




Abstract
Using the metabolomics-guided screening coupled to N-ethyl-N-nitrosourea-mediated mutagenesis, we identified mice that exhibited elevated levels of long-chain acylcarnitines. Whole genome homozygosity mapping with 262 SNP markers mapped the disease gene to chromosome 5 where candidate genes Hadha and Hadhb, encoding the mitochondria trifunctional protein (MTP) α- and β-subunits, respectively, are located. Direct sequencing revealed a normal α-subunit, but detected a nucleotide T-to-A transversion in exon 14 (c.1210T>A) of β-subunit (Hadhb) which resulted in a missense mutation of methionine to lysine (M404K). Western blot analysis showed a significant reduction of both the α- and β-subunits, consistent with reduced enzyme activity in both the long-chain 3-hydroxyacyl-CoA dehydrogenase and the long-chain 3-ketoacyl-CoA thiolase activities. These mice had a decreased weight gain and cardiac arrhythmias which manifested from a prolonged PR interval to a complete atrio-ventricular dissociation, and died suddenly between 9 and 16 months of age. Histopathological studies showed multifocal cardiac fibrosis and hepatic steatosis. This mouse model will be useful to further investigate the mechanisms underlying arrhythmogenesis relating to lipotoxic cardiomyopathy and to investigate pathophysiology and treatment strategies for human MTP deficiency.
INTRODUCTION
Fatty acids are the major source of energy for the heart and skeletal muscles and play an essential role in intermediary metabolism in the liver (1,2). Long-chain fatty acids are the primary fatty acids found in the diet. Their utilization requires oxidation which involves the activation of free fatty acids to acyl-CoA species, transport into mitochondria and processing through cyclic β-oxidation in the matrix to breakdown the long-chain fatty acids into acetyl-CoA. β-Oxidation is initiated by a catalytic reaction mediated by a long-chain acyl-CoA dehydrogenase, followed by the mitochondrial trifunctional protein (MTP), which is a heterooctamer multienzyme complex consisting of four α- and four β-subunits that catalyses the next three steps of β-oxidation of long-chain fatty acids (3–5). The α-subunit (hydroxyacyl-CoA dehydrogenase alpha, HADHA) contains the long-chain enoyl-CoA hydratase and long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) that catalyze the second and third step, respectively, and the β-subunit (hydroxyacyl-CoA dehydrogenase beta, HADHB) contains the long-chain 3-ketoacyl-CoA thiolase (LKAT) that catalyzes the fourth step (3–5).
An important group of inherited metabolic disorders in human that present in infancy or childhood with life-threatening metabolic crisis is caused by defects in the MTP complex (5). Isolated LCHAD deficiency is commonly associated with the α-subunit E474Q mutation that is located directly within the catalytic region of the LCHAD domain (6,7). General MTP deficiency is characterized by a reduction in all three enzyme activities and can be caused by mutations in either α- or β-subunit genes (6,8,9). Clinically, isolated LCHAD deficiency leads predominantly to Reye-Like syndrome, cardiomyopathy or sudden unexplained death, while general MTP deficiency has a broader clinical spectrum ranging from severe neonatal manifestations including metabolic derangement, cardiac arrhythmia, cardiomyopathy and death, to a moderate to severe infantile presentation with hypoglycemia and hepatic manifestations, to a mild peripheral neuropathy with episodic rhabdomyolysis (10,11). A viable animal model of MTP deficiency is currently not available; an α-subunit knock-out mouse model that exhibits neonatal lethality has been reported (12).
Treatment with N-ethyl-N-nitrosourea (ENU) efficiently generates single-nucleotide mutations in mice which can be screened for the disease phenotypes of interest. A systematic metabolomics-guided, genome-wide and phenotype-driven mouse mutagenesis program for gene function studies has been described (13–15) and using tandem mass spectrometry (MS/MS) to screen ENU mice for abnormalities in the pathways of amino acid and fatty acid metabolism, we have previously identified mice with mitochondrial branched-chain aminotransferase deficiency, resembling human maple syrup urine disease (15). Using the same approach, we have now generated mice with defective MTP due to a mutation in the gene encoding the β-subunit (Hadhb). These mice survived to adulthood despite a decreased weight gain, hepatic steatosis and cardiomyopathy, but eventually they developed cardiac conduction defects followed by sudden death.
RESULTS
Mutant mice with an abnormal acylcarnitine profile
We used tandem mass spectrometry to screen fatty acid intermediate metabolites, detected as acylcarnitine species, in 6.25 µl of a total of 25 µl whole blood spotted on the Guthrie card. In a recessive screening of 2300 G3 mice from 61 families at 2–3 months of age, we identified three G3 mice of the same G1 offsprings (3/21 in this family) with elevated blood long-chain acylcarnitines (C12–C18), especially C16-OH, C18-OH and C18:1-OH carnitines (10- to 12-fold elevation), C12-OH, C14-OH and C16:1-OH (four- to six-fold) and C16:1, C18 and C18:1 (two- to three-fold) (Table 1). All short- and medium-chain acylcarnitines as well as blood amino acids level were normal. The pattern of elevated long-chain acylcarnitines was confirmed in a second sample from the same mice obtained 4 weeks later. These G3 affected mice were then mated with C57BL/6 (B6) to generate F1, and the F2 offsprings were generated from F1 brother and sister crossmating. Of 85 F2 mice, 20 (23.5%) showed a similar elevation of long-chain acylcarnitine levels (Table 1), and all F1 were normal indicating that this biochemical abnormality is heritable and transmitted as an autosomal recessive mode.
