




Abstract
Motor neuron degeneration is the predominant pathological feature of spinal muscular atrophy (SMA). In patients with severe forms of the disease, additional sensory abnormalities have been reported. However, it is not clear whether the loss of sensory neurons is a common feature in severe forms of the disease, how many neurons are lost and how loss of sensory neurons compares with motor neuron degeneration. We have analysed dorsal root ganglionic sensory neurons in Smn−/−;SMN2 mice, a model of type I SMA. In contrast to lumbar motor neurons, no loss of sensory neurons in the L5 dorsal root ganglia is found at post-natal days 3–5 when these mice are severely paralyzed and die from motor defects. Survival of cultured sensory neurons in the presence of NGF and other neurotrophic factors is not reduced in comparison to wild-type controls. However, isolated sensory neurons have shorter neurites and smaller growth cones, and β-actin protein and β-actin mRNA are reduced in sensory neurite terminals. In footpads of Smn-deficient mouse embryos, sensory nerve terminals are smaller, suggesting that Smn deficiency reduces neurite outgrowth during embryogenesis. These data indicate that pathological alterations in severe forms of SMA are not restricted to motor neurons, but the defects in the sensory neurons are milder than those in the motor neurons.
INTRODUCTION
Autosomal recessive spinal muscular atrophy (SMA) is caused by the loss of the telomeric copy of the survival of motor neuron gene (SMN1) on human chromosome 5q13 (1). Despite the widespread expression of SMN in various tissues and neuronal cell types, motor neurons are predominantly affected, and motor neuron degeneration is the leading pathological alteration in this disease (2). The severity of the SMA phenotype is modified by the number of centromeric SMN2 copies (3–5). In the most severe form of the disease (type I SMA) according to Munsat and Davies (6) and Roberts et al. (7), sensory defects have also been observed (8). In particular in type I SMA patients, reduced nerve conduction velocity has been recorded from mixed and sensory nerves (9), and severely affected SMA type I patients with congenital muscular hypotonia which progressed to early death showed inexcitability of sensory nerves (10). More detailed analysis of sensory nerves from seven type I SMA patients revealed axonal degeneration in sural nerves (11). In type I SMA patients, many empty myelin sheaths and atrophic axons in the sensory nerves correlated with typical signs of sensory axonopathy in electrophysiological analyses. Interestingly, SMA II and SMA III patients did not show sensory nerve pathology.
On the basis of these findings, we analysed the loss of sensory neurons in the L5 dorsal root ganglia (DRGs) of post-natal Smn−/−;SMN2 mice. These mice serve as a model for human type I SMA (12). We investigated dorsal root sensory neurons at different developmental periods. E14 was chosen to investigate defects in neurite outgrowth at a stage when sensory nerve fibres normally reach their target, the skin. Survival of sensory neurons was investigated at post-natal days 3–5, when the developmental period of physiological cell death is over and neuronal losses due to pathological processes become apparent. We also investigated survival, neurite growth and growth cone morphology in cultured sensory neurons from E14 mouse embryos. Isolated sensory neurons from such embryos need trophic support to survive in cell culture, and neurite growth resembles initial fibre outgrowth and not a regenerative response resembling that of adult neurons after axotomy.
The number of cell bodies in the L5 dorsal root ganglia (DRGs) of Smn−/−; SMN2 mice is not reduced at post-natal day 5, when these mice are severely paralyzed due to defects in motor neurons. The sensory nerve endings in the skin of 14-day-old Smn−/−;SMN2 embryos appear smaller in comparison to controls. In isolated sensory neurons, β-actin mRNA and protein are reduced in distal neurites and growth cones. Growth cones of Smn-deficient cultured sensory neurons are smaller and their neurites are shorter. These findings suggest that sensory neurons are also affected by Smn deficiency, but the pathological changes are more discrete than those in motor neurons.
RESULTS
Quantification of neurons in L5 DRGs
Previous studies have shown that the number of spinal motor neurons is reduced in various models of SMA (12–15). Three-to-five-day-old Smn−/−;SMN2 mice that serve as a model for type I SMA exhibit a 17% loss of motor neurons in the lumbar spinal cord (12). At that stage, these mice are severely paralyzed, and they normally do not survive longer than 5–7 days after birth. To investigate whether sensory neurons are also affected by the disease, we counted the number of neurons in L5 DRGs from Smn−/−;SMN2 mice. Smn−/−;SMN2 mice and age-matched controls were perfused at post-natal days 1, 3 and 5. The spinal cord together with the DRGs was dissected, and 15 µm paraffin serial sections were prepared and Nissl stained. We counted the nuclei of neuronal cell bodies in every 10th section of the L5 DRGs (Fig. 1A). No significant difference was observed between Smn−/−;SMN2 and control DRGs (Fig. 1B) at any stage, even at post-natal day 5 when the mice are severely paralyzed, indicating that survival of sensory neurons in Smn−/−;SMN2 mice is not reduced when the motor neuron loss becomes apparent in this disease.
