




Abstract
Pitt–Hopkins syndrome (PHS) is a rare syndromic mental disorder, which is mainly characterized by severe motor and mental retardation including absent language development, a characteristic facial gestalt and episodes of hyperventilation. We report on a female patient with PHS showing severe mental retardation with absent speech, pronounced muscular hypotonia, ataxia, distinctive facial features, such as a coarse face, a broad nasal bridge and a wide mouth, and hyperventilation attacks. In this patient, genomic profiling by array-based comparative genomic hybridization and fluorescence in situ hybridization studies detected and confirmed a de novo 0.5 Mb deletion in 18q21.2 containing a single gene, the basic helix-loop-helix transcription factor TCF4 . cDNA and genomic analyses in the patient and her parents demonstrated TCF4 haploinsufficiency as the underlying cause of the disease. Analysis of the embryonal expression pattern of the Danio rerio ortholog, tcf4 , by whole-mount in situ hybridization showed a highly specific expression domain in the pallium of the telencephalon during late somitogenesis, when the patterning of the zebrafish brain is advanced and neural differentiation commences. Later expression domains were restricted to several regions in the central nervous system, including continued expression in the pallium of the telencephalon, and starting expression in the diencephalon (thalamus, ventral thalamus and posterior tuberculum), the midbrain tegmentum, the hindbrain and the branchial arches. This expression pattern correlates with the clinical phenotype. Our results show that haploinsufficiency of TCF4 causes PHS and suggest that D. rerio is a valuable model to study the molecular pathogenesis of PHS and the role of TCF4 in brain development.
INTRODUCTION
Pitt–Hopkins syndrome (PHS) is a syndromic type of severe motor and mental retardation (MR) of unknown molecular cause, which was first described by Pitt and Hopkins in 1978 ( 1 ). PHS patients display a characteristic combination of symptoms consisting of (i) severe MR with absent speech, (ii) a characteristic facial gestalt (a coarse face with a broad and beaked nasal bridge, a large mouth with a bow-shaped upper lip, everted lower lip and cup-shaped, fleshy ears) and (iii) typical breathing abnormalities with episodes of voluntary hyperventilation and apnea starting in childhood. In addition, subtle structural brain abnormalities, seizures, postnatal microcephaly and growth retardation, muscular hypotonia and motor retardation, intestinal problems and a happy disposition have been found in PHS patients ( 2–5 ). Although it is a potentially recognizable clinical entity, PHS seems to be underdiagnosed with just a very limited number of patients described so far, and there is currently no MIM entry for this disorder.
Clinically, PHS is similar to common MR syndromes like Angelman syndrome (MIM 105830), Rett syndrome (MIM 312750), or Mowat-Wilson syndrome (MIM 235730). Thus, to reliably distinguish PHS from these syndromes, the elucidation of the underlying molecular alteration is of great importance because this will allow genetic testing. Moreover, the knowledge of the causative gene could help to better understand the molecular (patho)mechanisms of severe MR, speech development and breathing regulation. PHS is thought to be an autosomal dominant disorder caused by de novo mutations. Thus, it is a plausible hypothesis that larger genomic rearrangements may cause the disease.
We were now able to identify a de novo microdeletion of ∼0.5 Mb on chromosome 18q21.2 by array-based comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH) analysis in a patient with typical PHS. The only known gene within this deletion is the transcription factor 4 ( TCF4 ; MIM 602272, also known as E2-2 , ITF2 , ME2 or SEF2 ). This gene should not be confused with the T-cell transcription factor 4 on human chromosome 10q25.3 (MIM 602228), which was formerly termed TCF4 , but is now designated TCF7L2 . The TCF4 gene on human chromosome 18 encodes a member of the basic helix-loop-helix (bHLH) transcription factor family ( 6 ) bHLH transcription factors are able to bind DNA as homo- or heterodimers and are key players in a variety of developmental processes including the differentiation of the vertebrate nervous system and the development of the cortex ( 7 , 8 ). They bind to the Ephrussi-box (‘E-box’) consensus binding site (‘CANNTG’). This is a motif that was first identified in immunoglobulin enhancers, but also critically involved in muscle- and neuron-specific gene expression. tcf4 is expressed during mouse brain development and in the adult mouse brain ( 9 ) and interacts with the rat tyrosine hydroxylase enhancer ( 10 ), which is well compatible with symptoms observed in PHS patients. We could show by RT–PCR analysis that the microdeletion in our patient leads to a functional TCF4 haploinsufficiency, confirming TCF4 as the PHS gene. This finding was very recently described by two other groups ( 11 , 12 ). In addition to the genetic data, we were interested in the expression pattern of TCF4 in early brain development. Therefore, we subcloned a part of the formerly unknown zebrafish ortholog of TCF4 and analyzed the expression by whole-mount in situ hybridization (WISH) on 1, 2 and 3 day post-fertilization (dpf) embryos. Interestingly, tcf4 showed a very early expression at 1 dpf in a highly specific manner in the pallium of the telencephalon, which in evolution is the precursor of the cortex, and later on in additional brain regions and the branchial arches, which may explain the severe MR and facial gestalt in PHS patients.