Blood acylcarnitine concentrations in ENU mice
| Acylcarnitine concentration (µm)a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C12-OH | C14:1-OH | C14-OH | C16:1 | C16 | C16:1-OH | C16-OH | C18:2 | C18:1 | C18 | C18:1-OH | C18-OH | |
| G3 ENU mice (n=1314) | 0.02±0.05 | 0.03±0.05 | 0.02±0.05 | 0.12±0.25 | 1.44±0.71 | 0.04±0.10 | 0.04±0.05 | 0.17±0.23 | 0.43±0.47 | 0.40±0.22 | 0.03±0.05 | 0.03±0.09 |
| Affected (n=20) | 0.08±0.05 | 0.07±0.04 | 0.11±0.08 | 0.34±0.12 | 1.80±0.43 | 0.22±0.24 | 0.45±0.15 | 0.41±0.17 | 1.08±0.36 | 0.80±0.21 | 0.43±0.26 | 0.32±0.12 |
| Acylcarnitine concentration (µm)a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C12-OH | C14:1-OH | C14-OH | C16:1 | C16 | C16:1-OH | C16-OH | C18:2 | C18:1 | C18 | C18:1-OH | C18-OH | |
| G3 ENU mice (n=1314) | 0.02±0.05 | 0.03±0.05 | 0.02±0.05 | 0.12±0.25 | 1.44±0.71 | 0.04±0.10 | 0.04±0.05 | 0.17±0.23 | 0.43±0.47 | 0.40±0.22 | 0.03±0.05 | 0.03±0.09 |
| Affected (n=20) | 0.08±0.05 | 0.07±0.04 | 0.11±0.08 | 0.34±0.12 | 1.80±0.43 | 0.22±0.24 | 0.45±0.15 | 0.41±0.17 | 1.08±0.36 | 0.80±0.21 | 0.43±0.26 | 0.32±0.12 |
aValues are mean±SD.
Blood acylcarnitine concentrations in ENU mice
| Acylcarnitine concentration (µm)a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C12-OH | C14:1-OH | C14-OH | C16:1 | C16 | C16:1-OH | C16-OH | C18:2 | C18:1 | C18 | C18:1-OH | C18-OH | |
| G3 ENU mice (n=1314) | 0.02±0.05 | 0.03±0.05 | 0.02±0.05 | 0.12±0.25 | 1.44±0.71 | 0.04±0.10 | 0.04±0.05 | 0.17±0.23 | 0.43±0.47 | 0.40±0.22 | 0.03±0.05 | 0.03±0.09 |
| Affected (n=20) | 0.08±0.05 | 0.07±0.04 | 0.11±0.08 | 0.34±0.12 | 1.80±0.43 | 0.22±0.24 | 0.45±0.15 | 0.41±0.17 | 1.08±0.36 | 0.80±0.21 | 0.43±0.26 | 0.32±0.12 |
| Acylcarnitine concentration (µm)a | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C12-OH | C14:1-OH | C14-OH | C16:1 | C16 | C16:1-OH | C16-OH | C18:2 | C18:1 | C18 | C18:1-OH | C18-OH | |
| G3 ENU mice (n=1314) | 0.02±0.05 | 0.03±0.05 | 0.02±0.05 | 0.12±0.25 | 1.44±0.71 | 0.04±0.10 | 0.04±0.05 | 0.17±0.23 | 0.43±0.47 | 0.40±0.22 | 0.03±0.05 | 0.03±0.09 |
| Affected (n=20) | 0.08±0.05 | 0.07±0.04 | 0.11±0.08 | 0.34±0.12 | 1.80±0.43 | 0.22±0.24 | 0.45±0.15 | 0.41±0.17 | 1.08±0.36 | 0.80±0.21 | 0.43±0.26 | 0.32±0.12 |
aValues are mean±SD.
Identification of the mutated gene.
To map the gene causing the elevation of long-chain acylcarnitines, G3 affected mice were outbred to C3H/HeJ (C3H) mice to generate N1 (B6 and C3H hybrid). A panel of 262 SNPs was used for whole genome SNP homozygosity mapping using eight affected mice from the N1×N1 offspring. One region located between 44 and 97 Mb of chromosome 5 was found to have complete homozygosity in consecutive SNPs (Fig. 1, circled region). This region contains Hadha and Hadhb, which are likely candidate genes as they encode MTP α- and β-subunit, respectively. Direct DNA sequencing of the two MTP genes in affected mice identified a homozygous nucleotide T to A transversion in exon 14 (c.1210T>A) of Hadhb which resulted in a missense mutation of methionine to lysine (M404K) (Fig. 2A). The parents were heterozygous for this mutation. Further study of siblings showed the homozygous M404K mutation (−/−) was completely segregated with the abnormal phenotypes of elevated long-chain acylcarnitines. Siblings as well as parents who were heterozygous for M404K mutation (+/−) exhibited normal acylcarnitine profile. No other mutations were found in the remaining exons of Hadhb or in its promoter region. Sequencing of Hadha and very long-chain acyl-CoA dehydrogenase (Acadvl) genes detected no sequence variation in these genes (data not shown).