Neurite growth is reduced in Smn-deficient sensory neurons
We then investigated whether reduced Smn levels influence neurite growth in sensory neurons at E14. Previous studies with isolated motor neurons from Smn−/−;SMN2 mice (16) and a zebrafish model (17) in which Smn was repressed by Morpholino knockdown have shown that Smn deficiency leads to defects in neurite outgrowth in motor neurons. Lumbar DRGs contain various types of neurons, including proprioceptive, fast- and slow-conducting sensory neurons. These subpopulations differ in their response to different neurotrophic factors (NFs). NT-3 predominantly supports survival of proprioceptive neurons (18,19), whereas BDNF acts on neurons which are responsive to tactile stimuli (20,21). Nerve growth factor (NGF) predominantly supports small pain-conducting neurons (21), and GDNF acts more broadly on these different subtypes of neurons (19,22). Therefore, to investigate whether Smn deficiency differentially affects these subpopulations, NGF, GDNF, BDNF and NT-3 were separately added at a concentration of 10 ng/ml each as survival factors to the sensory neuronal cultures. In a first attempt, we investigated whether NGF, NT-3, BDNF and GDNF responsiveness is altered in Smn−/−;SMN2 sensory neurons. Survival of DRG neurons did not differ in the presence of these different NFs (Fig. 2A and B). Without NFs, neurite outgrowth is normally very low (Fig. 2C), and there was no difference between neurite-bearing cells in mutants and controls (data not shown). We then investigated the size of cell bodies in these different groups. Although the subpopulations of NT-3 responsive neurons showed larger cell bodies (Fig. 2D and F) both in mutant and in control cultures, no differences were observed between control and Smn−/−;SMN2 sensory neurons.
To investigate neurite outgrowth, the cells were fixed with 4% paraformaldehyde (PFA) and immunostained with antibodies against phosphorylated Tau protein (red) and MAP-2 (green) after 24 h in culture (data not shown). The neuronal processes of Smn-deficient cells were slightly but significantly shorter when compared with wild-type controls (Fig. 2I). No significant difference was observed with respect to the survival factor added to these cultures. More than 200 cells in three independent experiments were measured and the reduction was significant (P<0.05) for each group, as shown in Figure 2I. To investigate whether longer culture periods lead to normalization of neurite outgrowth, Smn−/−;SMN2 sensory neurons were cultured with NGF for 48 h. The difference in neurite length between mutant and control cultures persisted, indicating that the effect of Smn deficiency cannot be explained by a delay of neurite growth that is compensated within 48 h.
Subcellular distribution of Smn, hnRNP-R and β-actin in cultured sensory and motor neurons
To investigate and compare the distribution of Smn, hnRNP-R and β-actin protein in sensory and motor neurons, both types of neurons were prepared from E14 embryos and taken into culture. Sensory neurons were plated on Laminin-111 (according to (23)) and cultured in the presence of 10 ng/ml NGF for 24 h. Motor neurons were grown on the same substrate for 7 days in the presence of BDNF and CNTF (10 ng/ml each). After fixation, sensory and motor neurons were stained with antibodies against Smn, β-actin and hnRNP-R, respectively. Staining against hnRNP-R was combined with antibodies against β-actin (Fig. 3A, E and I) and Smn (Fig. 3C, G and K) in motor and sensory neurons (Fig. 3B, F and J and D, H and L). The accumulation of hnRNP-R, Smn and β-actin in the distal part of motor axons was more pronounced in wild-type motor neurons than in wild-type sensory neurons (Fig. 3I and J and K and L, arrow).
