX39. (F) Normalized tail current amplitudes for HERG, HERG-G965X, HERG-R1014PfsX39 and HERG-V1038AfsX21 were plotted against the respective test potentials. A Boltzmann function was fitted to the data to obtain values for half-maximal activation and slope (Table 1). (G) Time constants of activation, τact, for HERG and HERG-G965X, HERG-R1014PfsX39 and HERG-V1038AfsX21 for test potentials of −20, 0 and +20 mV. Absence of error bars indicates errors smaller than symbol size.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/19/10.1093/hmg/ddl230/2/m_ddl23003.jpeg?Expires=1716594673&Signature=4L9kd5JsxMP2o3EJ39Kk-~XXHtHP66ydy3EbmkgfGP8sXLgOjHbMkmdDTiVPTAnzmDhdYesadgxX8KCBr1tVLctgkRbRSF8QVASfMBQd-lLohIjde5afSHpArwZYTRF70o29U6B~ZXeJnCtelCyi1SKeWAnBVcTjaLy8mNpdlTwtAZtHg88yCELV-EEHvvpl1Ng9DDokrUK47HvpruPXCrJMvfMBPf2LPLN8764Uo1oeePDdNCu47A4BZaMoVBztlr9USh3sq1dnO4ge5Tmqqmflkm7dBabHMdd9DvjQezP3BXYVKC0ddwedMs8ikjMf~oCO-dSEJRywY-0DDW2HHA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Voltage-dependence and activation kinetics of HERG and mutant HERG subunits harboring either mutation R1014PfsX39, V1038AfsX21 or G965X. Potassium currents measured by 3.5-s depolarizing test pulses from a holding potential of −80 mV to test potentials between −50 and +50 mV, followed by repolarization to −40 mV [protocol as depicted in (A, bottom)]. (A) Representative current traces from wild-type HERG-transfected cells. (B–D) Representative current traces obtained from HERG-G965X, HERG-R1014PfsX39 or HERG-V1038AfsX21-transfected cells. (E) Bell-shaped current–voltage relationships for HERG, HERG-G965X, HERG-V1038AfsX21 and HERG-R1014PfsX39. (F) Normalized tail current amplitudes for HERG, HERG-G965X, HERG-R1014PfsX39 and HERG-V1038AfsX21 were plotted against the respective test potentials. A Boltzmann function was fitted to the data to obtain values for half-maximal activation and slope (Table 1). (G) Time constants of activation, τact, for HERG and HERG-G965X, HERG-R1014PfsX39 and HERG-V1038AfsX21 for test potentials of −20, 0 and +20 mV. Absence of error bars indicates errors smaller than symbol size.
Activation properties of wild-type and mutant HERG subunits in absence or presence of 14-3-3ϵ
| V1/2 (mV) | Slope (mV) | τact (at 0 mV) (*s*) | N | |
|---|---|---|---|---|
| HERG | −6.28±0.62 | 6.79±0.22 | 1.30±0.09 | 12 |
| HERG and 14-3-3ϵ | −17.1±1.0***† | 6.91±0.10 | 0.57±0.04***a | 8 |
| G965X | −4.04±0.95 | 9.47±0.50 | 1.18±0.10 | 7 |
| G965x and 14-3-3ϵ | −6.91±1.6 | 8.49±0.47 | 1.01±0.10 | 7 |
| R1014PfsX38 | −7.63±0.71 | 8.50±5.39 | 1.07±0.07 | 7 |
| R1014PfsX38 and 14-3-3ϵ | −6.75±1.20 | 8.42±0.25 | 1.17±0.13 | 5 |
| V1038AfsX20 | −7.11±0.77 | 8.42±0.35 | 1.11±0.07 | 7 |
| V1038AfsX20 and 14-3-3ϵ | −1.42±1.30**† | 9.27±4.30 | 1.57±0.16 | 8 |
| V1/2 (mV) | Slope (mV) | τact (at 0 mV) (*s*) | N | |
|---|---|---|---|---|
| HERG | −6.28±0.62 | 6.79±0.22 | 1.30±0.09 | 12 |
| HERG and 14-3-3ϵ | −17.1±1.0***† | 6.91±0.10 | 0.57±0.04***a | 8 |
| G965X | −4.04±0.95 | 9.47±0.50 | 1.18±0.10 | 7 |
| G965x and 14-3-3ϵ | −6.91±1.6 | 8.49±0.47 | 1.01±0.10 | 7 |
| R1014PfsX38 | −7.63±0.71 | 8.50±5.39 | 1.07±0.07 | 7 |
| R1014PfsX38 and 14-3-3ϵ | −6.75±1.20 | 8.42±0.25 | 1.17±0.13 | 5 |
| V1038AfsX20 | −7.11±0.77 | 8.42±0.35 | 1.11±0.07 | 7 |
| V1038AfsX20 and 14-3-3ϵ | −1.42±1.30**† | 9.27±4.30 | 1.57±0.16 | 8 |
Activation properties of wild-type and mutant HERG subunits in absence or presence of 14-3-3ϵ
| V1/2 (mV) | Slope (mV) | τact (at 0 mV) (*s*) | N | |
|---|---|---|---|---|
| HERG | −6.28±0.62 | 6.79±0.22 | 1.30±0.09 | 12 |
| HERG and 14-3-3ϵ | −17.1±1.0***† | 6.91±0.10 | 0.57±0.04***a | 8 |
| G965X | −4.04±0.95 | 9.47±0.50 | 1.18±0.10 | 7 |
| G965x and 14-3-3ϵ | −6.91±1.6 | 8.49±0.47 | 1.01±0.10 | 7 |
| R1014PfsX38 | −7.63±0.71 | 8.50±5.39 | 1.07±0.07 | 7 |
| R1014PfsX38 and 14-3-3ϵ | −6.75±1.20 | 8.42±0.25 | 1.17±0.13 | 5 |
| V1038AfsX20 | −7.11±0.77 | 8.42±0.35 | 1.11±0.07 | 7 |
| V1038AfsX20 and 14-3-3ϵ | −1.42±1.30**† | 9.27±4.30 | 1.57±0.16 | 8 |
| V1/2 (mV) | Slope (mV) | τact (at 0 mV) (*s*) | N | |
|---|---|---|---|---|
| HERG | −6.28±0.62 | 6.79±0.22 | 1.30±0.09 | 12 |
| HERG and 14-3-3ϵ | −17.1±1.0***† | 6.91±0.10 | 0.57±0.04***a | 8 |
| G965X | −4.04±0.95 | 9.47±0.50 | 1.18±0.10 | 7 |
| G965x and 14-3-3ϵ | −6.91±1.6 | 8.49±0.47 | 1.01±0.10 | 7 |
| R1014PfsX38 | −7.63±0.71 | 8.50±5.39 | 1.07±0.07 | 7 |
| R1014PfsX38 and 14-3-3ϵ | −6.75±1.20 | 8.42±0.25 | 1.17±0.13 | 5 |
| V1038AfsX20 | −7.11±0.77 | 8.42±0.35 | 1.11±0.07 | 7 |
| V1038AfsX20 and 14-3-3ϵ | −1.42±1.30**† | 9.27±4.30 | 1.57±0.16 | 8 |
We used a voltage ramp protocol mimicking a cardiac action potential (26) to determine whether shape of the current trace or charge transfer across CHO cell membranes differed between wild-type and mutant HERG channels (Fig. 4). Shapes of normalized mean currents through mutant HERG channels elicited by this protocol (Fig. 4B–D) were similar to those mediated by wild-type channels (Fig. 4A). Charge transfers through mutant channels normalized to cell capacitance were not significantly different from wild-type, except for HERG-G965X channels. These channels showed a reduction in this parameter by ∼40% (P<0.05, Fig. 4E), which was consistent with the observation of a reduced amplitude for G965X steady-state currents (Fig. 3E).