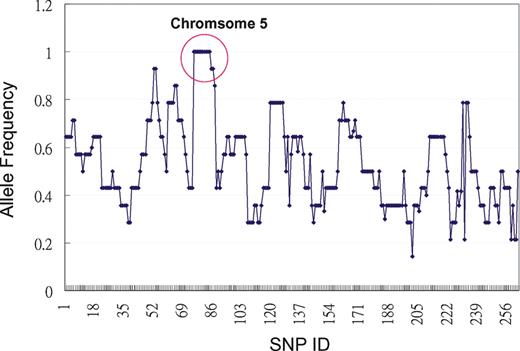
Mapping of the gene responsible for the abnormal long-chain acylcarnitine phenotype. Region that shows complete homozygosity is circled, which consists of consecutive SNPs between SNP rs13478227 (05.044.930, 44 Mb) to SNP rs13478418 (05.097.286, 97 Mb) on mouse chromosome 5.
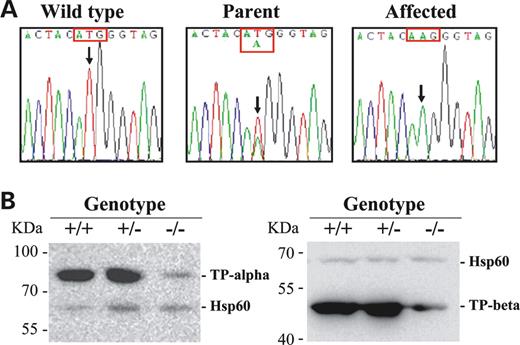
Molecular analyses of the mutant mice. (A) DNA sequence analysis of mouse Hadhb gene. Nucleotide sequences in exon 14 showing that the affected mouse was homozygous for A at the position c.1210 (arrow); parent was T/A and wild-type T/T at the same position. (B) Western blot analysis of HADHA and HADHB protein levels. Forty micrograms of mitochondrial protein from heart of wild-type (+/+), carrier (+/−) and affected (−/−) mice were applied to each lane. Antibodies raised against mouse HADHA, HADHB or HSP 60 (mitochondrial heat shock protein 60) was used; the latter was used as an internal control.
Western blot and enzyme activity assay.
To investigate whether the missense mutation M404K causes a decrease in HADHB protein level and/or its activity, we performed western blot on both α- (HADHA) and β- (HADHB) subunits of the MTP complex and measured the acyl-CoA dehydrogenase and 3-ketothiolase enzyme activities. Western blots showed that the levels of both HADHB and HADHA were greatly reduced in the heart mitochondria of the affected mice (Fig. 2B). Enzyme activity measurements demonstrated that LCHAD and LKAT levels were reduced to 37 and 23% of the control levels, respectively (Table 2). There was no significant difference between affected and control mice in the short-chain acyl-CoA dehydrogenase and short-chain 3-ketothiolase activities (Table 2). Similar findings were observed in the liver tissues (data not shown).
Enzymatic activities in the mouse heart mitochondrial fraction
| Control (n=4) | Affected (n=4) | |
|---|---|---|
| Acetoacetyl-CoA thiolase | 125.29±58.91 | 158.71±55.29 |
| Long chain 3-ketothiolase | 65.81±10.97 | 15.36±7.07 |
| Long chain hydroxyacyl-CoA dehydrogenase | 1046.89±84.13 | 387.90±126.92 |
| Short chain hydroxyacyl-CoA dehydrogenase | 1259.27±237.19 | 1318.70±469.83 |
| Control (n=4) | Affected (n=4) | |
|---|---|---|
| Acetoacetyl-CoA thiolase | 125.29±58.91 | 158.71±55.29 |
| Long chain 3-ketothiolase | 65.81±10.97 | 15.36±7.07 |
| Long chain hydroxyacyl-CoA dehydrogenase | 1046.89±84.13 | 387.90±126.92 |
| Short chain hydroxyacyl-CoA dehydrogenase | 1259.27±237.19 | 1318.70±469.83 |
Mean±SD (nmole/min/mg protein).