HnRNP-R and β-actin distribution in neurites and growth cones of primary cultured sensory neurons
We have previously observed that Smn, hnRNP-R, β-actin and β-actin mRNA accumulation is reduced in axon terminals of cultured Smn-deficient motor neurons (16). Therefore, we investigated the distribution of these proteins in sensory neurons cultured from control and Smn-deficient mice. In addition, we compared the growth cone size of Smn-deficient and wild-type sensory neurites (Fig. 4). After 24 h in the presence of NT-3, BDNF, GDNF and NGF, the sensory neurons were fixed and stained with monoclonal antibodies against β-actin and hnRNP-R. HnRNP-R staining in growth cones of sensory neurons is restricted to the pars compacta (Fig. 4F, arrow). Growth cone size was significantly reduced in sensory neurons (P<0.0001) (Fig. 4M). Immunoreactivity of β-actin protein appeared highly reduced in the distal part of the neurites (Fig. 4D and L). No differences in growth cone size were observed in various subgroups of sensory neurons, which survive in the presence of NGF, NT-3, BDNF or GDNF (Fig. 4M).
β-Actin mRNA is reduced in growth cones of cultured sensory neurons from Smn-deficient embryos
On the basis of the finding that growth cones of Smn-deficient sensory neurons contain less β-actin immunoreactivity than wild-type controls, we investigated whether β-actin mRNA is also reduced. For this purpose, we performed in situ hybridization with an antisense probe against actin mRNA. Light microscopic analysis revealed a reduced signal for actin mRNA in the distal part of Smn-deficient sensory neurons (Fig. 5B and D) in contrast to wild-type cells (Fig. 5A and C). An actin mRNA sense probe was used as a negative control (Fig. 5E).
Morphological characterization of sensory nerve endings in footpads of Smn-deficient mice
The observation that Smn-deficient cultured sensory neurons do not show any alterations with respect to cell survival but exhibit defects in neurite elongation and reduced growth cone size prompted us to examine sensory nerve endings in vivo at a stage when these neurites grow out and make contact with their targets. For this purpose, we isolated footpads from E14 Smn−/−;SMN2 embryos and control littermates. After fixation, 10 µm thick cryosections were cut and stained with a polyclonal antibody against neurofilament-M. Sections were analysed under a confocal microscope for morphological alterations of the nerve endings in the epidermis of the footpads.
In comparison to controls (Fig. 6A and C), nerve endings from Smn-deficient embryos do not reach the outer epidermal layer (Fig. 6B and D). Higher magnification of innervating nerves revealed that sensory neuron endings were prominent in control embryos and showed a typical globe-like structure (Fig. 6E and F). In contrast, the nerve endings were much thinner in Smn−/−;SMN2 embryos (Fig. 6G and H). We have also quantified the number of sensory nerve endings in Smn−/−;SMN2 and controls. No difference was observed in the number of nerve endings per footpad (data not shown).
DISCUSSION
SMA is generally considered as a disease exclusively affecting motor neurons. Nevertheless, several clinical observations have been made with type I SMA patients that sensory neurons are also affected. Abnormal sensory conduction velocity has been reported (9–11), and analysis of sural nerve biopsies revealed various degrees of axonal degeneration (11). It is not known how generalized these effects are or whether the axonal loss observed in the nerve biopsy material correlates with enhanced cell death of the corresponding sensory neurons in the DRGs.
We have analysed the number of sensory neurons in the L5 DRG and the morphology of sensory nerve terminals in the skin of Smn-deficient mouse embryos. Although no significant loss of sensory neurons was observed, sensory neurons from the severely affected SMA mouse model do not develop properly. Their terminals in the skin are much smaller at embryonic day 14. This correlates with defects in neurite growth and growth cone morphology in sensory neurons isolated from mouse embryos at the same developmental stage. Growth cones of these sensory neurons were significantly smaller in cultured Smn-deficient neurons. Accumulation of the Smn interaction partner hnRNP-R is less pronounced in the distal part of the sensory nerve processes. Moreover, β-actin protein and mRNA levels are reduced in growth cones of sensory neurites, indicating that similar pathophysiological processes as those observed in Smn-deficient motor neurons are responsible for these defects.
Sensory defects have so far only been reported in severe SMA, particularly in patients with pre-natal disease onset. For example, a patient described by Rudnik-Schoneborn et al. (11), who was ventilated from birth on with no motor function at all, showed a fibre density of 3500/mm2 in the sural nerve, whereas age-matched controls have more than 20 000 fibres per mm2. This finding indicates that sensory defects develop early in severe SMA, at the same time when motor defects become apparent. Similarly, isolated sensory neurons from 14-day-old Smn−/−;SMN2 mouse embryos show reduced neurite growth, and the growth cones in the skin of these mice are smaller than in controls. Surprisingly, these alterations did not result in the loss of sensory cell bodies in the L5 DRG at post-natal days 3–5, indicating that reduced SMN levels do not affect survival of sensory neurons in culture or in vivo.