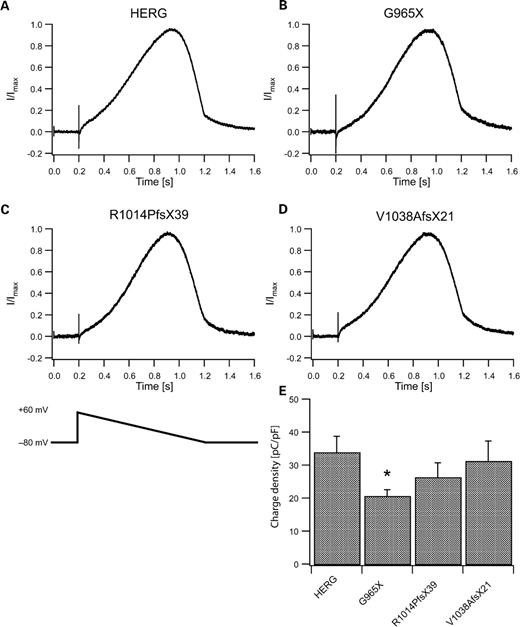
Currents elicited by a voltage ramp protocol that mimics a cardiac action potential. (A) Normalized mean current through wild-type HERG channels (N=12) elicited by the ramp protocol shown in (C, bottom). (B–D) Mean currents mediated by G965X (N=8), R1014PfsX39 (N=9) and V1038AfsX21 (N=9) show similar time courses and shapes as those of wild-type HERG currents. (E) Mean charge densities as calculated from charge transfer divided by cell capacitance obtained from experiments shown in (A–D). (*P<0.05).
C-terminal sequences in HERG have been implicated in subunit stability, maturation and trafficking (17–20). Since all three mutations led to C-terminal truncation, we examined protein levels and maturation of wild-type and mutant HERG subunits by immunoblotting of epitope-tagged subunits expressed in CHO cells. In agreement with previous reports (27–29), we observed at least three immunoreactive bands for wild-type HERG subunits corresponding to an immature, core-glycosylated form of about 130 kDa and to two mature, fully glycosylated forms larger than 185 kDa (Fig. 5A). Wild-type and mutant HERG subunits showed a similar band pattern, but mutant subunits were smaller in molecular weight because of C-terminal truncations (Fig. 5A). The overall signal intensities observed for mutant subunits HERG-R1014PfsX39 and HERG-1038fsX20 were comparable to those of wild-type subunits, although a slight increase was observed for HERG-G965X. To further test the stability of mutant HERG subunits, we utilized the Tet-Off system to suppress transcription of transfected cDNAs at defined time points by supplementing the culture media with doxycycline (30). As before, expression levels were normalized to co-transfected EGFP expressed independently of the Tet-Off system. Two time points were selected for comparison: 24 h after transfection to assess initial protein levels (0 h) and 16 h after addition of doxycycline (Fig. 5C). Although wt HERG and HERG-G965X subunits showed similar levels at 16 h, HERG-R1014PfsX39 and HERG-1038fsX20 subunits showed a faster decay in immunoreactivity, demonstrating decreased stability of these homomeric mutant channel complexes (Fig. 5C). However, this difference, disappeared when we analyzed heteromeric channel complexes of co-expressed wt and mutant HERG subunits. This finding indicates that the presence of the wt HERG subunit stabilized the heteromeric channel and, therefore, argues against a significant pathophysiological role for reduced mutant subunit stability, as the affected patients were heterozygous mutation carriers.
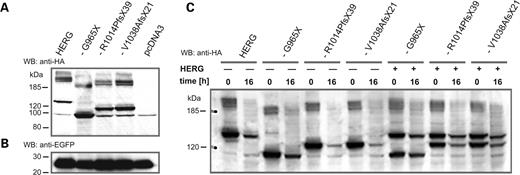
WB analysis of wild-type and mutant HERG subunits. (A) WB analysis of HA epitope-tagged wild-type and mutant HERG subunits transiently expressed in CHO cells, as indicated on top of each lane. Anti-HA antibodies stained three immunoreactive bands corresponding to immature, core-glycosylated HERG protein migrating at about 130 kDa, and to two mature, glycosylated bands migrating above 185 kDa. A similar band pattern was observed for the mutant HERG subunits carrying either G965X, R1014PfsX39 or V1038AfsX21. Smaller molecular sizes correspond to C-terminal truncations of the mutant subunits (Fig. 2). Note the unspecific band, migrating at about 100 kDa, which was also present in control samples of cells transfected with pcDNA3 vector alone. (B) Co-transfected EGFP was used as internal control for transfection efficacy. (C) WB analysis of the stability of homo- and heteromeric HERG channel complexes expressed in CHO cells using the Tet-Off system (30). Cell extracts were prepared either 24 h after transfection (time 0 h) or after a 16-h period (time 16 h) of doxycycline-mediated suppression of cDNA expression. Amounts of lysate used in (A and C) were normalized to EGFP controls. Size markers in kilodaltons are given on the left.
Modulation of mutant HERG subunits by 14-3-3ϵ
Since transient expression of mutant subunits in CHO cells did not reveal the mechanism by which LQTS was caused in most of our patients, we were looking for alternative explanations. In previous experiments, it was shown that binding of 14-3-3ϵ modulates the voltage dependence of in vitro-expressed HERG channels by induction of a hyperpolarizing shift in V1/2 that was critically dependent on the presence of two PKA phosphorylation sites in HERG (S283 and S1137, Fig. 2) (2). We hypothesized that the interaction of HERG with 14-3-3ϵ would be different for wild-type HERG and C-terminally truncated mutant HERG channels. We therefore transiently co-expressed HA epitope-tagged HERG subunits along with 14-3-3ϵ in CHO cells. The expression of both polypeptides was verified by western blotting using either anti-14-3-3ϵ or anti-HA antibodies. The amount of cell lysate used in the following experiments was normalized to 14-3-3ϵ immunoreactivity (Fig. 6B). The presence of HERG/14-3-3ϵ complexes was analyzed by immunoprecipitation of 14-3-3ϵ protein with anti-14-3-3ϵ antibodies, followed by western blotting of the precipitate and by detection of HA-tagged HERG subunits using the anti-HA antibodies. These co-immunoprecipitation experiments clearly showed that mutant HERG subunits, like wild-type HERG subunits, interacted with 14-3-3ϵ (Fig. 6A), presumably by binding to the phosphorylated N-terminus.