Enzymatic activities in the mouse heart mitochondrial fraction
| Control (n=4) | Affected (n=4) | |
|---|---|---|
| Acetoacetyl-CoA thiolase | 125.29±58.91 | 158.71±55.29 |
| Long chain 3-ketothiolase | 65.81±10.97 | 15.36±7.07 |
| Long chain hydroxyacyl-CoA dehydrogenase | 1046.89±84.13 | 387.90±126.92 |
| Short chain hydroxyacyl-CoA dehydrogenase | 1259.27±237.19 | 1318.70±469.83 |
| Control (n=4) | Affected (n=4) | |
|---|---|---|
| Acetoacetyl-CoA thiolase | 125.29±58.91 | 158.71±55.29 |
| Long chain 3-ketothiolase | 65.81±10.97 | 15.36±7.07 |
| Long chain hydroxyacyl-CoA dehydrogenase | 1046.89±84.13 | 387.90±126.92 |
| Short chain hydroxyacyl-CoA dehydrogenase | 1259.27±237.19 | 1318.70±469.83 |
Mean±SD (nmole/min/mg protein).
Clinical phenotypes of the affected mice.
The affected mice appeared normal at birth and survived to adulthood. The only noticeable abnormal physical finding before 6 months of age was a decreased weight gain which became more obvious after ∼3–4 months of age with the affected mice weighed, on average, 15% less than the unaffected siblings (Fig. 3). These mice also had a shortened life span (373±87 days, range 272–489 days, n=8, compared with the non-affected sibs that live for more than 720 days). The cause of the early death was not immediately apparent; they died suddenly in the absence of preceding illness, undue stress or fasting condition. In addition to the relatively uneventful post-natal life, there was no gestational loss of affected mice as mating of the carriers (n=30) produced the expected number of progeny (22%, 46/209 offspring were affected). Pregnancies of carrier mothers appeared normal without complications; no hypoglycemia or elevated serum glutamic-oxaloacetic transaminase (SGOT) and serum glutamate pyruvate transaminase (SGPT) were observed at 18–20 days of gestation (n=4).
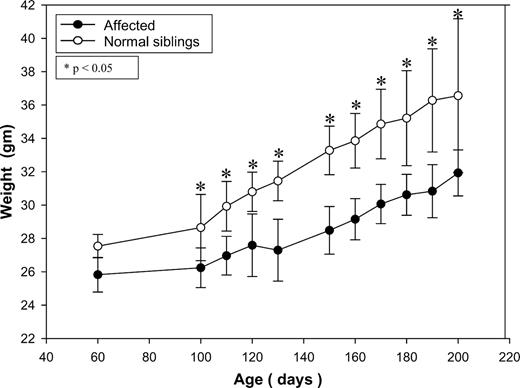
Body weight changes of mice as a function of time. Only male mice are shown: open circles denote affected mice (n=6); closed circles denote normal siblings (+/+ and +/−, total n=8). Data are expressed as mean±SD. *P<0.05.
Blood chemistry (non-fasting) and complete blood counts obtained at 4 and 12 months of age were normal (n=9); there was no hypoglycemia, no elevation of SGOT and SGPT and the creatine phosphokinase (CPK) was normal. However, 24 h fasting resulted in the decline of blood glucose in the affected mice from baseline of 188.8±13.6 mg/dl (range 174–202) to 79±15.8 mg/dl (range 58–99) (P=0.0004), while control mice maintained fasting blood glucose levels at 117.9±34 mg/dl (range 91–201) (P=0.0076, when compared with the fasting levels of the affected).
Because of the phenomenon of sudden death in affected mice, we performed detailed cardiac examination using echocardiography (ECHO) and electrocardiograms (ECG). Seven of nine affected mice examined were found to have cardiac arrhythmias as early as 6 months of age. Their ECHO examination revealed abnormal cardiac rhythmic tracing in both the m-mode cardiac echo and pulse wave Doppler mapping, but no signs of heart failure were detected (Fig. 4A). ECG studies demonstrated a prolonged PR interval as early as 6 months of age and later these mice developed complete atrio-ventricular dissociation as early as 9 months of age (Fig. 4B) implying a defect in the cardiac conduction system.
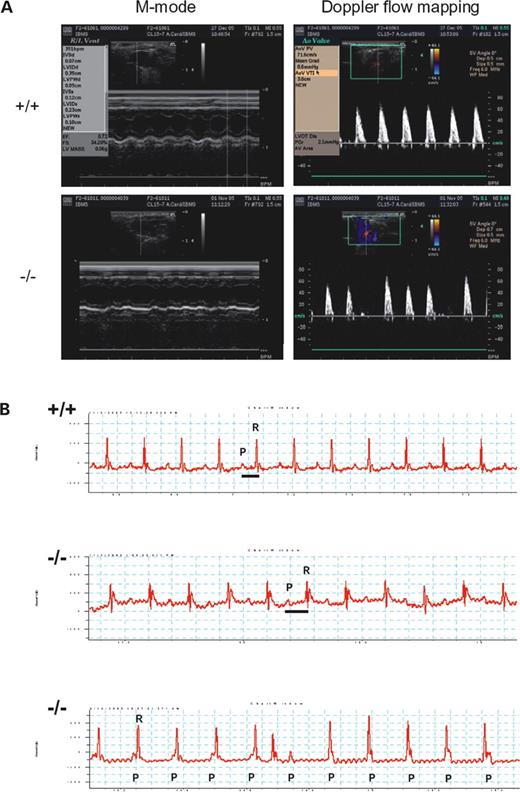
(A) ECHO assessment of wild-type (+/+) and mutant (−/−) mice. Representative M-mode ECHO (left) and Doppler flow mapping of aortic velocity profiles (right) in wild-type and mutant mice, respectively. The mutant mice revealed frequent irregular heart rate indicating cardiac arrhythmia. (B) ECG analysis of wild-type (+/+) and ENU mutant (−/−) mice. Mutant mice (6 months old) have prolonged PR intervals (middle tracing, with PR/RR ratio of 0.58) than wild-type mice (upper tracing, with PR/RR ratio of 0.35). The lower tracing showed further progression of PR prolongation to complete AV dissociation in the mutant mice (9 months old, P wave marching-in to R wave and then marching-out to R wave implying complete AV dissociation).
