




Abstract
Huntington disease is an adult-onset neurodegenerative disorder that is caused by the expansion of a polyglutamine tract within the Huntingtin (htt) protein. Wild-type htt has been shown to be involved in transcription, transport and cell survival. Here, we demonstrate that increased expression of full-length wild-type htt in mice is associated with a dose-dependent increase in body weight which results from an increase in both total fat mass and fat-free mass. Conversely, we show that a reduction in the levels of wild-type htt is associated with decreased body weight. Examination of individual organ weights revealed that the weight of the heart, liver, kidneys, lungs and spleen increased with the over-expression of wild-type htt, whereas the brain and testis were unaltered. On the basis of these initial findings, we examined mice that over-express full-length mutant htt to determine the effect of polyglutamine expansion on this novel function of wild-type htt. We found that over-expression of full-length mutant htt, but not an N-terminal fragment of mutant htt, also increased body weight and organ weight, except in the brain and testis where mutant htt appears to be toxic. In these mice, the majority of weight gain could be accounted for by increases in total fat mass. Further investigation of the weight gain phenotype revealed that the increases in weight were not accounted for by increased food consumption relative to body weight. Overall, we demonstrate that increased levels of both wild-type and mutant full-length htt are associated with increased body weight.
INTRODUCTION
Huntington disease (HD) is an adult-onset neurodegenerative disorder caused by a CAG expansion in the HD gene which codes for the huntingtin (htt) protein. The disease is postulated to result from a gain of toxic function in the mutant htt protein combined with a loss of wild-type htt function (1,2). As a result of the CAG expansion, HD patients heterozygous for the disease mutation have levels of wild-type htt that are decreased by 50% from birth, whereas HD patients homozygous for CAG expansion do not express wild-type htt at all. Accordingly, it is important to define the normal functions of the wild-type protein and to determine how these functions are modulated by polyglutamine expansion.
Htt is essential for embryonic development and normal function in adult mice [see (3) for review of normal huntingtin function]. Mice homozygous for a targeted inactivation of the mouse HD gene die as embryos at day E8.5 (1,4,5), whereas mice heterozygous for the targeted inactivation of the mouse HD gene show behavioural abnormalities and neuronal loss (1,6). As htt levels are decreased below 50% of normal, the phenotypic abnormalities become more severe, manifesting in gross brain abnormalities and early death (7,8). The continued importance of htt function in adulthood is indicated by the neurologic phenotype that results from the inactivation of htt expression in mature mice (9).
Wild-type htt appears to be involved in cellular survival, transcription and intracellular transport. Wild-type htt has been shown to protect neurons from toxic insults both in vitro and in vivo (10–14). Htt has also been shown to interact with several transcription factors (15) and to specifically increase the transcription of genes under the control of a neuron restrictive silencer element including BDNF (16,17). The interaction of htt with multiple proteins involved in trafficking and endocytosis (15) suggests a role for htt in intracellular transport. This is supported by functional evidence demonstrating that htt facilitates fast axonal transport of vesicles and mitochondria within the cell (18–21).
Although polyglutamine expansion disrupts many assayable functions of wild-type htt (10,14,16,20), even highly expanded mutant htt is able to rescue mice homozygous for the targeted inactivation of the mouse HD gene from embryonic lethality (12,22,23). This suggests the possibility that some of the most critical functions of htt are not disrupted by polyglutamine expansion and may be unknown. This concept is supported by studies which show that the loss of wild-type htt in mouse models of HD results in only mild exacerbation of HD-like phenotypes (23–25).
We have previously generated mice that over-express full-length wild-type htt in order to study htt function (26). These YAC18 mice express human htt, with 18 glutamines, under its endogenous regulatory elements and show the same pattern of tissue-specific htt expression as WT mice. These mice were used to demonstrate the neuroprotective function of wild-type htt (13,14). Similarly, we have generated YAC128 mice which express full-length mutant htt with 120 glutamines in order to study how htt function is affected by polyglutamine expansion (27). These mice recapitulate the motor, cognitive and neuropathological abnormalities of HD (27,28). In this study, we find that the levels of full-length htt influence body weight in both YAC18 and YAC128 mice.
RESULTS
Increased expression of wild-type huntingtin results in increased body weight in a dose-dependent manner
We previously generated two lines of YAC18 mice to study wild-type htt function. The B60 line expresses low levels of wild-type htt, whereas Line 212 expresses wild-type htt at about 2–3 times endogenous levels (26). Examination of these mice for overt phenotypes resulting from the over-expression of wild-type htt revealed that aged YAC18 mice were physically larger than WT mice. To quantify this difference, we monitored body weight in a cohort of mice from 2 to 11 months of age and found that total body weight increased with full-length htt expression (genotype: F(2,36)=18.2, P<0.001). Line 212 mice weighed significantly more than Line B60 mice and Line B60 mice weighed significantly more than WT mice (Fig. 1A; 2 months—WT: 25.5±1.1 g, Line B60: 26.8±0.7 g, Line 212: 30.0±1.0 g; 11 months—WT: 34.5±1.6 g, Line B60: 40.6±1.1 g, Line 212: 46.5±1.4 g). Comparison of the slopes of weight gain over time for Line 212 and WT mice as a measure of growth rate suggests that the differences in body weight result from increased weight gain prior to 6 months of age, after which the slopes for YAC18 and WT mice are similar (Fig. 1A; Slope 2–6 months—WT: 0.32±0.06 g/week, YAC18, Line 212: 0.75±0.05 g/week, P<0.001; Slope 6–11 months—WT: 0.19±0.03 g/week, YAC18, Line 212: 0.27±0.03 g/week, P=0.09). Western blotting for htt expression in whole brain lysates confirmed that Line B60 mice express more wild-type htt than WT mice and Line 212 mice express more wild-type htt than Line B60 mice (Fig. 1B). Thus, higher levels of htt expression are associated with increased body weight. The demonstration of dose-dependent increases in total body weight in two different lines of YAC18 mice indicates that the increased weight is not a result of the site of integration and suggests the possibility that altered htt levels are causative of this finding.