Interestingly, motor neuron loss in the same mice is small at birth and increases during the following 3–5 days (12). When motor neurons are isolated at E14, axon growth is reduced during a period of 7 days in culture. This indicates that axon pathology precedes neuronal cell death in motor neurons. Indeed, when isolated motor and sensory neurons are grown in culture in the presence of NFs, cell death is not enhanced, although the defects in neurite growth are clearly apparent under these in vitro conditions. This suggests that the cell death of motor neurons is a consequence of loss of contact and subsequent loss of trophic support from skeletal muscle. The finding that treatment with cardiotrophin-1, neurotrophic factor that is secreted from developing skeletal muscle to innervating motor neurons, can reduce motor neuron loss in Smn-deficient mice (24) supports this hypothesis.
The neurite growth defect observed with isolated sensory neurons is much less pronounced in comparison to motor neurons. Neurites from sensory neurons grow much faster than from isolated motor neurons under similar culture conditions. Within 24 h, they grow distances up to 300 or 400 µm, whereas axons from cultured motor neurons need 7 days for the same distance. Growth of sensory and motor axons in cell culture differs by the frequency of growth cone collapses, changes in growth directions and turns, which are much more frequent in motor than in sensory axons (Fig. 3A and B). The growth cone plays an essential role in axon guidance (25), and β-actin dynamics regulate axon growth direction (26). The growth cone in isolated sensory neurons is smaller than in motor axons, and this reflects differences in neurite growth between these two types of cells. The finding that defects in motor axon guidance are a dominant feature after RNAi knockdown of Smn in zebrafish (17) is in line with this idea.
In summary, Smn−/−;SMN2 mice exhibit specific alterations in sensory neurons, which are less prominent than defects in motor neurons. Neurites are shorter and growth cones are smaller in sensory neurons from Smn-deficient embryos. Reduced levels of β-actin mRNA and protein in sensory growth cones point to a similar pathomechanism in both cell types, indicating that Smn deficiency might result in more widespread changes in the nervous system, particularly in type I SMA.
MATERIALS AND METHODS
Quantification of neurons per DRG
Mice were deeply anaesthetized and transcardially perfused with 4% PFA at post-natal days 1, 3 and 5. The spinal cord with the attached DRGs was prepared, and 15 µm paraffin serial sections were cut. Neurons in DRGs were counted in every 10th section from L5 spinal cord segments. The raw counts were corrected for double counting of split nucleoli, as described (27).
Sensory neuronal culture from mouse embryos
L1 to L5 DRGs were dissected from E14 embryos in parallel with the ventral part of the spinal cord. The DRGs were transferred to phosphate-buffered saline (PBS) and incubated with trypsin (0.05% in HBSS) for 30 min. Trypsin digestion was stopped by the addition of F14 medium (Gibco) containing 10% horse serum and 35 mm KCl. The cell suspension was pre-plated for 3–4 h to suppress growth of non-neuronal cells. The supernatant was centrifuged (10 min at 400g), and the cell pellet was resuspended in F14 medium containing 10% horse serum and 35 mm KCl. The cells were counted and plated at 2000 cells per cm2 on polyornithine-coated plates on Laminin-111. The cells were incubated for 24 or 48 h, respectively, at 37°C and 5% CO2.
Immunocytochemistry and data analysis
Sensory neurons grown for 24 or 48 h on glass cover slips were fixed with 4% PFA. After treatment with 10% bovine serum albumin (BSA), the fixed cells were incubated O/N at +4°C with the following primary antibodies: rabbit antibodies against phospho-Tau (Sigma, 1 µg/ml) and hnRNP-R [(28), 1:1000] and a monoclonal antibody against Smn (Transduction Laboratories, 1:1000), β-actin (Abcam, 1:1000) and Map-2 (Sigma, 1:1000). Cells were then washed three times with Tris-buffered saline (TBS)-T and incubated for 1 h at room temperature with Cy2- and Cy3-conjugated secondary antibodies (Dianova, 1:200). After washing with TBS-T, cover slips were embedded in Mowiol. For the quantification of neurite length and growth cone area, phospho-Tau-positive processes and β-actin-positive growth cones were scored. Images recorded at the confocal microscope (Leica) were analysed using the Scion Image software package. Data were analysed using the Student's t-test for significance of differences.