Histopathological analyses.
We studied the histopathological changes in affected mice sacrificed at 6, 8 and 12 months of age. The hepatocytes in affected liver appeared foamy and nuclei were central (Fig. 5B) as compared with normal hepatocytes extended radically from the central vein in normal liver (Fig. 5A). An increased number and size of lipid droplets in hepatocytes was detected as early as 6 months of age, which further increased in the 9- and 12-month-old mice (Fig. 5B, insert). The brown adipose tissues in the affected mice also had a more abundant lipid accumulation than those of the normal control mice (Fig. 5C and D). Examination of the heart showed cardiomyopathy with increased fat accumulation and multifocal fibrosis (Fig. 5F) where bundles of myocardial fibers embedded in the dense fibrous tissue (Fig. 5H and J). Cardiomyocytes within the bundles exhibited occasionally small vacuoles and loss of sarcomere. Remarkably, the focal fibrosis was noted in both ventricles (5H and 5J) and the interventricular septum (5F) and this can be seen as early as 6 months of age with lesser degree.
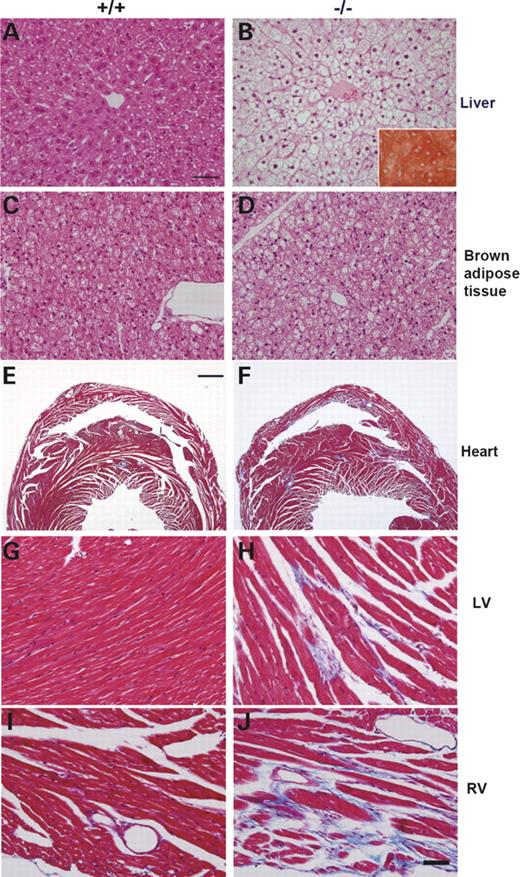
Histopathological analyses of livers, brown adipose tissues and hearts of mice sacrificed at 12 months of age. Livers were stained with hematoxylin and eosin (A and B) and with Oil-Red O (insert in B). Hepatocytes in normal liver showed normal architecture without any sign of hepatic steatosis (A), while those in the affected liver appeared foamy with lipid accumulation and central nuclei (B). Normal brown adipocytes had a considerable volume of cytoplasm with round nuclei located almost centrally (C), while those in affected mice (−/−) contained much larger volume of lipid droplets and peripheral nuclei (D). Hearts were sectioned coronally through the left and right ventricle (E–J). The connective tissue proliferation (multifocal fibrosis) was detected by blue coloration in Masson Trichrome staining. Fibrosis was prominent in myocardium of left ventricle (H), right ventricle (F and J) and septum (F) in the affected heart. Bars=50 µm (A–D and G–J) and 1 mm (E and F).
Examination of skeletal muscle showed an otherwise normal histopathology except for a small increase of the lipid in the muscle fibers (data not shown). Retinal epithelium appeared normal.
DISCUSSION
In this study, we describe for the first time the generation of a viable mouse model of human MTP deficiency. This is also the first animal model of cardiac conduction defects due to a block in a lipid metabolic pathway. The mice exhibited a decreased weight gain, hepatic steatosis, fasting intolerance, cardiac myopathy and arrhythmia, and sudden death, all features that can be seen in human MTP deficiency (10,11,16).