To further test the hypothesis that wild-type htt influences body weight, we sought to determine if decreasing the levels of wild-type htt would result in decreased body weight. Our laboratory has generated mice heterozygous for the targeted inactivation of the mouse HD gene (Hdh +/− mice) that express htt protein at half of wild-type levels (1). We examined the weight of a cohort of WT and Hdh +/− mice from 2 to 11 months of age and found that Hdh +/− mice weigh significantly less than WT mice beginning before 2 months of age (Fig. 1C; 2 months—WT: 24.1±0.4 g, Hdh +/−: 21.0±0.8 g, P=0.003). Although the difference was maintained until 11 months of age, the difference appeared to be greatest closer to 2 months of age suggesting that the difference in weight may stem from altered development in Hdh +/− mice (genotype: F(1,20)=4.2, P=0.05). This is supported by the fact that the slopes of the weight gain graphs are similar from 2 to 11 months of age (slope 2–12 months—WT: 0.26±0.02g/week, Hdh +/−: 0.23±0.02 g/week, P=0.3). Western blotting for htt protein in brain confirmed that Hdh +/− mice express htt at half of wild-type levels (Fig. 1D). In combination with our findings in mice over-expressing wild-type type htt, it is clear that expression levels of full-length wild-type htt influence body weight.
Increased expression of wild-type huntingtin results in increased peripheral organ weights
In order to investigate the underlying cause of increased body weight in YAC18 mice, we measured organ weights in 12-month-old YAC18, Line 212 mice. We found that organ weight was significantly increased in YAC18 mice when compared with WT mice in the heart, liver, lungs, kidneys and spleen (Fig. 2A; Heart—WT: 187±4 mg, YAC18: 231±7 mg, P<0.001; Liver—WT: 1.94±0.06 g, YAC18: 2.59±0.11 g, P<0.001; Lungs—WT: 420±12 mg, YAC18: 485±15 mg, P=0.002; Kidneys—WT: 673±13 mg, YAC18: 848±26 mg, P<0.001; Spleen—WT: 108±2 mg, YAC18: 147±11, P=0.002). The increases in organ weight were seen in both male and female mice with similar magnitude except in the spleen where the percentage increase was greater in males (Supplementary Material, Fig. S1). In contrast to all of the other organs tested, the brain and testis show no increase in weight (Fig. 2A; Brain—403±3 mg, YAC18: 407±4 mg, P=0.4; Testis—WT: 161±4 mg, YAC18: 152±7 mg, P=0.3). It is noteworthy that the organs were not grossly oedematous, nor were there pericardial, pleural or peritoneal effusions. We also examined the organs from YAC18 mice for gross pathology and compared these sections with those of WT mice. In sections from the liver, heart, spleen, kidneys and lungs of YAC18 mice the cytoarchitecture and histologic appearance was normal (see Supplementary Material, Fig. S1 for sample sections). We found no evidence of abnormal pathology including no accumulations of vacuoles or lipids, no increased extracellular matrix deposition and no infiltration of tissues by inflammatory or other cells.
Next, we examined the level of htt expression in each of the organs studied to see if there was any correlation between the relative increases in weight and the level of htt expression. We found that, as previously reported, expression of full-length htt was highest in the brain and testis (Fig. 2B and C; Htt expression as percentage of brain—Brain: 100±11%, Heart: 10±7%, Liver: 34±15%; Lungs: 70±21%; Kidney: 0±2%; Spleen: 79±16%; Testis: 110±9%). Among the remaining peripheral tissues htt expression was highest in the lungs, which showed very little increase in weight, and the spleen, which showed the largest increase in weight in male mice. We were unable to detect any full-length htt in the kidneys. Thus, although overall body weight increases with htt expression, increases in organ weight do not appear to be directly correlated with the level of htt expression in each organ. In fact, the brain and testis express the highest levels of htt, yet, neither show any increase in weight.
To determine whether the increases in organ weight resulted from an increase in cell number, we measured the volume and nuclear density of the liver and kidney. As expected from the increase in weight, the volume of organs from YAC18 mice were significantly larger from WT mice (Kidney—WT: 938±49 mm3, YAC18: 1281±54 mm3, P<0.001; Liver—WT: 2375±72 mm3, YAC18: 3054±82 mm3, P<0.001). Using counts of nuclei as an estimate of cell number, we found that there were also significantly more cells in the YAC18 organs than in WT organs (Kidney—WT: 360±21 million nuclei, YAC18: 457±23 million nuclei, P=0.01; Liver—WT: 369±32 million nuclei, YAC18: 494±34 million nuclei, P=0.02).
Similar to wild-type huntingtin, mutant huntingtin expression increases body weight and organ weight except in the brain and testis
As many functions of wild-type htt are disrupted by polyglutamine expansion, we examined the effect of mutant htt over-expression on weight to determine how this function is affected by polyglutamine expansion. For this purpose, we used the YAC128 mouse model of HD which expresses mutant htt at levels that are approximately three-quarters of endogenous levels of wild-type htt (23,27). As in the YAC18 mice, we found that YAC128 mice show significantly increased body weight when compared with WT mice from 2 to 11 months of age (Fig. 3A; genotype: F(1,49)=33.6; P<0.001; 2 months—WT: 25.6±0.6 g, YAC128: 28.3±0.5 g, P=0.001; 12 months—WT: 35.5±1.1 g, YAC128: 45.9±0.8 g, P<0.001). The difference in weight appears to result from increased weight gain up until 6 months of age, after which the slopes of weight versus time are similar between YAC128 and WT mice (slope 2–6 months—WT: 0.34±0.04 g/week, YAC128: 0.73±0.03 g/week, P<0.001; slope 6–11 months—WT: 0.16±0.04 g/week, YAC128: 0.20±0.02 g/week, P=0.4). Western blotting with whole brain lysates was used to confirm that YAC128 mice express wild-type htt at a level similar to WT mice and mutant htt at a level that is slightly less than endogenous levels (Fig. 3B). Total htt expression in YAC128 mice is greater than in YAC18 Line B60 mice but less than in YAC18 Line 212 and the weight curve for YAC128 mice is intermediate between the two. As with wild-type htt, increased expression of full-length mutant htt is associated with increased body weight.