In situ hybridization
Cells grown on glass cover slips were fixed with 4% PFA in PBS for 15 min at room temperature and then washed with PBS containing 0.1% active DEPC for 10 min at room temperature. Cells were then permeabilized with 0.3% (v/v) Triton in PBS for 20 min at room temperature, and endogenous peroxidase activity was quenched through incubation in 0.3% (v/v) H2O2 in methanol for 40 min at room temperature. Following a wash in 5× SSC, cover slips were pre-incubated in hybridization buffer [4× SSC, 20% dextran sulphate, 50% formamide, 0.25 mg/ml poly(A), 0.25 mg/ml salmon sperm DNA, 0.25 mg/ml tRNA, 0.1 m dithiothreitol (DTT), 0.5× Denhardt's] for 1 h at 37°C. Then, fresh hybridization solution containing 3′ biotinylated sense or antisense actin oligonucleotide (200 ng/ml, GeneDetect) was applied to the cover slips at 200 ng/ml. Hybridization was carried out for 24 h at 37°C. Two low stringency washes in 1× SSC, 10 mm DTT were performed for 15 min at 55°C and followed by two washes in 0.5× SSC, 10 mm DTT for 15 min at 55°C. Finally, a wash in 0.1× SSC, 10 mm DTT for 10 min at 55°C was performed. A hybridized probe was detected through DAKO GenPoint, a Tyramide Signal Amplification System for in situ hybridization with biotinylated probes (DAKO), following the manufacturer's instruction. Finally, cover slips were counterstained with haematoxylin, dehydrated and mounted with Vitro-clud (Legenbrick). Images were acquired with an Axiophot microscope (Zeiss) equipped with a CCD camera using Axioplan 2 Software (Zeiss).
Cryosections and NF-M antibody stainings of footpads from E14 embryos
Distal limbs from E14 embryos were prepared and frozen in Tissue-Tek. Cross-sections (10 µm) from the footpad area were cut, mounted on gelatine-coated glass slides and pre-incubated with 10% BSA in 1× TBS-T. After BSA treatment, the neurofilament-M antibody (Abcam, 1:200) staining was performed O/N. Sections were washed three times with TBS-T and incubated for 1 h at room temperature with Cy3-conjugated secondary antibodies (Dianova, 1:200). After washing with TBS-T, cover slips were embedded in DABCO. Images were recorded with a confocal microscope (Leica).
ACKNOWLEDGEMENTS
We thank Christine Schneider for skilful technical assistance. This work was supported by grants from DFG, SFB 581, TP B1, the EU through the APOPIS project, the SMA Foundation and the Schilling Stiftung.
Conflict of Interest statement. None declared.
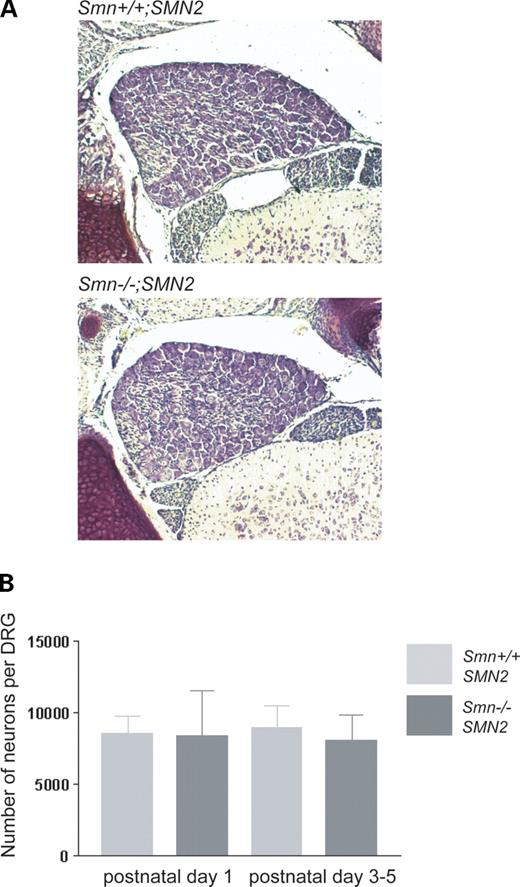
Figure 1. Quantification of neuronal cell bodies in L5 DRGs of Smn−/−;SMN2 and Smn+/+;SMN2 mice. Cell numbers were counted at post-natal days 1, 3 and 5 in sectioned L5 DRGs from Smn−/−;SMN2 mice and age-matched controls (A). Neurons were counted in every 10th section. The counts are depicted as bar charts (B). No difference in cell number was observed between Smn−/−;SMN2 (P1 n=3; P3–5 n=4; black) and controls (P1 n=3; P3–5 n=4; grey). Data represent mean±SD.