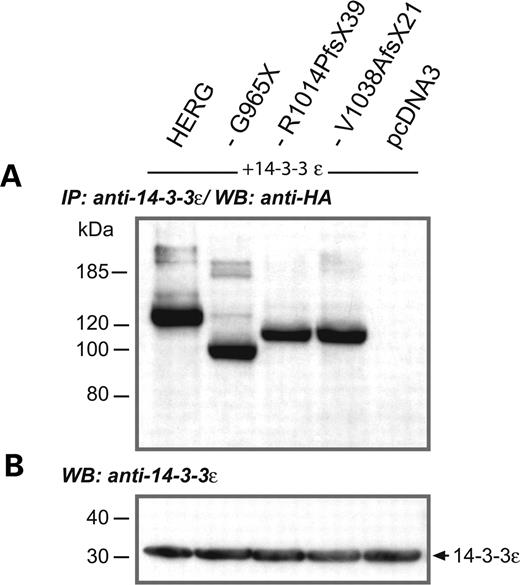
Interaction of 14-3-3ϵ with wild-type and mutant HERG subunits. (A) Immunoprecipitation (IP) of 14-3-3ϵ protein co-expressed with HA epitope-tagged wild-type or mutant HERG subunits as indicated on top of each lane, followed by immunoblotting (WB) and detection of HERG subunits with anti-HA antibodies, reveals that all subunits still interacted with 14-3-3ϵ, regardless of C-terminal truncation. Again, we observed a comparable pattern of immunoreactive bands for all subunits analyzed. (B) The amount of protein used in (A) was always adjusted to the levels of 14-3-3ϵ detected in a control immunoblot (WB) with anti-14-3-3ϵ antibodies. Size markers in kilodaltons are given on the left. Arrow indicates the molecular weight of 14-3-3ϵ.
Next, we investigated the effect of 14-3-3ϵ on the biophysical properties of HERG-G965X, HERG-R1014PfsX39 and HERG-V1038AfsX21 channels expressed together with 14-3-3ϵ in CHO cells. Expression of 14-3-3ϵ was linked to EGFP expression using a bicistronic expression vector (see Materials and Methods). This ensured the presence of 14-3-3ϵ in EGFP-positive cells selected for electrophysiological analysis. Upon co-expression of HERG with 14-3-3ϵ, V1/2 showed a hyperpolarizing shift to −17.1±1.03 mV (N=8, P<0.001; Fig. 7A, first and second panels). In agreement with a previous report (2), the test potential applied to obtain a peak-current amplitude had shifted by about −10 mV (Fig. 7A, third panel). Furthermore, current activation was accelerated (2) (Fig. 7A, right panel). In contrast, co-expression with 14-3-3ϵ did not affect V1/2 or activation time constants of HERG-G965X (N=7) or HERG-R1014PfsX39 (N=5) currents (Fig. 7B and C, Table 1). HERG-V1038AfsX21 channels (N=8) even showed a significant depolarizing shift in V1/2 and a concomitant slowing of activation when co-expressed with 14-3-3ϵ (P<0.01, Fig. 7D and Table 1).
![Co-expression of HERG, HERG-G965X, HERG-R1014PfsX39 and HERG-V1038AfsX21 with 14-3-3ϵ. Representative current traces (left column) from wild-type or mutant HERG-transfected cells in the absence (top trace in each row) or presence of 14-3-3ϵ (bottom trace in each row). (A) The voltage-dependence of activation for cells co-expressing HERG with 14-3-3ϵ showed a hyperpolarizing shift compared with expression of HERG alone (first and second panel). The bell-shaped current–voltage relationship (third panel) for HERG with 14-3-3ϵ also showed a hyperpolarizing shift compared with expression of HERG alone. Furthermore, HERG channel activation was faster in the presence of 14-3-3ϵ as reflected in smaller activation time constants (right panel). (B) Voltage-dependencies of current activation, bell-shaped current–voltage relationships and time constants of current activation were similar for HERG-G965X channel in the absence or presence of 14-3-3ϵ. (C) HERG-R1014PfsX39 and 14-3-3ϵ-co-expressing cells showed no significant difference with respect to the voltage-dependence of activation and the current–voltage relationship when compared with cells expressing only HERG-R1014PfsX39. Furthermore, co-expression of 14-3-3ϵ did not accelerate channel activation. (D) Comparison of cells expressing HERG-V1038AfsX21 and cells co-expressing HERG-V1038AfsX21 with 14-3-3ϵ. A significant depolarizing shift was observed for voltage-dependence of activation and current–voltage relationship. Activation time constants at −20 and 0 mV were significantly increased in the presence of 14-3-3ϵ. (E) Comparison of the V1/2 shifts shows a highly significant hyperpolarizing shift for HERG, a significant depolarizing shift for HERG-V1038AfsX21, but no change for HERG-G965X and HERG-R1014PfsX39. Dashed lines show wild-type HERG values in the absence (dotted-dashed line) or presence (dashed line) of 14-3-3ϵ. Significance levels [(A)–(D): Duncan post hoc test (43); (E): Student's t-test]: *P<0.05; **P<0.01; ***P<0.001; horizontal scale bar: 500 ms, vertical scale bars: 500 pA.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/19/10.1093/hmg/ddl230/2/m_ddl23007.jpeg?Expires=1716594673&Signature=tV4-2lhymOjFDP1MYA~ThvT0MG3zVPs-opuMAMzcT2FOU4t8zLAPZw0qpUHbcD53drw-NuBRYdnH-Mkm~Gl-Fh9UeFqXQlpKU2HRCTXj2xnGVTSG1dxNPZV23sA9-1fYyVDl2NSiX764b6IM0Un3CscWaE90Impv9uS63JbGLwDpqAkGMgmzvoE4kTWK2xNJzQRn4w315RKJIYy3bv-BIhsiTBlmJR1Gm6YB6kai-x4bhQMnQdXtazewJtv4t5lnMTJjHlctLsCVxbD5L~4vxnO~TwKrUUDfzQPbD4nPMfHVtVn9iCLpt0ILJLl7yK6fhgNXgdofHs1abKa1uE5fJQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Co-expression of HERG, HERG-G965X, HERG-R1014PfsX39 and HERG-V1038AfsX21 with 14-3-3ϵ. Representative current traces (left column) from wild-type or mutant HERG-transfected cells in the absence (top trace in each row) or presence of 14-3-3ϵ (bottom trace in each row). (A) The voltage-dependence of activation for cells co-expressing HERG with 14-3-3ϵ showed a hyperpolarizing shift compared with expression of HERG alone (first and second panel). The bell-shaped current–voltage relationship (third panel) for HERG with 14-3-3ϵ also showed a hyperpolarizing shift compared with expression of HERG alone. Furthermore, HERG channel activation was faster in the presence of 14-3-3ϵ as reflected in smaller activation time constants (right panel). (B) Voltage-dependencies of current activation, bell-shaped current–voltage relationships and time constants of current activation were similar for HERG-G965X channel in the absence or presence of 14-3-3ϵ. (C) HERG-R1014PfsX39 and 14-3-3ϵ-co-expressing cells showed no significant difference with respect to the voltage-dependence of activation and the current–voltage relationship when compared with cells expressing only HERG-R1014PfsX39. Furthermore, co-expression of 14-3-3ϵ did not accelerate channel activation. (D) Comparison of cells expressing HERG-V1038AfsX21 and cells co-expressing HERG-V1038AfsX21 with 14-3-3ϵ. A significant depolarizing shift was observed for voltage-dependence of activation and current–voltage relationship. Activation time constants at −20 and 0 mV were significantly increased in the presence of 14-3-3ϵ. (E) Comparison of the V1/2 shifts shows a highly significant hyperpolarizing shift for HERG, a significant depolarizing shift for HERG-V1038AfsX21, but no change for HERG-G965X and HERG-R1014PfsX39. Dashed lines show wild-type HERG values in the absence (dotted-dashed line) or presence (dashed line) of 14-3-3ϵ. Significance levels [(A)–(D): Duncan post hoc test (43); (E): Student's t-test]: *P<0.05; **P<0.01; ***P<0.001; horizontal scale bar: 500 ms, vertical scale bars: 500 pA.