To determine whether the expression of an N-terminal fragment of htt is sufficient to increase weight, we examined body weights of shortstop mice at 12 months of age. Shortstop mice express exons 1 and 2 of mutant htt with ∼120 glutamines from the same YAC as YAC128 mice (29). Importantly, shortstop mice express the mutant htt fragment at levels that are greater than the expression level of full-length mutant htt in YAC128 mice. We found that the weight of shortstop mice was equivalent to that of WT mice (WT: 37.8±1.2 g, Shortstop: 36.4±1.6 g, P=0.5). The absence of weight increase in these mice was not a result of toxicity of the short mutant htt fragment as shortstop mice do not show any neurodegeneration (29).
We have previously shown atrophy of the brain and testis of YAC128 mice resulting from mutant htt expression (23,27). To determine the effect of the expression of full-length mutant htt on organ weights, we weighed organs from perfused, 12-month-old YAC128 mice. Both the brain and testis showed significant atrophy in YAC128 mice when compared with WT mice (Fig. 4A; Brain—WT: 400±3 mg, YAC128: 386±3 mg, P<0.001; Testis—WT: 161±4 mg, YAC128: 126±8 mg, P<0.001). In contrast, the heart, liver, kidneys and spleen from YAC128 mice all weighed significantly more than from WT mice suggesting a lack of toxicity in these organs (Fig. 4A; Heart—WT: 183±4 mg, YAC128: 216±3 mg, P<0.001; Liver—WT: 1.90±0.06 g, YAC128: 2.34±0.06 g, P<0.001; Kidneys—WT: 655±15 mg, YAC128: 807±12 mg, P<0.001; Spleen—WT: 106±2 mg, YAC128: 132±4 mg, P<0.001). Although the magnitude of the changes differed in some cases, both male and female YAC128 mice showed increased organ weights when compared with WT mice (Supplementary Material, Fig. S2). As with the organs from YAC18 mice, these organs were not grossly oedematous and microscopic examination of sections from the heart, liver, kidneys, lungs and spleen revealed no obvious pathology. There was no accumulation of vacuoles or lipids, no alterations in extracellular matrix deposition and no infiltration of tissue by inflammatory cells.
In order to gain insight into why mutant htt caused atrophy in the brain and testis but increased the weight of the heart, liver, kidneys and spleen, we examined mutant htt expression in each of the organs to see if there was a correlation between mutant htt expression and either weight gain or toxicity. We found that mutant htt was most highly expressed in the brain and testis, thereby providing a possible explanation for why these two organs showed atrophy (Fig. 4B and C; Htt expression as percentage of brain—Brain: 100±4%, Heart: 9±2%, Liver: 29±6%; Lungs: 13±7%; Kidney: 0±0%; Spleen: 20±6%; Testis: 52±5%). Among the remaining tissues, mutant htt expression was highest in the liver, spleen and lungs (Fig. 4B and C). We were unable to detect any full-length mutant htt in the kidney of YAC128 mice (Fig. 4B and C). As in YAC18 mice, increases in htt expression did not correlate with increases in organ weight but toxicity was observed in the two organs expressing the highest levels of mutant htt.
Huntingtin expression does not affect food consumption
To further characterize the weight gain phenotype in YAC18 and YAC128 mice and also to determine whether the underlying mechanism is similar in these two transgenic mice, we examined the food consumption and weight change in 3-month-old animals, when the differences in growth rate appeared to be most pronounced. On the basis of previous reports in the R6/2 mouse model of HD showing increased fat accumulation (30,31), we also examined body composition. Finally, we completed a fasting-refeeding experiment as a basic measure of metabolic rate (32).
We found that YAC18 mice gained weight more rapidly than WT mice (Fig. 5A; genotype—F(1,14)=31.9, P<0.001). However, during this period, food consumption per gram of lean body mass was not significantly different between the genotypes (Fig. 5B; genotype—F(1,14)=0.3, P=0.6). During a 24 h fast, the body weight of YAC18 mice did not decrease at a faster rate than WT mice, suggesting that the metabolic rates are not different (Fig. 5C). The rate of weight recovery following the 24 h fast was also similar between WT and YAC18 mice (Fig. 5C) and as a result, the percentage of lost weight that was recovered did not differ between the genotypes.
Following full recovery of body weight, we performed body composition scans to determine what percentage of the weight increase was accounted for by increases in lean weight and fat accumulation (a sample scan is shown in Supplementary Material, Fig. S3). We found that both fat-free mass and total fat mass were significantly increased in YAC18 mice when compared with WT (Fig. 5D; fat-free mass: genotype—F(1,18)=11.0, P=0.004; total fat mass: genotype—F(1,18)=19.0, P<0.001). At this age, the organ weights in YAC18 mice were already significantly greater than in WT mice (not shown).
To determine the effect of polyglutamine expansion on the weight gain phenotype, we completed the same analysis in YAC128 mice. In contrast to the YAC18 mice, we observed that YAC128 mice did not gain weight more rapidly than WT mice relative to their body weight (Fig. 6A; genotype—F(1,14)=0.19, P=0.7). As with the YAC18 cohort, there was no difference between the genotypes in food consumption during this period (Fig. 6B, genotype—F(1,14)=2.7, P=0.1). There was also no significant difference in the rate of weight loss during a 24 h fast or weight gain afterwards between the YAC128 and WT mice (Fig. 6C).
Scans of body composition revealed that although the fat-free mass was not significantly different between YAC128 and WT mice. YAC128 mice showed a significant increase in total fat mass which accounted for the majority of the weight difference from WT mice at this age (Fig. 6D; fat-free mass: genotype—F(1,14)=0.2, P=0.6; total fat mass: genotype—F(1,14)=15.4, P=0.02). This may result from mutant htt disrupting the partitioning of calories to lean tissues. An increase in fat accumulation is also seen in the R6/2 mouse model despite their overall loss of body weight (30). At this age there was a trend towards increased organ weight in YAC128 mice when compared with WT mice which only reached significance in the spleen (not shown).