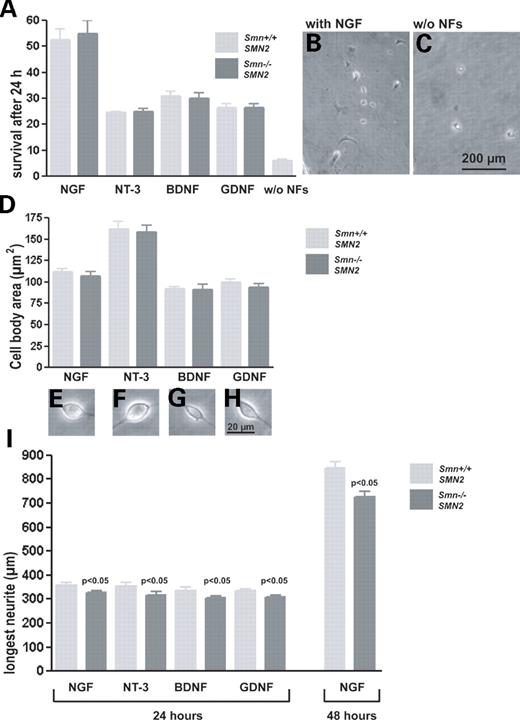
Figure 2. Survival, size of cell bodies and neurite length in cultured dorsal root ganglionic neurons from Smn−/−;SMN2 and control embryos. Cells were cultured for 24 h in the presence of NGF, NT-3, BDNF and GDNF (10 ng/ml each) and for 48 h in the presence of NGF to support the survival of various sensory neuronal subtypes. Cells were counted immediately after plating and after 24 h. Survival of cells treated with different NFs and without NFs (A), morphology of NGF-treated cultures (B) and controls without NFs (C), cell body size (D) and morphology (E–H). No differences in cell survival and size of cell bodies were observed between Smn-deficient sensory neurons and controls. Neurite length was measured; data are shown as bar charts (I). After 24 h, neurites of Smn-deficient sensory neurons (black bar) are shorter than controls (grey bar) in the presence of NGF (−9%, P<0.05), NT-3 (−11%, P<0.05), BDNF (−8%, P<0.05) and GDNF (−7%, P<0.05). Neurite length reduction of Smn-deficient cells increases to 14% in the presence of NGF after 48 h of culturing (results from three independent experiments). Data represent mean±SEM.
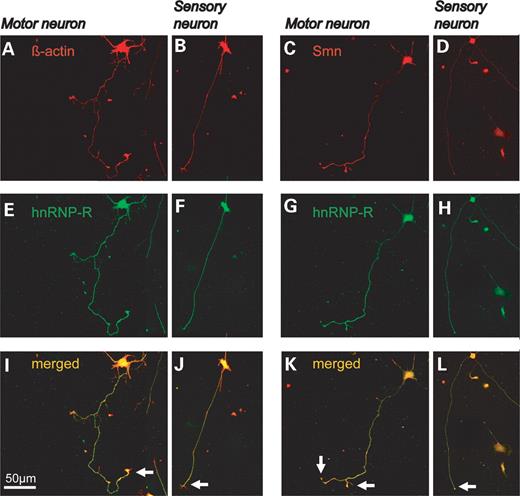
Figure 3. Smn, hnRNP-R and β-actin distribution in cultured sensory and motor neurons from Smn wild-type embryos. DRGs were isolated from 14-day-old Smn wild-type embryos and cultured for 24 h in the presence of NGF (10 ng/ml). The cells were then fixed with 4% PFA and stained with antibodies against hnRNP-R (green), β-actin and Smn (red). In parallel, motor neurons were isolated from the same mouse embryos and cultured for 7 days. In motor neurons, hnRNP-R, β-actin and Smn strongly accumulate in the distal part of the axon (A, C, E and G, arrows in I and K). In sensory neurons, the distribution of Smn, hnRNP-R and β-actin is similar, but the accumulation at the neuron terminal is less pronounced (B, D, F and H, arrows in J and L).