Since the three HERG mutations occurred in a heterozygous context, we first tested whether wt and mutant HERG subunits formed heteromers, as already suggested by the data in Figure 5C. Co-immunoprecipitation experiments with flag epitope-tagged wt HERG subunits and HA epitope-tagged mutant subunits indeed showed that all three mutant subunits interacted with wt HERG (Fig. 8A). In order to simulate the heterozygous state and reliably co-express all cDNAs with 14-3-3ϵ, we generated a bicistronic expression plasmid containing full-length wild-type cDNAs and either one of the mutant HERG cDNAs (Fig. 8B). In addition, 14-3-3ϵ expression was coupled with EGFP expression to allow selection of transfected cells (see Materials and Methods). Since the expression of wild-type and mutant HERG cDNAs was driven by different promoters (Fig. 8B), we investigated the relative protein abundance in transiently transfected CHO cells (Fig. 8C). As a result of the lower efficiency of the CMV promoter, which controlled the expression of mutant HERG cDNAs, compared with that of the EF-1α promoter, we obtained higher levels of wild-type HERG subunits (Fig. 8C) (31). A significant hyperpolarizing V1/2 shift was again found for homomeric wild-type HERG channels (N=12, P<0.001, Fig. 8D). However, analysis of 14-3-3ϵ effects on V1/2 of heteromeric channels revealed that co-expression of wild-type with mutant subunits prevented a 14-3-3ϵ-induced hyperpolarizing shift in V1/2 of HERG current (N=7–8, P>0.05 for all comparisons). These data indicate that mutant HERG subunits exert their effects in a dominant-negative manner (Fig. 8D).
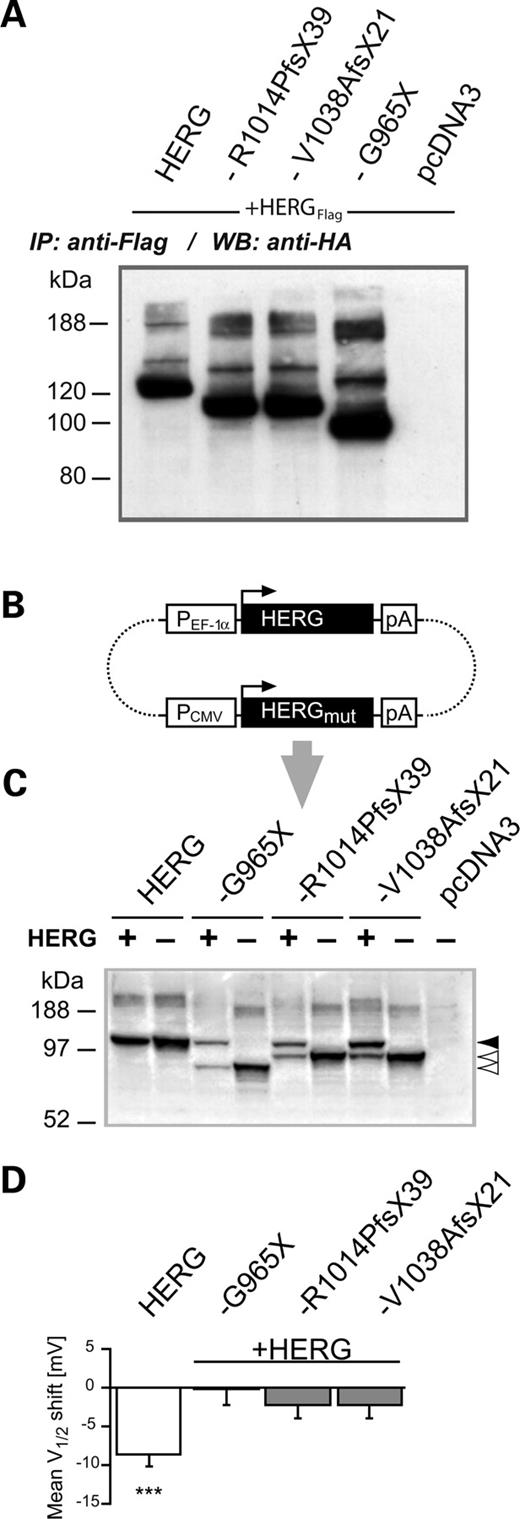
Co-expression of wild-type HERG with HERG-G965X, HERG-R1014PfsX39 or HERG-V1038AfsX21 in the absence or presence of 14-3-3ϵ. (A) Immunoprecipitation of Flag epitope-tagged wt HERG subunits co-expressed with HA epitope-tagged wild-type or mutant HERG subunits as indicated on top of each lane, followed by immunoblotting (WB) and detection with anti-HA antibodies, revealed that all mutant subunits formed heteromers with wt HERG subunits. (B) Diagram of a bicistronic expression construct used for co-expression experiments of wild-type and HERG mutants in the absence or presence of 14-3-3ϵ. Wild-type and mutant HERG cDNAs are located on the same plasmid (indicated with dotted lines), and initiation codons of open reading frames (ORFs) are indicated with horizontal arrows. Structural elements such as promoters (PCMV, PEF-1α) and polyadenylation signals (pA) are shown in open boxes. (C) WBs used to estimate relative abundance of wild-type and mutant HERG subunits in CHO cell homogenates obtained from cells transiently transfected with one of the bicistronic expression plasmids shown in (A) (lanes labeled with +). For comparison, CHO cell lysates from pcDNA3-driven transient expression of homomeric wild-type and mutant HERG subunits were included (lanes labeled by –). HERG protein was stained with antibody directed against the HERG N-terminus (see Materials and Methods). The arrows on the right indicate the position of the core-glycosylated forms of wild-type (filled arrow) or mutant (open arrows) HERG subunits. (D) Analysis of 14-3-3ϵ-induced V1/2 shifts in co-expression experiments of wild-type and mutant HERG subunits. The significant hyperpolarizing V1/2 shift observed for homomeric HERG channels (N=12) was not observed when mutant HERG subunits were present (N=7–8). Significance levels: ***P<0.001.