DISCUSSION
Wild-type htt has been shown to function in neuroprotection, transcription and transport within the cell (10,16,20). Here, we demonstrate a novel function of wild-type htt. We show that over-expression of full-length wild-type htt is associated with a dose-dependent increase in body weight as both the fat-free mass and total fat mass are increased in YAC18 mice. This increase does not appear to result from increased food consumption or decreased metabolic rate. Examination of YAC128 mice revealed that the expression of full-length mutant htt also results in increased body weight. The increased weight in YAC128 mice did not appear to result from increased food consumption or decreased metabolism. In contrast to YAC18 mice, most of the difference in weight resulted from an increase in the total fat mass. Similarly, an increase in fat accumulation has been reported in R6/2 mice which express an N-terminal fragment of mutant htt (30).
Increasing expression of full-length htt also resulted in increased organ weights with the exception of the brain and testis, the two organs that express the highest levels of htt (33). A possible explanation for this is that the high levels of htt present in the brain and testis are already sufficient for maximum growth and that additional htt has little impact. Interestingly, over-expression of full-length mutant htt also resulted in increased organ weights except in the brain, testis and lungs. In agreement with our previous findings mutant htt expression in the brain and testis resulted in significant atrophy when compared with WT mice (12,23,27). The apparent toxicity of mutant htt in the brain and testis may result from high expression of mutant htt in these two organs and/or an increased sensitivity to the effects of polyglutamine toxicity. It is also possible that these two organs are more affected as they are more dependent on wild-type htt function (33). Although full-length htt expression resulted in a dose-dependent increase in total body weight, there was no correlation between htt expression levels in organs and their relative increase in weight with htt over-expression. The levels of full-length htt varied widely between organs, yet, aside from the lungs, the increases in organ weights were roughly proportional to the increase in total body weight. This suggests the possibility that the increase in organ weight results from a central mechanism perhaps originating in the brain. Support for the levels of htt in brain affecting body weight comes from the fact that mice with reduced levels of wild-type htt in the adult forebrain shows decreased weight (9).
The activity of wild-type htt in transcription (16,17), transport (20) and neuroprotection (10,12) are all disrupted by polyglutamine expansion and polyglutamine expansion also alters htt's interaction with most htt interacting proteins (15). As many of the functions of wild-type htt have been defined by their loss in mutant htt, it follows that most known functions of wild-type htt are disrupted by polyglutamine expansion. The fact that mutant htt can rescue mice homozygous for the targeted inactivation of the mouse HD gene from embryonic lethality (12,22,23) suggests that the most critical functions of wild-type htt in development must be maintained in mutant htt. If the mechanism of weight increase is similar between YAC18 and YAC128 mice, it is possible that the effect of wild-type htt on weight represents one of these critical functions. In order to identify additional functions of wild-type htt it will be important to carefully characterize mice over-expressing wild-type htt in addition to defining those functions that are lost through polyglutamine expansion.
Our work and previous studies in mouse models support a role of htt in determining body weight. Here, we demonstrate that a 50% reduction in wild-type htt levels resulted in a decrease in body weight. Similarly, mice with severely reduced htt levels in adulthood show a 20% decrease in body mass with proportional decreases in organ weight (9). Chimeric mice, in which 20–75% of cells do not express wild-type htt, show decreases in body mass as great as 40% (34). Mice expressing only 10–20% levels of endogenous htt have also been shown to have markedly decreased weight when compared with controls (8). Thus, it is clear that decreases in wild-type htt expression are associated with decreased body weight. As we have previously shown that decreasing wild-type htt levels in culture results in decreased cellular proliferation, a similar decrease in proliferation in vivo may contribute to the observed decreases in weight (35). A role for htt altering cell proliferation is supported by the increased cell numbers we observe in the liver and kidneys of YAC18 mice when compared with WT mice.
Weight loss has also been reported in mouse models of HD. Two mouse models expressing a small N-terminal fragment of mutant htt have demonstrated progressive weight loss that precedes premature death (36,37). In one of these models, the transgenic expression of a mutant htt fragment resulted in the depletion of full-length wild-type htt beginning at 7 weeks of age which is followed closely by a decrease in body weight at 8 weeks (38). This finding suggests the possibility that decreasing levels of full-length htt in HD may contribute to the characteristic weight loss observed in late stages of the disease. Weight loss has also been demonstrated in a knock-in model of HD with 150 CAG repeats (25).
In contrast, weight loss has not been reported in transgenic mouse models of HD which express full-length mutant htt (22,27,39). These mice express two copies of full-length wild-type htt as well as one or more copies of full-length mutant htt. As a result, there is an over-expression of total full-length htt and we show here that both wild-type and mutant full-length htt act to increase body weight. We also show here that shortstop mice, which express an N-terminal fragment of mutant htt, do not show increased weight when compared with WT mice. This suggests that N-terminal fragments of htt do not modulate weight gain and offers a possible explanation for why mouse models of HD expressing N-terminal fragments of mutant htt exhibit weight loss.
The cause of weight loss in HD patients is currently unknown. Decreased body mass index is reported in the early stages of the disease with obvious thinning occurring in the later stages (40). Although some researchers have suggested that hypothalamic cell loss may lead to decreased food intake in HD patients (41), studies have shown that HD patients actually consume more calories than unaffected individuals (42,43). Others have proposed that the increased energy expenditure associated with chorea results in the decrease in weight. However, weight loss appears to worsen with the progression of the disease while chorea subsides. Furthermore, while sedentary energy expenditure is increased in HD patients, overall energy expenditure is not increased because HD patients engage in less voluntary physical activity again suggesting that energy expenditure does not account for differences in weight (44). It has also been suggested that mitochondrial dysfunction in HD leads to increased energy expenditure and decreased weight (45). However, sleeping metabolic rates of HD patients and unaffected individuals were not different suggesting that mitochondrial defects may not increase the basal metabolic rate in HD patients (44).