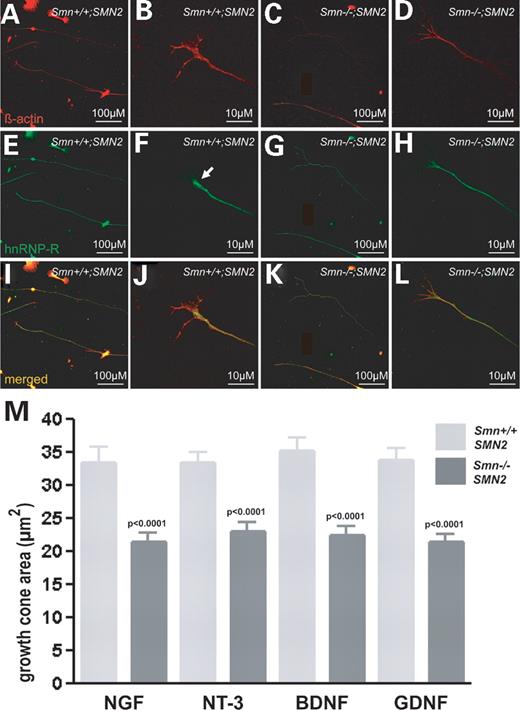
Figure 4. Reduced β-actin content in neurite terminals of isolated Smn-deficient sensory neurons. Dorsal root ganglionic sensory neurons from Smn−/−;SMN2 and Smn+/+;SMN2 embryos were cultured for 24 h, fixed with 4% PFA and stained with antibodies against β-actin (red) and hnRNP-R (green). HnRNP-R and β-actin are homogenously distributed throughout the cell bodies and proximal neurites in wild-type (A, B, E, F, I and J) and Smn-mutant sensory neurons (C, D, G, H, K and L). Staining in the distal parts of Smn-deficient neurites appears weaker. The area of the growth cones of Smn-deficient sensory neurons (black bar) and wild-type controls (grey bar) treated with NT-3, NGF, BDNF and GDNF was determined by staining against β-actin (B, D, J and L), followed by morphological analysis (M). The growth cone area of Smn-deficient sensory neurons in the presence of each survival factor is significantly reduced (P<0.0001).
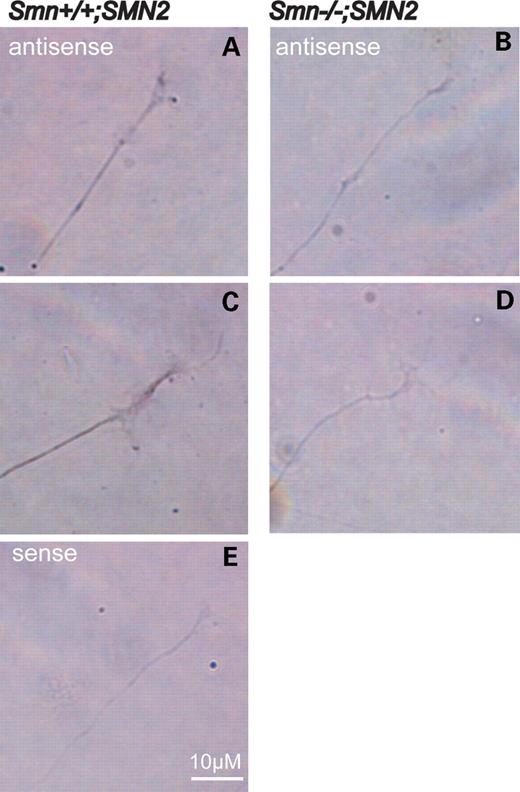
Figure 5. Reduction of actin mRNA in growth cones of Smn-deficient sensory neurons. In situ hybridization of cultured sensory neurons derived from Smn−/−;SMN2 (B and D) and Smn+/+;SMN2 mice with an antisense probe against actin mRNA (A and C). The signal for actin mRNA is reduced in the distal part of the neurites in Smn-deficient sensory neurons (B and D). An actin-sense probe was used as negative control and did not reveal a detectable signal (E).