Effects of the 14-3-3ϵ-induced V1/2 shift of HERG channels in a model ventricular myocyte
We next investigated the possible pathophysiological consequences of our main finding, the loss of a 14-3-3ϵ-induced hyperpolarizing shift in the voltage-dependence of HERG-current activation, on action potential properties in a model myocyte. The HERG channel model was based on a previously published Markov model fitted to the current HERG channel data (32) (Fig. 9A and B). Since modulation of HERG channel activity by 14-3-3ϵ depends on PKA-phosphorylation (2), which in turn is stimulated by β-adrenergic signaling, we used a cardiac myocyte β-adrenergic signaling model (6,33) to investigate regulation of the HERG channel. Assuming similar kinetics to those presented for ICaL, phospholamban (PLB) (33), and IKs (6), we incorporated into this model targeted enzymatic modification of HERG by PKA and protein phosphatase 1 (PP1).
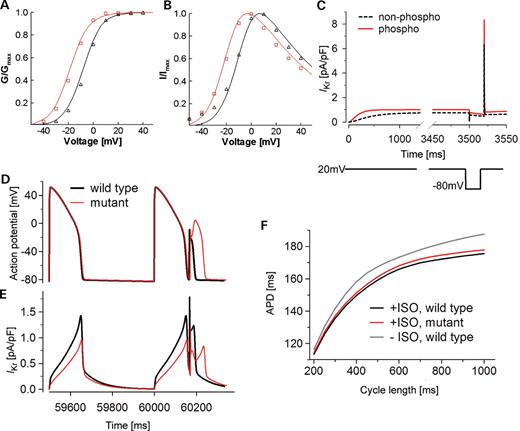
Analysis of the possible physiological role of the phosphorylation-induced V1/2 shift of HERG using voltage clamp and action potential simulations. Experimentally recorded (symbols) and simulated (lines) HERG channel kinetics. Red circles and red lines represent the phosphorylated HERG channel; black circles and black dashed lines represent the non-phosphorylated HERG channel. (A) Activation curves. (B) I–V relationship. The voltage clamp protocol is identical to that shown in Figure 3A. (C) The HERG model recapitulates the essential features of HERG channel gating (25,34). The voltage clamp protocol (lower panel) is as indicated: depolarization to +20 mV (from −80 mV) for 3.5 s, followed by repolarization to −80 mV for 20 ms and then by depolarization to +20 mV. (D) β-AR stimulation results in HERG phosphorylation and improved suppression of premature stimulation. Cells containing a mutant HERG channel that is not phosphorylated fail to suppress a premature beat. Cells containing the wild-type (black line) or the mutant (red line) HERG channels were paced at a fixed CL (500 ms) for 100 beats followed by a premature stimulus given after a very short diastolic interval to mimic an ectopic beat or phase-2 reentry (7.2 ms after 95% repolarization). In the wild-type cell, the phosphorylated HERG channel exhibits a large outward current in response to the premature stimulus and effectively suppresses the premature beat. In the mutant cell, the premature stimulus triggers a more substantial action potential. (E) The phosphorylated HERG current (black) rapidly repolarizes the membrane before Na+ and Ca2+ channels can be activated. In the mutant cell, phosphorylation is ablated and the HERG current (red) is too small to suppress the activation of depolarizing inward currents. (F) The effect of HERG phosphorylation on APD adaptation. Model cells containing the wild-type or the mutant HERG channels were paced at a fixed CL (ranging from 200 to 1000 ms) for 100 beats before or after β-AR stimulation (ISO). The APD of the 100th beat is recorded and plotted as a function of CL. Black line: wild-type cell paced after β-AR stimulation. Red line: mutant cell (no HERG phosphorylation, IKs and L-type Ca2+ channels are phosphorylated) paced after β-AR stimulation. Gray line: wild-type cell paced before β-AR stimulation.
The HERG model recapitulates essential features of HERG channel gating in both non-phosphorylated and phosphorylated forms as shown by the good fits of the model to the experimentally obtained steady-state activation and current–voltage (IV) relationships in Figure 9A and B, respectively. Importantly, the model also reproduces features unique to HERG gating, suggesting that the channel is a critical component in repolarization reserve and arrhythmia suppression (25,34), that is, extremely fast channel inactivation from the open state results in relatively small currents in response to an initial depolarization (Fig. 9C). However, rapid repolarization elicits large tail currents due to fast recovery of the channel from inactivation to the open state, followed by a slow transition from open to closed states (channel deactivation) (Fig. 9C) (32). Because of the large percentage of channels that recover from inactivation to the open state during brief repolarization, a large outward current is induced in response to the second depolarization. These open channels instantly conduct in response to the second depolarizing pulse. Both the non-phosphorylated (black line) and phosphorylated (red line) HERG channels respond to the second pulse with an increased current. However, because of increased transition rates and bigger conductance, the phosphorylated channel produces a larger current increase than the non-phosphorylated channel (Fig. 9C). Our simulation predicts that phosphorylated HERG suppresses more effectively a premature beat from an ectopic focus or early after depolarization. This may prevent propagation into neighboring excitable tissue and protect against development of lethal arrhythmias (25,34).
Most likely, β-adrenergic stimulation leads to PKA phosphorylation of several ionic targets in cardiac myocytes. The combined effects on cardiac excitability are difficult to predict. To test the effect of HERG phosphorylation on premature beat suppression in a more realistic setting, we next tested the effect of β-adrenergic stimulation and phosphorylation-mediated increase in HERG activity on action potential properties in an integrative Luo-Rudy model myocyte (26). Cells containing the wild-type (black line) or the mutant (red line) HERG channels were paced at a fixed cycle length (CL) (500 ms) for 100 beats followed by a premature stimulus given after a very short diastolic interval to mimic an ectopic beat or phase-2 reentry (7.2 ms after 95% repolarization) (Fig. 9D). In the wild-type cell, phosphorylated HERG channel mediated a large outward current in response to a premature stimulus, thereby effectively suppressing a premature beat (Fig. 9D and E). In the mutant cell, a premature stimulus triggered a more substantial action potential that might be sufficiently large to propagate into surrounding tissue and to lead to phase-2 reentry.