The clear impact of full-length htt expression on body weight that has been demonstrated here and elsewhere suggests the possibility that decreased levels of full-length htt contribute to weight loss in HD. At birth, HD patients express decreased levels of full-length wild-type htt by virtue of replacing one or both wild-type alleles with a mutant allele. Although full-length mutant htt is expressed in its place, our results suggest that wild-type htt has a greater impact on fat-free mass. An increased effect of wild-type htt on body weight is supported by the fact that mice expressing one copy of mutant htt and one copy of wild-type htt weigh more than mice expressing two copies of mutant htt (25). Furthermore, mutant htt mRNA has been shown to be expressed at lower levels than wild-type htt mRNA (46). In addition, the levels of total full-length htt are thought to be decreased during the course of the disease through aggregation and increased proteolysis. In support of this, decreased levels of full-length htt have been demonstrated in the striatum of HD patients (47) and have been shown to precede weight loss in a mouse model of the disease (38,48). Although further experiments will be required to elucidate the precise mechanism of weight loss in HD, our data suggests the possibility that a decrease in the level of full-length mutant or wild-type htt may contribute to this phenotype. Regardless, we demonstrate clearly that expression levels of full-length htt influence body weight in a dose-dependent manner.
MATERIALS AND METHODS
Mice
Experiments were carried out on YAC18 and YAC128 mice generated to express wild-type htt with 18 glutamines and mutant htt with 120 glutamines, respectively (26,27). We also examined mice heterozygous for the targeted inactivation of the mouse HD gene (1). All mice were maintained on the FVB/N strain background (Charles River, Wilmington, MA, USA) and group housed with littermates of mixed genotype. Mice were kept on a normal light/dark cycle where lights were turned off at 8:00 PM and on at 6:00 AM. Experimenters were blind to the genotype of the mice. For longitudinal measurement of body weight in YAC18 mice we used five female WT, four male WT, nine female YAC18, Line B60, five male YAC18, Line B60, nine female YAC18, Line 212, and seven male YAC18, Line 212 mice. For longitudinal measurement of body weight in YAC128 mice, we used 12 female WT, 11 male WT, 18 female YAC128 and 13 male YAC128 mice. For measurement of organ weights, we used 18 female WT, 16 male WT, 8 female YAC18, 10 male YAC18, 22 female YAC128 and 15 male YAC128 mice.
Body weights and organ weights
Body weights were taken at 9:00 AM every two weeks from 2 to 11 months of age. At 12 months of age mice were perfused with 3% paraformaldehyde. All of the mice were perfused for exactly 10 min under a constant flow of 5 ml/min in order to ensure equivalent perfusion. Following perfusion, the organs were checked for firmness and loss of colour to confirm adequate perfusion. Subsequently, the organs were dissected out carefully by the same experimenter for each mouse in order to ensure consistency. The feeder blood vessels were trimmed to the junction with the organ parenchyma and all fat associated with the organs was removed. After excision and trimming, the organs were post-fixed overnight in perfusate and equilibrated in phosphate-buffered saline for 48 h. Prior to weighing, organs were blotted thoroughly on kimwipes with gentle pressure until the organs were dry enough not to dampen a dry area of kimwipe under mild pressure. In the case of the heart and lungs, additional pressure was applied to ensure that no solution was trapped in the cavities.
Organ volumes were determined by volume displacement in a graduated cylinder filled with water. Prior to measurement, the organs were dried thoroughly as described earlier. Counts of nuclei were performed using Stereoinvestigator stereology software (Microbrightfield, Williston, VA, USA). Sections measuring 10 µm were cut on a cryostat (Microm HM 500M, Richard-Allan Scientific, Calamazoo, MI, USA) and mounted directly onto slides. Sections were stained with toluidine blue (water for 1 min, toluidine blue for 3 min, three washes with water for 1 min, five dips in 95% ethanol, five dips in 100% ethanol, 30 s in xylene). Sections of liver and kidney were outlined and nuclei were counted in a 25 µm×25 µm counting frame with a grid size of 800 µm×800 µm. Cell number was estimated by multiplying nuclear density by organ volume and dividing by the volume of the counting frame.
Organs were also analyzed for gross pathology. Cryostat sections measuring 10 µm were stained with cresyl violet (70% ethanol for 2 min, water for 2 min, cresyl violet for 5 min, two dips in water, two dips in 70% ethanol with 1% glacial acetic acid, two dips in 100% ethanol with 1% glacial acetic acid, 100% ethanol for 1 min, xylenes for 1 min) and examined under the microscope. We specifically looked for accumulations of vacuoles or lipids, increased extracellular matrix deposition and infiltration of tissues by inflammatory or other cells.
Western blots
Western blots were performed on tissue samples frozen immediately after mice were asphyxiated with carbon dioxide. Protein lysates containing 100 µg of total protein were separated on a 7.5% acrylamide gel. Following transfer to a membrane, htt protein was detected using the htt-specific MAB2166 antibody (1:2000, 1 h, room temperature; Chemicon, Temecula, CA, USA) followed by incubation with a peroxidase-linked anti-mouse secondary antibody (1:5000) and enhanced chemiluminescent detection. Protein levels were quantified by measuring band density with Quantity One software (BioRad, Hercules, CA, USA).
Feeding experiments
Feeding and weight gain were monitored in 3-month-old YAC18 mice, YAC128 mice and WT littermates controls. Mice were singly housed for these measurements. Mice and food were weighed twice a week for a period of ∼6 weeks. In addition to weighing the food remaining in the hopper, the bedding was visually inspected for food particles that had been spilled or buried by the mice. These particles were added back to the leftover food prior to weighing. Female mice under these conditions were stressed and showed fluctuations in weight. Accordingly, we only analyzed data from male mice. For the YAC18 comparison, we examined four WT mice and six YAC18 mice, whereas the YAC128 comparison used eight WT mice and seven YAC128 mice. After 6 weeks, mice were fasted for 24 h and the weight lost during that period was used to estimate their metabolic rate. The mice were then given free access to food and the weight of the mice was measured after 24 h as a measure of their ability to recover.
One week after measuring food consumption, the body composition of the mice was determined by nuclear magnetic resonance imaging. Briefly, whole body fat measurements have been carried out on a 7T animal MRI scanner (Bruker, Germany). Awake mice were placed inside a restrainer, and the restrainer positioned inside the magnet. NMR signal from the entire body was acquired with a quadrature volume RF coil tuned to 300 MHz. Standard CPMG sequence (TE=2.377 ms, TR=10 s) was used to acquire 256 echoes from which the T2 decay curve was extracted. The decay curves were fit to a double exponential function using software procedure developed in house with Igor (WaveMetrics, OR, USA). The component corresponding to T2≈40 ms was identified as water in lean tissue, and the one with T2≈200 ms as body fat (49). The dc shift of the double exponential function was identified as a ‘free’ water component corresponding to body fluids, e.g. urine and CSF, with the typical amounts of less than 5% of the total signal. The ratio of lean tissue/body fat expressed as w/w was calculated from the NMR data as described by Kunnecke et al. (49).