The most striking phenotypes found in these mice were cardiac conduction defects, which ranged from a prolonged PR interval to a complete AV dissociation. No signs of heart failure were observed. Interestingly, the histopathology revealed multifocal cardiac fibrosis, including both ventricles and interventricular septum in which the AV conduction system is located. Arrhythmias and conduction defects as presenting symptoms in several fatty acid oxidation disorders have been documented in children (17); the cardiac findings in patients with TFP deficiency include conduction defects, atrial and ventricular tachycardia associated with cardiomyopathy. Most of these patients died in infancy due to associated severe metabolic derangement. The cause of sudden death in our mice was not clear; there were no preceding illness or undue stress. Furthermore, no death was observed when these mice were subjected to 24 h fasting.
Mouse models of disorders of mitochondrial fatty acid β-oxidation have been reported (18). Two models with defects in long-chain fatty acid oxidation showed cardiac lesions in histopathological studies: a Hadha knock-out mouse showed severe cardiac myocyte degeneration and necrosis but all mice died within 4 days of birth (12); targeted disruption of long-chain acyl-CoA dehydrogenase gene revealed that 10% of adult mice (14–16 weeks) having severe multifocal myocardial fibrosis (19). However, the neonatal death of the Hadha knockout mice precluded further electrophysiological studies and no AV conduction defects were demonstrated, as no ECHO and ECG were performed in the LCAD-deficient mice. Furthermore, neither study documented cardiac fibrosis in the interventricular septum where AV conduction system is located. The mice described here offer a valuable animal model to study cardiac arrhythmia in fatty acid oxidation disorders in human.
Excessive accumulation of lipids is known to be toxic to the cardiac myocytes and can cause cardiomyopathy leading to heart failure (20,21). Long-chain acylcarnitine and lysophosphatidylcholine have been shown to accumulate during acute myocardial ischemia and may play a role in the production of arrhythmia. In vitro studies in the isolated heart tissue indicated that the incorporation of long-chain acylcarnitine in the sarcolemma-elicited electrophysiological anomalies analogous to those seen in the ischemia heart in vivo (22). However, the exact cellular electrophysiological bases of proarrhythmia effects of long-chain acylcarnitine are not completely understood. The availability of a mouse model with AV conduction defects with accumulation of long-chain fatty acids will allow further investigation of the mechanisms underlying arrhythmogenesis at both organismic and cellular levels. Furthermore, our finding of focal cardiac fibrosis, suggest that certain areas of the heart or specialized cardiac cells, such as those involved in the conduction system, may be more active in lipid metabolism and are more susceptible to the block in the fatty acids β-oxidation.
The mice described here exhibit elevated long-chain hydroxyacyl carnitine levels and were homozygous for a missense mutation (M404K) in the gene encoding the β-subunit of the MTP while the α-subunit gene was intact. Methionine at position 404 is well conserved in human, mouse, frog and Caenorhabditis elegans and its substitution of methionine, a neutral amino acid with lysine, a basic amino acid may affect the LKAT enzyme activity. However, at the protein and enzyme activity levels, both α- and β-subunit proteins were reduced along with a reduction of both LCHAD and LKAT enzyme activities. Although it is not clear how a mutation in one subunit affects both subunits, this finding is consistent with observations in the human in that various mutations either in the α or in the β-subunit resulted in disappearance of both subunits and MTP deficiency. Presumably, mutations in either subunit can affect the stability of MTP complex by altering TFP complex expression and/or subunit turnover (7,10,11,23,24).
Tandem mass spectrometry for analysis of both amino acids and acylcarnitines has been automated and can reliably analyze more than 30 metabolites and thereby provides a comprehensive assessment of the entire fatty acid and selected amino acid metabolic pathways. Using this method, more than 20 metabolic disorders in human can be detected; and the method is in widespread use to screen newborns for these conditions (25,26). In addition to the present study and our previous report of the identification of mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease (15), we also identified mice with elevation of both propionyl (C3) and butyryl (C4) carnitines, mice with elevation of C3 carnitine, isobutyryl carnitine and 3-OH butyryl carnitine, mice with elevation of methylmalonyl carnitine and mice with hyperphenylalaninemia (data not shown). Taken together, our data indicate that metabolomics-guided screening coupled with ENU-mediated mutagenesis is a powerful approach to uncover novel enzyme deficiencies and recognize important pathways for genetic metabolic disorders.
In summary, we have identified mice with hepatic steatosis and cardiac conduction defects due to a missense mutation in the gene coding for the β-subunit of MTP. This mouse model will be useful for further investigation of mechanisms underlying arrhythmogenesis. Furthermore, these mice represent an important animal model for the study of abnormal long-chain fatty acid metabolism and its toxicity and to investigate pathophysiology and treatment strategies for human MTP deficiency.
MATERIALS AND METHODS
Generation of ENU-recessive mice.
C57BL/6J male mice were treated with ENU and bred according to the three-generation breeding scheme (27) to produce G3 offspring as previously described (15). Breeding and housing of the mice were conducted in the Mouse Mutagenesis Program Core facility of the Academia Sinica under specific pathogen-free conditions. The animal protocol was approved by the Institutional Animal Care and Use Committee.