We further tested the influence of HERG phosphorylation on APD adaptation. Model cells containing wild-type or mutant HERG channels were paced at a fixed CL (ranging from 200 to 1000 ms) for 100 beats before or after β-adrenergic stimulation (ISO). The APD of the 100th beat was recorded and plotted as a function of CL (Fig. 9F). As expected, we found that under conditions of β-AR stimulation cells containing the wild-type channels had shorter APD and stronger adaptation (more shortening at faster CLs) than cells without β-AR stimulation. In cells containing the mutant HERG channel (i.e. no HERG phosphorylation, IKs and ICaL channels were phosphorylated), APD was slightly longer and adaptation was reduced, in particular at longer CLs (Fig. 9F).
DISCUSSION
The present study provides clinical, genetic and functional evidence that HERG channel regulation by 14-3-3ϵ is of physiological significance in human heart. The importance of HERG in the repolarization process of ventricular cardiac action potentials is obvious, because carriers of HERG (LQT2) mutations, which reduce the activity of HERG channels in general, are at risk for developing triggered arrhythmias as a result of stimulation of the SNS (12). The novel LQT2 mutations described here underline the significance of the regulation of HERG channel activity by 14-3-3ϵ in providing a reserve of repolarizing outward current during β-adrenergic receptor-mediated stimulation of heart rate.
In the large multi-generation family carrying the mutation R1014PfsX39, genetic linkage was established between microsatellite markers at the KCNH2 gene locus and the LQTS phenotype (Fig. 1). The mutation was not found in family members who were negative for the disease haplotype. These results, along with the finding that mutation R1014PfsX39 is absent in normal control samples from unrelated individuals, provide compelling genetic evidence that R1014PfsX39 is indeed the pathogenic mutation associated with the LQTS phenotype in this family. Furthermore, the data point to an incomplete disease penetrance. Incomplete penetrance, which means that mutation carriers have no clinical signs, is found in various LQTS types. Recently, a study of a large genotyped LQTS population revealed that 19% of the LQT2 mutation carriers were clinically unaffected (35). Potential mechanisms may include age- and gender-specific aspects of disease manifestation, unknown genetic factors or non-genetic mechanisms.
The other families were too small to perform linkage analysis for carriers with other mutations (V1038AfsX21 or G965X). It is, however, unlikely that these mutations represent polymorphisms because they were not detected in more than 100 unrelated control samples.
The mutations G965X, R1014PfsX39 and V1038AfsX21 disrupt the HERG open reading frame and cause a truncation in the HERG C-terminus. The truncated C-termini retain the cNBD (Fig. 2) and an adjacent segment that has recently been shown to be involved in maturation and stability of HERG channels (20). When expressed in CHO cells, no differences were found in protein abundance and glycosylated forms of higher molecular weight, which indicates that there were no major alterations in maturation of mutant subunits. In contrast, stability of the homomeric channel complex was reduced in two of the mutant subunits. Surprisingly, these differences disappeared when wild-type subunits were co-expressed to mimic the heteromeric channel complexes that are most likely present in the patients’ myocytes. Furthermore, we found that HERG channel activity under basal conditions was only reduced for mutation G965X. In agreement with a recent report showing that deletion of up to 215 C-terminal amino acids had no significant effect on the properties of HERG channels expressed in Xenopus oocytes (16), our data demonstrate that mutant HERG channels mediated currents with kinetic properties that resembled those of wild-type channels. The truncations in the mutant HERG subunits eliminated a ‘tetramerizing coiled coil’ (TCC) domain, which has recently been implicated in ether-a-go-go (EAG) and ERG subunit assembly (17). Apparently, assembly of functional HERG channels does not require a TCC domain.
It has been proposed that only full-length HERG subunits are delivered to the plasma membrane because of an endoplasmic reticulum (ER) retention signal (‘RGR’; amino acids 1005–1007) in the HERG C-terminus that becomes masked by the C-terminal 104 amino acids (19). These amino acids are absent in our truncated mutants. Yet, all of these subunits produced functional channels and were thus delivered to the plasma membrane. Current densities, protein abundance or glycosylation patterns of HERG-R1014PfsX39 or HERG-V1038AfsX21 subunits indicated no major alterations in intracellular trafficking. The remaining C-terminal amino acids in the truncated HERG subunits (including novel amino acids introduced by shifts in the reading frame) may still be capable of masking the ER retention signal in the HERG C-terminus. In western blot (WB) analyses, we observed a slight increase in the amount of core-glycosylated subunits for HERG-G965X subunits lacking the ER retention motif (Fig. 5). However, unexpectedly, this increase was paralleled by a decrease in current density, suggesting altered maturation and/or trafficking of HERG-G965X subunits.
The truncation of our mutant HERG subunits caused a loss of a C-terminal PKA phosphorylation site (S1137). This site, together with an N-terminal PKA phosphorylation site (S283), plays an important role in the interaction of HERG channels with 14-3-3ϵ (2). This interaction leads to an acceleration of activation kinetics and to a concomitant hyperpolarizing shift in the half-maximal voltage dependence of HERG channel activation. As a result, HERG-channel availability is increased at physiologically significant membrane potentials (2). Although mutant HERG channels apparently bound 14-3-3ϵ, as did wild-type channels, our electrophysiological data revealed that mutants did not respond to 14-3-3ϵ with a hyperpolarizing shift in the voltage-dependence of current activation. The voltage-dependence of current activation determined for HERG-R1014PfsX39 was shifted to even more depolarized potentials. The functional 14-3-3ϵ effect depends on its association with both the N-terminal serine 283 and the C-terminal serine 1137 of HERG. In addition, 14-3-3ϵ dimerization is an important prerequisite for the 14-3-3ϵ effect on HERG channel activation (2). Under the assumption that 14-3-3ϵ is the mediator of β-adrenergic stimulation of HERG-channel activity, IKr amplitudes mediated by the mutant HERG channels are likely to be resilient to β-adrenergic stimulation and subsequent PKA activation.
β-Adrenergic signaling plays an important role in the regulation of heart rate and contraction force in response to stimulation of the SNS by, for example, exercise and emotional stress. The activation leads to increased cAMP synthesis and stimulation of PKA, which then regulates the activity of specific cardiac ion channels such as the L-type calcium channel Cav1.2 (36) and the KCNQ1/KCNE1 and HERG potassium channels (3,4,22). It has been suggested that PKA-dependent modulation of KCNQ1/KCNE1 channels leads to an increase in IKs, the slow outward current in ventricular myocytes. IKs is known to be affected by LQT1 and LQT5 mutations (8,9), which cause dysfunctional ventricular repolarization and prolongation of the QT interval. Whereas KCNQ1/KCNE1 channel activity appears to be directly affected by PKA phosphorylation and attenuated in the presence of LQT5 mutations (5), PKA phosphorylation of HERG channels influences the interaction with 14-3-3ϵ (2). This interaction, which may lead to a PKA-dependent increase in the rapid outward current IKr observed in ventricular myocytes (37,38), is affected by the novel LQT2 mutations described here and may thus increase the risk for ventricular tachyarrhythmias under conditions of a higher adrenergic tone.