Statistical analysis
Data are given as the mean±SEM. Longitudinal measurements of weight and food consumption were analyzed by repeated measures ANOVA. Comparisons of single outcome measures were analyzed by one-way ANOVA. In case of significant differences between genotypes, post hoc comparisons between genotypes at individual points were assessed with a Student's t-test.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We would like to thank M. Dawn McArthur for her advice. This work was supported by grants from the Michael Smith Foundation for Health Research, the Canadian Institutes of Health Research, the Huntington's Disease Society of America and the High Q Foundation. J.M.V.R. has been supported by the Canadian Institutes of Health Research, the Michael Smith Foundation for Health Research and the Huntington Society of Canada. W.T.G. is supported by a Clinician Investigator Award from the Canadian Institutes of Health Research, Institute of Genetics. B.R.L. and M.R.H. are supported by the Canadian Institutes of Health Research, the Huntington Society of Canada, the Hereditary Disease Foundation and the Canadian Genetic Diseases Network. M.R.H. is a Killam University Professor and holds a Canada Research Chair in Human Genetics.
Conflict of Interest statement. None declared.
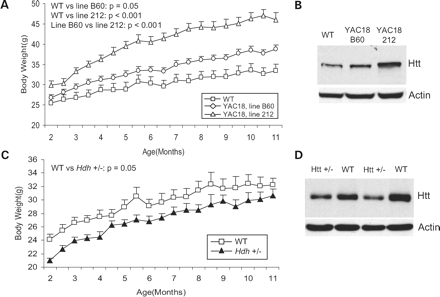
Figure 1. Wild-type huntingtin expression influences body weight. In order to identify overt phenotypes caused by over-expression of wild-type htt, we examined two lines of YAC18 mice that over-express htt with 18 glutamines. (A) Both YAC18, Line B60 mice and YAC18, Line 212 mice weighed significantly more than WT mice. (11 months—WT: 34.5±1.6 g, Line 212: 46.5±1.4 g, Line B60: 40.6±1.1 g, P<0.001, N=9 WT, 14 Line B60, 16 Line 212). (B) Examination of total htt expression in brain revealed that increased expression of htt resulted in a dose-dependent increase in body weight as Line B60 mice express more htt than WT and Line 212 mice express more htt than Line B60. (C) To determine if reductions in htt expression also affected body weight, we examined mice heterozygous for the targeted inactivation of the mouse HD gene (Hdh +/− mice) and found that Hdh +/− mice weighed significantly less than WT mice (2 months—WT: 24.1±0.4 g, Hdh +/−: 21.0±0.8 g, P=0.003, N=12 WT, 10 Hdh +/−). (D) Western blotting with whole brain lysates confirmed that Hdh +/− mice have reduced levels of wild-type htt expression. Thus, full-length wild-type htt levels have a dose-dependent effect on body weight. Error bars show standard error of the mean.
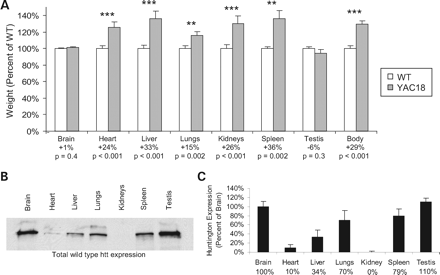
Figure 2. Wild-type huntingtin expression influences organ weight. Organs were collected from 12-month-old YAC18, Line 212 and WT mice and weighed. (A) Organ weight in the YAC18 mice was greater than WT for the heart, liver, lungs, kidneys and spleen (Heart—WT: 187±4 mg, YAC18: 231±7 mg, P<0.001; Liver—WT: 1.94±0.06 g, YAC18: 2.59±0.11 g, P<0.001; Lungs—WT: 420±12 mg, YAC18: 485±15 mg, P=0.002; Kidneys—WT: 673±13 mg, YAC18: 848±26 mg, P<0.001; Spleen—WT: 108±2 mg, YAC18: 147±11, P=0.002). In contrast, over-expression of wild-type htt did not increase the weight of the brain or testis (Brain—403±3 mg, YAC18: 407±4 mg, P=0.4; Testis—WT: 161±4 mg, YAC18: 152±7 mg, P=0.3). N=34 WT, 21 YAC18. (B) A western blot for total htt expression in organs of YAC18 mice revealed that htt expression was highest in the brain and testis, the two organs that did not show increased weight in the YAC18 mice. Among the other organs, the spleen and lungs had the highest level of htt expression. (C) These differences were confirmed by quantification of band densities from the western blot (Htt expression as percentage of brain—Brain: 100±11%; Heart: 10±7%; Liver: 34±15%; Lungs: 70±21%; Kidney: 0±2%; Spleen: 79±16%; Testis: 110±9%; N=4). Error bars show standard error of the mean. **P<0.01; ***P<0.001.
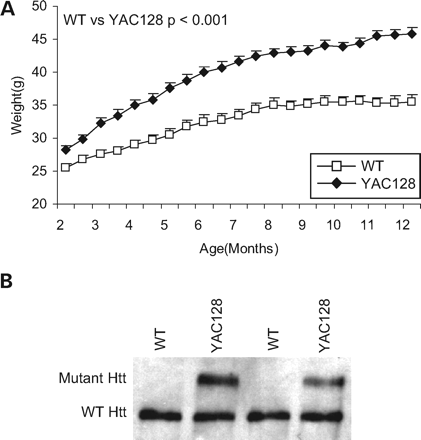
Figure 3. Mutant huntingtin over-expression increases body weight. (A) YAC128 mice over-expressing mutant huntingtin weigh significantly more than wild-type littermate controls starting at 2 months of age (2 months—WT: 25.6±0.6 g, YAC128: 28.3±0.5 g, P=0.001; 12 months—WT: 35.5±1.1 g, YAC128: 45.9±0.8 g, P<0.001; N=20 WT, 31 YAC128). (B) Examination of total htt expression in the brain confirms that htt levels are increased in YAC128 mice when compared with WT mice. Error bars show standard error of the mean.