Tandem mass spectrometry screening of amino acids and acylcarnitines.
Standardized filter papers (S&S 903; Schleicher & Shuell, Dassel, Germany) were impregnated with 25 µl whole blood from the tail vein of 2- to 3-month-old G3 mice. Analytes were eluted from punching of the blood spots (∼6.25 µl) and then subjected to tandem mass spectrometry analysis (Quattro Micro, Micromass, Beverly, MA, USA) using electrospray ionization in positive ion mode to analyze the targeted acylcarnitines and amino acids by means of a precursor ion scan of 85 kDa, and by neutral loss scans of 102, 119 and 161 kDa, respectively (25,26).
Mapping of the gene responsible for the abnormal long-chain acylcarnitine phenotype.
A genome-wide mouse SNPs dataset containing 10 915 SNPs of 48 mouse strains was kindly provided by Tim Wiltshire (Genomics Institute of the Novartis Research Foundation). A total of 262 SNPs were selected for genotyping according to the criterion that the genotypes of the mouse strain C3H/HeJ, DBA/2J or BALB/cByJ are different from that of C57BL6J. The average space between each SNP is 10 Mb. SNP genotyping using genomic DNA isolated from mouse tails was performed by high-throughput MALDI-TOF mass spectrometry (28,29). Primers and probes flanking the SNPs were designed in multiplex format using SpectroDESIGNER software (Sequenom, San Diego, CA, USA). PCRs were performed in a volume of 5 µl containing 0.15 U of Taq polymerase (HotStarTaq™, Qiagen, Valencia, CA, USA), 5.0 ng of genomic DNA, 1.0 pmol of each PCR primer and 2.5 nmol of dNTP. Thermocycling consisted of one cycle at 94°C for 15 min, 45 cycles of 94°C for 20 s, 56°C for 30 s, 72°C for 30 s and one final cycle of extension at 72°C for 3 min. Unincorporated dNTPs were dephosphorylated using 0.3 U of Shrimp Alkaline Phosphatase (Hoffman-LaRoche, Basel, Switzerland) followed by primer extension using 9 pmol of each primer extension probe, 4.5 nmole of the appropriate dNTP/ddNTP combination, and 1.28 U of Thermosequenase (Amersham Pharmacia, Piscataway, NJ, USA). Reactions were cycled at 94°C for 2 min, followed by 55 cycles of 94°C for 5 s, 52°C for 5 s and 72°C for 5 s. Following the addition of a cation exchange resin (SpectroCLEAN, Sequenom) to remove residual salt from the reactions, 15 nl of the purified primer extension reaction was spotted onto a 384-element silicon chip preloaded with 3-hydroxypicoloinic acid matrix (SpectroCHIP, Sequenom), using the SpectroPOINT (Sequenom). SpectroCHIPs were analyzed using a Bruker Biflex III MALDI-TOF SpectroREADER mass spectrometer (Sequenom) and spectra processed with SpectroTYPER (Sequenom). The affected mice were outcrossed to the mapping strain, C3He/HeJ, to generate the N1 offsprings, and were subsequently intercrossed to generate the N1F1. The N1 share the 1:1 ratio of C3He/HeJ (A) and C57BL/6 (B) alleles. For the N1 intercross, the mutated locus was associated with the homozygous C57BL/6 alleles and non-mutant locus represent the ratio of 1:2:1 (AA:AB:BB) in N1F1 affected mice. DNA was collected from eight affected N1F1 mice. To analyze the linkage between the mutant and homozygous phenotype for C57BL/6 alleles, the frequency of C57BL/6 alleles in all affected mice was evaluated using SNP genotyping.
PCR verification and DNA sequencing.
Genomic DNA was purified from tail using Puregene™ DNA purification kit (Gentra Systems, Minneapolis, MN, USA). All exons of candidate genes (Hadha, Hadhb, and very long-chain acyl-CoA dehydrogenase, Acadvl) were amplified and sequenced. Primers were designed by Primer3 PCR primer picking program (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi). The primers used in the detection of exon 14 mutation in the Hadhb gene were 5′-GCAGAGGCAGGAGGATTTC, sense strand in intron 13 and 5′-GACCTGCCTGCTTCTACCTC, anti-sense strand in intron 14 (25). The PCRs were performed in a final volume of 25 µl, containing 0.2 µm each primer, 10 mm Tris–HCl (pH 8.3), 50 mm KCl, 1.5 mm MgCl2, 0.2 mm dNTPs and 1 U TaKaRa Taq™ (Takara, Shiga, Japan). Amplification conditions consisted of an initial denaturation of 3 min at 94°C, followed by 20 cycles of touchdown PCR in 30 s at 94°C, 30 s at 65°C (decrease 0.5°C per cycle), 40 s at 72°C; and a final 20 cycles in 30 s at 94°C, 30 s at 55°C, 40 s at 72°C. All amplified PCR fragments were digested with shrimp alkaline phosphatase and ExoI to remove unincorporated primers and sequenced using the BigDye Terminator Cycle Sequencing Kit v1.1/3.1 (Applied Biosystems, Foster City, CA, USA) following the manufacturer's instructions. Sequencing products were separated on either ABI PRISM 3100 Genetic Analyzer or ABI PRISM 3700 DNA Analyzer (Applied Biosystems). Raw sequencing data was analyzed by the DNA Sequencing Analysis Software v3.7 (Applied Biosystems).