An increase in repolarizing HERG current may have two important physiological functions: (i) to improve the rate-dependent adaptation of cells to increased heart rates by shortening APD. This preserves sufficiently long diastolic intervals to allow for ventricular filling; and (ii) to enhance HERG contribution to a ‘repolarization reserve’, since HERG channels recover from inactivation through an open conducting state during repolarization. Our simulations in a model ventricular myocyte suggest that APD adaptation, albeit diminished, is not the main parameter affected by the mutations. Rather, it is the ability of the cell to suppress a premature beat from an ectopic focus or early after depolarization and to prevent propagation of excitation that leads to arrhythmia suppression (25,34). In this scenario, non-phosphorylated HERG channels may not effectively protect against cardiac arrhythmia. This finding distinguishes mutations in HERG from mutations in other K+ channels, such as, for example, in IKs, that primarily affect APD adaptation.
The simulations further suggest that a combination of β-adrenergically enhanced repolarizing currents, IKr and IKs, is required to counterbalance the larger inward depolarizing current of the L-type Ca2+ current that is also enhanced with β-adrenergic stimulation. This produces a substantial increase in outward current sufficient to suppress premature stimulation. Since both LQT1 patients (with an attenuated IKs increase at higher heart rates) and LQT2 patients are at risk for ventricular tachyarrhythmias under conditions of an increased adrenergic tone, this notion is of particular interest. Schwartz et al. (12) reported that in 13% of LQT2 patients physical stress and in 43% emotional stress were triggering factors. Some patients had syncope after arousal due to sudden acoustic noise, which appears to be a typical finding in LQT2. These data are in agreement with our observation that most of the HERG R1014PfsX39 mutation carriers in our study had episodic cardiac events induced through physical activity or emotional stress.
The identification of C-terminal HERG mutations that led to disruption of 14-3-3ϵ-dependent regulation of HERG channels described here represents a novel pathophysiological mechanism for adrenergically mediated arrhythmias and underlines the importance of phosphorylation-mediated rapid regulation of ion channel activity in the control of cardiac electrical activity.
MATERIALS AND METHODS
Clinical and genetic analysis
We investigated unrelated patients with congenital LQTS for mutations in the KCNH2 gene and followed the nomenclature recommendations for mutations as proposed by den Dunnen et al. (39). LQTS was diagnosed using a standard 12-lead baseline ECG (without concomitant drug exposure or electrolyte disturbances) according to previously proposed diagnostic criteria (40). QT interval was corrected for heart rate with Bazett's equation. Informed consent—as approved by the Ethics Committees of the involved universities and/or by institutional guidelines—was obtained from each individual before participation in this study and was in accordance with the Declaration of Helsinki (41).
Genetic analysis was performed using previously published KCNH2 primers and reaction conditions (42). Microsatellite markers (D7S483 and D7S636), both closely flanking the LQT2 locus, were used for co-segregation analysis in a large LQT family (Fig. 1).
Expression plasmids
Mutant HERG cDNAs were generated using PCR-based site-directed mutagenesis and subcloned into the eukaryotic expression vectors pcDNA3 (Invitrogen, Karlsruhe, Germany) and pTre-Tight (Clontech, Mountain View, CA, USA). For co-expression experiments of wild-type and mutant channels, (Fig. 8) expression plasmids were constructed that contained one of the mutant HERG cDNAs in the first polylinker of pBud-CE4.1 (under control of PCMV) and the wt HERG cDNA in the second polylinker (under control of PEF-1α; Fig. 8A). CHO cells were transiently transfected with either wild-type, mutant or one of the co-expression HERG cDNA constructs. Cells were routinely co-transfected with an expression plasmid encoding ‘enhanced green fluorescent protein’ (EGFP, Clontech, Palo Alto, CA, USA) to allow identification of transfected cells by EGFP fluorescence. Wild-type or mutant HERG cDNAs in pTre-Tight were co-transfected with pTet-Off (30) to analyze subunit stability using doxycycline-mediated suppression of cDNA transcription (Fig. 5C). Twenty-four hours after transfection, cells were either harvested immediately (Fig. 5C, time 0) or supplemented with doxycycline (1 µg/ml, Sigma, Taufkirchen, Germany) and harvested after 16 h (Fig. 5C, time 16).
For co-expression of HERG constructs with 14-3-3ϵ, the human 14-3-3ϵ cDNA (GenBank accession no. U43430) and EGFP were subcloned into the bicistronic eukaryotic expression vector pBudCE4.1 (Invitrogen) to couple 14-3-3ϵ and EGFP expression.
In order to detect HERG subunits in immunoprecipitation and WB experiments, a hemagglutinin (HA) epitope tag (NH2-YPYDVPDYAS-COOH) was added to the N-terminus of wild-type or mutant HERG cDNAs. In addition, a wild-type HERG cDNA was generated that carried a C-terminal flag epitope tag (NH2-DYKDDDDK-COOH).
Cell culture and transfection
CHO cells were cultured under standard conditions in MEM (Invitrogen) supplemented with 10% fetal calf serum, penicillin (100 U/ml, Invitrogen), and streptomycin (100 µg/ml, Invitrogen). For electrophysiological studies and biochemical analyses, transient transfections were performed with 3.5–4.0 µl of Lipofectamine (Invitrogen) within 24 h after plating in 35 mm plastic dishes.
WB and co-immunoprecipitation experiments were carried out using cells (5.5×105 cells in a 5 cm dish) that were transiently transfected with 4.5 µl of Fugene (Roche, Basel, Switzerland).
Electrophysiology
CHO cells were recorded 24–36 h after transfection at room temperature (20–24°C). Whole-cell patch clamp studies were performed with an EPC9 amplifier and Pulse software (HEKA Electronik, Lambrecht, Germany). Series resistances were below 5 MΩ and electronically compensated (80–90%).