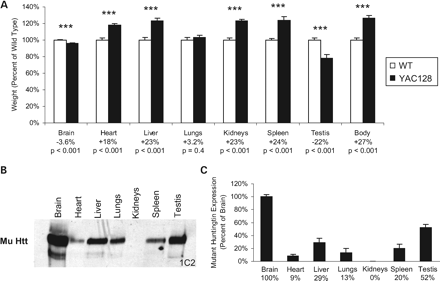
Figure 4. Mutant huntingtin expression increases organ weight except in the brain and testis. (A) The weight of the heart, liver, kidneys and spleen of YAC128 mice were significantly increased when compared with WT mice, whereas lung mass was similar (Heart—WT: 183±4 mg, YAC128: 216±3 mg, P<0.001; Liver—WT: 1.90±0.06 g, YAC128: 2.34±0.06 g, P<0.001; Lungs—WT: 412±1 mg, YAC128: 425±1 mg, P=0.4; Kidneys—WT: 655±15 mg, YAC128: 807±12 mg, P<0.001; Spleen—WT: 106±2 mg, YAC128: 132±4 mg, P<0.001; N=34 WT, 37 YAC128). In contrast, both the brain and testis of YAC128 mice showed significant atrophy when compared with WT mice (Brain—WT: 400±3 mg, YAC128: 386±3 mg, P<0.001, N=32 WT, 35 YAC128; Testis—WT: 161±4 mg, YAC128: 126±8 mg, P<0.001, N=12 WT, 12 YAC128). (B) Examination of htt expression in each organ revealed the highest levels of mutant htt expression in the brain and testis, the two regions demonstrating toxicity. (C) Differences in expression levels were confirmed by quantification of band densities (Htt expression as percentage of brain—Brain: 100±4%; Heart: 9±2%; Liver: 29±6%; Lungs: 13±7%; Kidney: 0±0%; Spleen: 20±6%; Testis: 52±5%; N=4). Error bars show standard error of the mean. ***P<0.001.
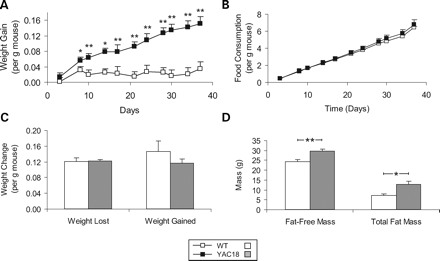
Figure 5. Characterization of weight gain phenotype in YAC18 mice. Three-month-old YAC18 and WT mice were examined. (A) YAC18 mice gain weight significantly faster than WT mice. (B) This does not result from increased food consumption as the curves of total food consumption over time are almost exactly parallel. (C) During a 24 h fast, YAC18 and WT mice lose weight at the same rate, indicating similar metabolic rates of lean tissue. During the refeeding period there is also no significant difference in the rate at which YAC18 and WT mice regain weight. (D) Analysis of body composition reveals that the increased weight in YAC18 mice is a result of increases in both fat-free mass and in total fat mass. N=4 WT, 6 YAC18. Error bars indicate standard error of the mean. *P<0.05, **P<0.01.
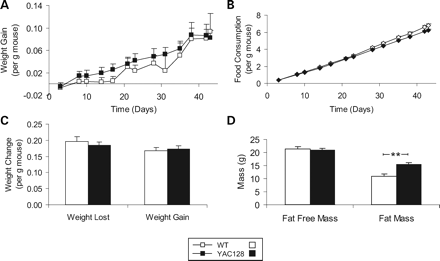
Figure 6. Characterization of weight gain phenotype in YAC128 mice. Three-month-old YAC128 and WT mice were examined. (A) Weight gain relative to body weight is equivalent between YAC128 and WT mice. (B) Food consumption was also equivalent between YAC128 and WT mice as indicated by parallel lines in the graph of food consumption over time. (C) During a 24 h fast and a 24 h refeeding period the rate of weight loss and weight gain were similar between YAC128 and WT mice suggesting no difference in the metabolic rate of lean tissue. (D) Fat-free mass was equivalent between YAC128 and WT mice, whereas YAC128 mice had significantly more fat than WT mice. N=8 WT, 7 YAC128. Error bars indicate standard error of the mean. **P<0.01.