Western blot analysis.
Western blot analysis was performed following 10% SDS–PAGE according to the procedures of Laemmli (30) using rabbit polyclonal antibodies raised against the mouse HADHB (TP-beta) amino acid residues 241–259 (EQDEYALRSH SLAKKAQDEG) and the mouse HADHA (TP-alpha) (a generous gift by Arnold W. Strauss of Vanderbilt University School of Medicine). Goat anti-human Hsp60 polyclonal antibody (Stressgen Biotechnologies Corporation, San Diego, CA, USA) was used as an internal control. Mitochondrial fractions were isolated from mouse heart and liver using a mitochondria isolation kit, MITO-ISO1 (Sigma–Aldrich, St Louis, MO, USA). The immunoreactive protein bands were visualized using the ECL substrate system detected by the Image Systems LAS-3000 (Fuji Photo Film Co. Ltd., Tokyo, Japan).
Enzyme assays.
The activities of short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD), LCHAD, acetoacetyl-CoA thiolase and LKAT were measured in the mitochondrial fractions as previously described with minor modification in the reaction mixtures (31–33). The substrates were acetoacetyl-CoA (Sigma–Aldrich) for the short-chain assay and 3-ketopalmitoyl-CoA (custom synthesis by Universidad Autónoma de Madrid, Madrid, Spain) for long-chain assay. The reaction mixture used in the thiolase assay contained 0.1 m Tris–HCl (pH 8.3), 1 mm MgCl2, 0.1 mm acetoacetyl-CoA or 3-ketopalmitoyl-CoA, 0.2 mm CoASH (Sigma–Aldrich) and 10 µg tissue extract proteins in a total reaction mixture of 250 µl and the reactions were followed by the change of absorbance at 303 nm. The reaction mixture used in the dehydrogenase assay contained 0.1 m potassium phosphate buffer (pH 7.2), 0.2 mm NADH, 0.075 mm acetoacetyl-CoA or 3-ketopalmitoyl-CoA and 1 µg tissue extract proteins in a total reaction mixture of 250 µl and the reactions were followed by the change of absorbance at 340 nm. All reactions proceeded at 37°C for up to 5 min.
Laboratory analyses.
All blood samples were obtained through an incision of the tail artery or by cardiac puncture at the time of sacrifice. Plasma was analyzed using FUJI DRI-CHEM SYSTEM 3500s (Fuji Photo Film Co. Ltd.) for measurement of glucose, SGOT, SGPT, CPK, creatinine, triglycerides and cholesterol. Whole blood was analyzed for the complete blood counts using Cell-Dyn 3700 (Abbott Laboratories, Illinois, USA).
Echocardiography.
Mice were anesthetized with Pentobarbital (66 mg/kg I.P) for echo examination. The ECHO system (model HDI 5000, Philips, Washington) equipped with a 15-MHz linear transducer, was used in this study for 2D, M-mode and Doppler flow mapping. Fractional shortening, left ventricular ejection fraction, systolic and diastolic chamber diameters, mitral inflow with diastolic performance, aortic and pulmonary blood flow were measured and recorded.
Electrocardiograms.
Mice were sedated by intraperitoneal tribromoethanol (TBE; Avertin, with 12 µl/g of 2.5% solution) and a standard Lead II (limb leads) ECG was recorded using Dual Bio Amp amplifiers (ADInstruments, New South Wales, Australia) and PowerLab (ADInstruments) for data analysis. Parameters such as PR, QT, QTc, RR interval, P wave, QRS duration and ST-segment were measured and recorded.
Histopathology.
Three affected female mice of 6-, 8- and 12-month-old and age-matched control mice were sacrificed for the histopathological examinations. In addition, one mouse that died suddenly at the age of 9 months was also included in the study. The mice were perfused through heart with fixative (4% paraformaldehyde, buffered at pH 7.4). Liver, heart, kidney, stomach, intestine, spleen, brain, retina, skin, white and brown adipose tissues, and leg muscles were then removed, embedded in paraffin, cut into 5 µm sections, and stained with hematoxylin–eosin for a general pathological evaluation and Masson's trichrome (Azan) for the detection of connective tissue proliferation. The liver and heart were also frozen, sectioned into 5 mm in cryostat and stained with Oil red O to detect lipid content.
ACKNOWLEDGEMENTS
We thank the Academia Sinica Mouse Mutagenesis Core for providing ENU mice and National Genotyping Core for gene mapping. We thank Dr Arnold Strauss for providing the anti-Hadha antibody. This work was supported by the National Science and Technology Program for Genomic Medicine from the National Science Council, Taiwan, the Genomics and Proteomics Program from the Academia Sinica, Taiwan and a grant from the National Science Council (94-2320-B-039-041).
Conflicts of Interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}