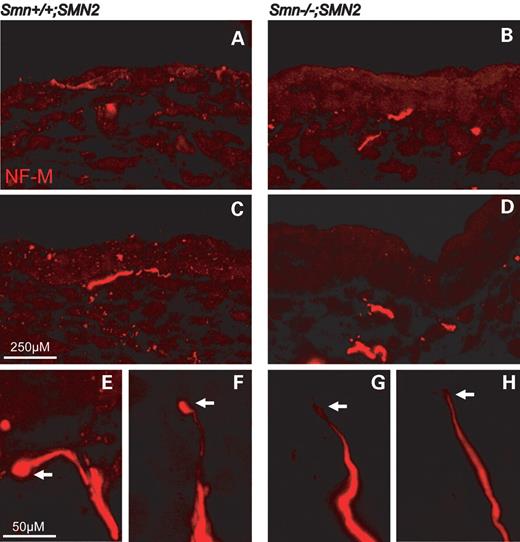
Figure 6. Nerve endings of sensory neurons in footpads from E14-old-Smn-deficient mouse embryos. Cryosections of footpads from Smn-wild-type and Smn-deficient E14 embryos were prepared. Each section was stained with a polyclonal antibody against neurofilament-M (red) and analysed by confocal microscope. Distal nerve endings from Smn-deficient sensory neurons do not reach the outer epidermal layer (B and D) in contrast to sensory nerve endings from control embryos (A and C). Nerve endings of sensory neurons from wild-type embryos were prominent, and the terminals showed a typical globe-like structure (E and F, arrow). In contrast, the nerve endings in Smn−/−;SMN2 embryos were much thinner (G and H, arrow).
References
Lefebvre, S., Burglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., Benichou, B., Cruaud, C., Millasseau, P., Zeviani, M. et al. (
Crawford, T.O. and Pardo, C.A. (
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., Dreyfuss, G. and Melki, J. (
Lorson, C.L., Hahnen, E., Androphy, E.J. and Wirth, B. (
Monani, U.R., Lorson, C.L., Parsons, D.W., Prior, T.W., Androphy, E.J., Burghes, A.H. and McPherson, J.D. (
Munsat, T.L. and Davies, K.E. (
Roberts, D.F., Chavez, J. and Court, S.D. (
Korinthenberg, R., Sauer, M., Ketelsen, U.P., Hanemann, C.O., Stoll, G., Graf, M., Baborie, A., Volk, B., Wirth, B., Rudnik-Schoneborn, S. and Zerres, K. (
Anagnostou, E., Miller, S.P., Guiot, M.C., Karpati, G., Simard, L., Dilenge, M.E. and Shevell, M.I. (
Omran, H., Ketelsen, U.P., Heinen, F., Sauer, M., Rudnik-Schoneborn, S., Wirth, B., Zerres, K., Kratzer, W. and Korinthenberg, R. (
Rudnik-Schoneborn, S., Goebel, H.H., Schlote, W., Molaian, S., Omran, H., Ketelsen, U., Korinthenberg, R., Wenzel, D., Lauffer, H., Kreiss-Nachtsheim, M., Wirth, B. and Zerres, K. (
Monani, U.R., Sendtner, M., Coovert, D.D., Parsons, D.W., Andreassi, C., Le, T.T., Jablonka, S., Schrank, B., Rossol, W., Prior, T.W., Morris, G.E. and Burghes, A.H. (
Frugier, T., Tiziano, F.D., Cifuentes-Diaz, C., Miniou, P., Roblot, N., Dierich, A., Le Meur, M. and Melki, J. (
Jablonka, S., Schrank, B., Kralewski, M., Rossoll, W. and Sendtner, M. (
Hsieh-Li, H.M., Chang, J.G., Jong, Y.J., Wu, M.H., Wang, N.M., Tsai, C.H. and Li, H. (
Rossoll, W., Jablonka, S., Andreassi, C., Kroning, A.K., Karle, K., Monani, U.R. and Sendtner, M. (
McWhorter, M.L., Monani, U.R., Burghes, A.H. and Beattie, C.E. (
Wright, D.E., Zhou, L., Kucera, J. and Snider, W.D. (
Baudet, C., Mikaels, A., Westphal, H., Johansen, J., Johansen, T.E. and Ernfors, P. (
Carroll, P., Lewin, G.R., Koltzenburg, M., Toyka, K.V. and Thoenen, H. (
Lewin, G.R., Ritter, A.M. and Mendell, L.M. (
Matheson, C.R., Carnahan, J., Urich, J.L., Bocangel, D., Zhang, T.J. and Yan, Q. (
Aumailley, M., Bruckner-Tuderman, L., Carter, W.G., Deutzmann, R., Edgar, D., Ekblom, P., Engel, J., Engvall, E., Hohenester, E., Jones, J.C. et al. (
Lesbordes, J.C., Cifuentes-Diaz, C., Miroglio, A., Joshi, V., Bordet, T., Kahn, A. and Melki, J. (
Gundersen, R.W. and Barrett, J.H. (
Challacombe, J.F., Snow, D.M. and Letourneau, P.C. (
Masu, Y., Wolf, E., Holtmann, B., Sendtner, M., Brem, G. and Thoenen, H. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}