References
Nasir, J., Floresco, S.B., O'Kusky, J.R., Diewert, V.M., Richman, J.M., Zeisler, J., Borowski, A., Marth, J.D., Phillips, A.G. and Hayden, M.R. (
Cattaneo, E., Rigamonti, D., Goffredo, D., Zuccato, C., Squitieri, F. and Sipione, S. (
Cattaneo, E., Zuccato, C. and Tartari, M. (
Zeitlin, S., Liu, J.P., Chapman, D.L., Papaioannou, V.E. and Efstratiadis, A. (
Duyao, M.P., Auerbach, A.B., Ryan, A., Persichetti, F., Barnes, G.T., McNeil, S.M., Ge, P., Vonsattel, J.P., Gusella, J.F. and Joyner, A.L. (
O'Kusky, J.R., Nasir, J., Cicchetti, F., Parent, A. and Hayden, M.R. (
White, J.K., Auerbach, W., Duyao, M.P., Vonsattel, J.P., Gusella, J.F., Joyner, A.L. and Macdonald, M.E. (
Auerbach, W., Hurlbert, M.S., Hilditch-Maguire, P., Wadghiri, Y.Z., Wheeler, V.C., Cohen, S.I., Joyner, A.L., Macdonald, M.E. and Turnbull, D.H. (
Dragatsis, I., Levine, M.S. and Zeitlin, S. (
Rigamonti, D., Bauer, J.H., De Fraja, C., Conti, L., Sipione, S., Sciorati, C., Clementi, E., Hackam, A., Hayden, M.R., Li, Y. et al. (
Ho, L.W., Brown, R., Maxwell, M., Wyttenbach, A. and Rubinsztein, D.C. (
Leavitt, B.R., Guttman, J.A., Hodgson, J.G., Kimel, G.H., Singaraja, R., Vogl, A.W. and Hayden, M.R. (
Zhang, Y., Ona, V.O., Li, M., Drozda, M., Dubois-Dauphin, M., Przedborski, S., Ferrante, R.J. and Friedlander, R.M. (
Leavitt, B.R., Van Raamsdonk, J.M., Shehadeh, J., Fernandes, H.B., Graham, R.K., Murphy, Z., Raymond, L. and Hayden, M.R. (
Li, S.H. and Li, X.J. (
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B.R., Goffredo, D., Conti, L., Macdonald, M.E., Friedlander, R.M., Silani, V., Hayden, M.R. et al. (
Zuccato, C., Tartari, M., Crotti, A., Goffredo, D., Valenza, M., Conti, L., Cataudella, T., Leavitt, B.R., Hayden, M.R., Timmusk, T. et al. (
Szebenyi, G., Morfini, G.A., Babcock, A., Gould, M., Selkoe, K., Stenoien, D.L., Young, M., Faber, P.W., Macdonald, M.E., McPhaul, M.J. and Brady, S.T. (
Gunawardena, S., Her, L.S., Brusch, R.G., Laymon, R.A., Niesman, I.R., Gordesky-Gold, B., Sintasath, L., Bonini, N.M. and Goldstein, L.S. (
Gauthier, L.R., Charrin, B.C., Borrell-Pages, M., Dompierre, J.P., Rangone, H., Cordelieres, F.P., De Mey, J., Macdonald, M.E., Lessmann, V., Humbert, S. and Saudou, F. (
Trushina, E., Dyer, R.B., Badger, J.D., Ure, D., Eide, L., Tran, D.D., Vrieze, B.T., Legendre-Guillemin, V., McPherson, P.S., Mandavilli, B.S. et al. (
Hodgson, J.G., Agopyan, N., Gutekunst, C.A., Leavitt, B.R., LePiane, F., Singaraja, R., Smith, D.J., Bissada, N., McCutcheon, K., Nasir, J. et al. (
Van Raamsdonk, J.M., Pearson, J., Rogers, D.A., Bissada, N., Vogl, A.W., Hayden, M.R. and Leavitt, B.R. (
Wheeler, V.C., White, J.K., Gutekunst, C.A., Vrbanac, V., Weaver, M., Li, X.J., Li, S.H., Yi, H., Vonsattel, J.P., Gusella, J.F. et al. (
Lin, C.H., Tallaksen-Greene, S., Chien, W.M., Cearley, J.A., Jackson, W.S., Crouse, A.B., Ren, S., Li, X.J., Albin, R.L. and Detloff, P.J. (
Hodgson, J.G., Smith, D.J., McCutcheon, K., Koide, H.B., Nishiyama, K., Dinulos, M.B., Stevens, M.E., Bissada, N., Nasir, J., Kanazawa, I. et al. (
Slow, E.J., van Raamsdonk, J., Rogers, D., Coleman, S.H., Graham, R.K., Deng, Y., Oh, R., Bissada, N., Hossain, S.M., Yang, Y.Z. et al. (
Van Raamsdonk, J.M., Pearson, J., Slow, E.J., Hossain, S.M., Leavitt, B.R. and Hayden, M.R. (
Slow, E.J., Graham, R.K., Osmand, A.P., Devon, R.S., Lu, G., Deng, Y., Pearson, J., Vaid, K., Bissada, N., Wetzel, R. et al. (
Fain, J.N., Del Mar, N.A., Meade, C.A., Reiner, A. and Goldowitz, D. (
Hurlbert, M.S., Zhou, W., Wasmeier, C., Kaddis, F.G., Hutton, J.C. and Freed, C.R. (
Qian, S., Chen, H., Weingarth, D., Trumbauer, M.E., Novi, D.E., Guan, X., Yu, H., Shen, Z., Feng, Y., Frazier, E. et al. (
Sharp, A.H., Loev, S.J., Schilling, G., Li, S.H., Li, X.J., Bao, J., Wagster, M.V., Kotzuk, J.A., Steiner, J.P. and Lo, A. (
Reiner, A., Del Mar, N., Meade, C.A., Yang, H., Dragatsis, I., Zeitlin, S. and Goldowitz, D. (
Metzler, M., Helgason, C.D., Dragatsis, I., Zhang, T., Gan, L., Pineault, N., Zeitlin, S.O., Humphries, R.K. and Hayden, M.R. (
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S.W. and Bates, G.P. (
Schilling, G., Becher, M.W., Sharp, A.H., Jinnah, H.A., Duan, K., Kotzuk, J.A., Slunt, H.H., Ratovitski, T., Cooper, J.K., Jenkins, N.A. et al. (
Zhang, Y., Li, M., Drozda, M., Chen, M., Ren, S., Mejia Sanchez, R.O., Leavitt, B.R., Cattaneo, E., Ferrante, R.J., Hayden, M.R. and Friedlander, R.M. (
Reddy, P.H., Williams, M., Charles, V., Garrett, L., Pike-Buchanan, L., Whetsell, W.O., Jr, Miller, G. and Tagle, D.A. (
Djousse, L., Knowlton, B., Cupples, L.A., Marder, K., Shoulson, I. and Myers, R.H. (
Morales, L.M., Estevez, J., Suarez, H., Villalobos, R., Chacin, d.B. and Bonilla, E. (
Sanberg, P.R., Fibiger, H.C. and Mark, R.F. (
Pratley, R.E., Salbe, A.D., Ravussin, E. and Caviness, J.N. (
Beal, M.F. (
Dixon, K.T., Cearley, J.A., Hunter, J.M. and Detloff, P.J. (
Schilling, G., Sharp, A.H., Loev, S.J., Wagster, M.V., Li, S.H., Stine, O.C. and Ross, C.A. (
Ona, V.O., Li, M., Vonsattel, J.P., Andrews, L.J., Khan, S.Q., Chung, W.M., Frey, A.S., Menon, A.S., Li, X.J., Stieg, P.E. et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}