The extracellular solution was (in mm): 150 NaCl, 1.8 CaCl2, 4 KCl, 1 MgCl2, 5 glucose and 10 HEPES, pH 7.4 (with NaOH). The (intracellular) pipette solution was (in mm): 126 KCl, 4 Mg-ATP, 2 Mg2SO4, 5 EGTA, 0.5 CaCl2 and 25 HEPES, pH 7.3 (with KOH) (2). Voltage activation data were plotted as peak tail current amplitudes against the test potential values, and a Boltzmann function was fitted to the data: I=a/(1+exp[(V1/2−V)/k])−b, with V1/2 being the voltage at half-maximal activation and k being the slope factor. Charge transfer density (Fig. 4E) was calculated by integrating the area under the current trace for the duration of the depolarizing ramp (Pulsfit, HEKA Electronik). Data are given as mean±SEM. The Student's t-test (Microsoft Excel, Microsoft, Redmond, WA, USA) was used for statistical analysis of V1/2, slope factor and current densities. Time constants for inactivation and deactivation were compared using two-way ANOVA for repeated measurements and Duncan's multiple range post hoc test (Statistica 5, Statsoft, Tulsa, OK, USA), which is a multiple comparison procedure, and a variant of the Student–Newman–Keuls method (43).
Immunoblotting and co-immunoprecipitation
Transiently transfected CHO cells were processed 24 h after transfection according to the following protocol: Cells were rinsed twice with ice-cold PBS, incubated for 1.5 h in 250 µl lysis buffer [50 mm Hepes pH 7.4, 150 mm NaCl, 0.5% Triton-X100, 2.5 µl proteinase inhibitor mix (Sigma)] and centrifuged at 10 000g for 10 min. The supernatant (5–15 µl) was separated under reducing conditions by SDS-PAGE using NuPage™ 8–12% gradient gels (Invitrogen). Subsequently, proteins were transferred onto Protran nitrocellulose membranes (Schleicher and Schüll, Dassel, Germany). Membranes were blocked for 1 h in blocking solution (5% non-fat dry milk powder in PBS, 0.05% Tween-20) before adding primary antibodies [anti-HA (3F10, Roche), anti-EGFP (Roche) and anti-14-3-3ϵ (Upstate, Waltham, MA, USA) diluted 1:500 in antibody solution (1% non-fat dry milk powder in PBS, 0.05% Tween-20)]. After incubation for 1 h at room temperature, membranes were washed three times in PBS, 0.05% Tween-20, followed by incubation with secondary antibodies diluted in antibody solution as follows: peroxidase-conjugated affinity-purified goat anti-rat IgG (H+L) (Dianova, Hamburg, Germany) 1:5000, peroxidase-conjugated affinity-purified goat anti-mouse IgG (H+L) (Dianova) 1:10 000 and peroxidase-conjugated affinity-purified anti-rabbit IgG (H+L) (Vector, Burlingame, CA, USA) 1:5000.
For co-immunoprecipitation experiments, we first performed a 14-3-3ϵ or EGFP WB from CHO cell lysates (see above) in order to ensure transfection efficiency and to correct the amount of protein used for immunoprecipitation accordingly. Immunoprecipitation was done overnight at 4°C (continuous rotation) with 80–100 µl cell lysate, 10 µl anti-14-3-3ϵ antibodies (Upstate, Dundee, UK) or 5 µl anti-Flag epitope antibodies (Rockland, Gilbertsville, PA, USA) and lysis buffer up to a final volume of 110 µl. The next day, cell lysates were incubated for 2 h with 20 µl of equilibrated protein-G beads (Dynal, Hamburg, Germany); antigen/antibody complexes were precipitated according to the manufacturer's protocol. Precipitates were washed twice in five volumes of lysis buffer and were then incubated for 10 min at 70°C in reducing NuPage™ sample buffer (Invitrogen). Separation, blotting and immunodetection (anti-HA antibody; Roche) were performed as described earlier. Immunodetection of untagged HERG subunits was carried out as described earlier, but with a polyclonal antiserum directed against the N-terminus of HERG (Santa Cruz).
Computational methods
Channel kinetics.
The HERG channel model was based on a previously published Markov model fitted to the current HERG channel data (32). Rate constants were determined via empirical fits to the experimental data of non-phosphorylated and phosphorylated channel kinetics.
Rate constants in the non-phosphorylated state
Changed rate constants in the phosphorylated state
β-Adrenergic stimulation model.
We used a cardiac myocyte β-adrenergic signaling model to investigate its regulation of the HERG channel based on previously published models (6,33). We incorporated targeted enzymatic modification of HERG by PKA and PP1 into this model, and assumed similar kinetics to those presented for ICaL and PLB by Saucerman et al. (33) and Iks by Terrenoire et al. (6).
Previously, the complete signaling, calcium handling and electrophysiology model was implemented with the stiff solver in Berkeley Madonna (6,33). In our simulations, we solved the complete signaling with added HERG regulation using a combined approach. Ordinary differential equations were solved by Rosenbrock stiff solver, and algebraic equations were solved by Newton–Raphson non-linear solver. This new approach increases both accuracy and stability of the solution, allowing for variable concentrations of β-adrenergic agonists (and variable time courses) to be used in the simulation. Maximal receptor stimulation was assumed for the simulations presented here. The signaling program was implemented in C/C++. All source codes used in the simulations can be obtained by E-mailing Colleen E. Clancy at clc7003@med.cornell.edu.
IHERG regulation module
Cell model-action potential simulations.
We used the Luo-Rudy guinea pig ventricular integrative cell model for all of the action potential simulations (44). Modifications to ICal, ICat and IKs were made according to Terreniore et al. (6). The IKr model was developed based on the data in the present study as described earlier. The naturally occurring HERG long-QT mutations characterized in this study were shown to disrupt the transduction of the HERG phosphorylation from the β-adrenergic signaling cascade. Hence, mutant HERG channels were simulated as ‘non-phosphorylated’ channels, while L-type Ca2+ channels and IKs phosphorylation remained intact. The model also incorporated ‘baseline’ phosphorylation of channels before β-AR by receptor activation. Baseline phosphorylation rates were: Fraction of L-type Ca2+ channel β- subunit phosphorylation=0.20, fraction of L-type Ca2+ channel alpha-subunit phosphorylation=0.23, fraction of IKr channel phosphorylation=0.13, fraction of IKs channel phosphorylation=0.13. After β-AR stimulation: Fraction of L-type Ca2+ channel beta-subunit phosphorylation=0.57, fraction of L-type Ca2+ channel alpha-subunit phosphorylation= 0.53, fraction of IKr channel phosphorylation=0.61, fraction of IKs channel phosphorylation=0.61.
ACKNOWLEDGEMENTS
We would like to express our thanks to the LQT probands and family members for their participation, and to Andrea Zaisser, Stefan Schillemeit, Marielies Hesse and Ellen Schulze-Bahr for expert technical assistance. We thank Sven Hartmann for help with cell culture and transfections. Furthermore, we would like to thank the anonymous reviewers for their helpful comments. Our study was supported by grants from the Deutsche Forschungsgemeinschaft, Bonn, Germany (Graduiertenkolleg 255/II, Po137/35-1, DFG-FOR 604 Schu1082/3-1, SFB 656-C1), by the Ernst und Berta Grimmke-Stiftung, Düsseldorf, Germany, and by Fondation Leducq, Paris, France.
Conflict of Interest statement. The authors declare no conflict of interests.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}