RESULTS
Molecular cytogenetic and molecular genetic analyses in the index patient with PHS, her parents and TCF4 mutation analysis in patients with idiopathic MR (IMR)
By genome-wide array-based CGH, we detected a deletion comprising two adjacent BAC clones, RP11-7L24 and RP11-397A16, on chromosome 18q21.2 in the index patient with PHS (Fig. 1 B). This alteration was independently confirmed by FISH analysis using the same BAC clones on patient's metaphase chromosomes (Fig. 1 C, left). FISH studies on her parents' metaphase chromosomes demonstrated that the microdeletion had occurred de novo (Fig. 1 C, middle and right). To better characterize the extent of the aberration, we performed further FISH experiments with adjacent BAC probes. Both the proximal and the distal breakpoints were finemapped with altogether six overlapping BAC clones, and the deletion was shown to be ∼0.5 Mb in size. On the centromeric boundary, clone RP11-746K23 was not deleted; on the telomeric side, BAC clone RP11-659K3 showed two signals on the patient's chromosomes (Fig. 2 ). This deletion is much smaller than those described in other patients with PHS ( 11 , 12 ), and is the only structural alteration just including TCF4 , but no other known genes (Fig. 2 ). In fact, the centromeric breakpoint seemed to be within the TCF4 gene thereby deleting the 5′-end of the gene, whereas the 3′-part was suspected to be intact leaving the possibility of a shortened transcript/protein with dominant negative effect.
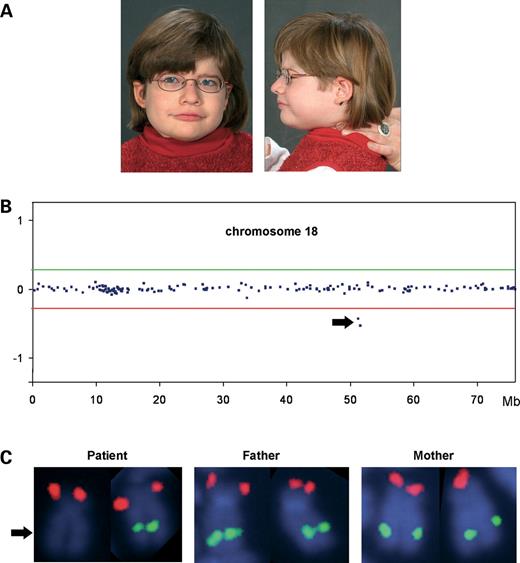
Index patient and results of the molecular cytogenetic analyses. ( A ) Facial appearance of the index patient at the age of 6 years. Note the coarse face with broad nasal bridge, large mouth with bow-shaped upper lip and the slightly dysmorphic ears with anteverted earlobes. Owing to extreme muscular hypotonia, head had to be supported by a parent. ( B ) Profile of chromosome 18 in the index patient showing the microdeletion detected by array-based CGH with an 8k large insert clone DNA microarray. Midpoints of all clones on the chromosome are plotted in genomic order from the p- to the q-terminal arm on the x -axis against their normalized log 2 test to reference ratio on the y -axis. Diagnostic thresholds used were ± 7 standard deviations from the mean, and are given as horizontal lines (red line, lower threshold for deleted clones; green line, upper threshold for duplicated clones). The two deleted clones are indicated by an arrow. ( C ) Deletion confirmation in the index patient by FISH showing a deleted BAC probe RP11-397A16 (from 18q21.2, green) on one homolog of chromosome 18 (arrow). RP11-324G2 (from 18p, red) served as a control probe and showed signals on both chromosomes 18 in the index patient. A combination of the same BAC probes hybridized to her parents' metaphase chromosomes displayed regular signal patterns on both chromosomes 18, thus demonstrating the de novo occurrence of the deletion.
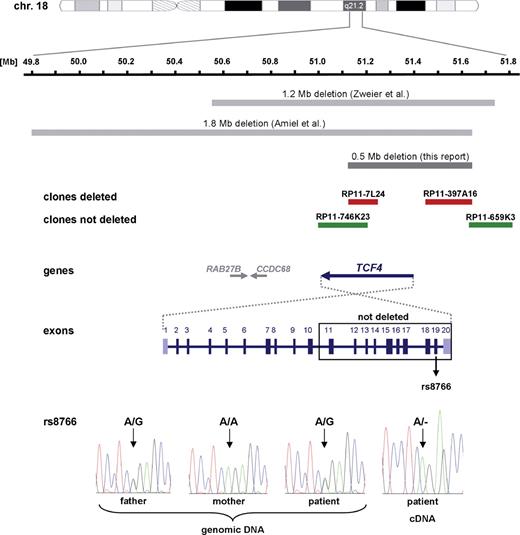
Graphical overview of the deleted region on chromosome 18q21.2 in this study (dark gray bar) and in the reports published recently by Zweier et al. ( 11 ) and Amiel et al. ( 12 ) (light gray bars). Deleted BAC probes are depicted as red bars and non-deleted as green bars. In the schematically drawn genomic structure of the TCF4 gene, coding exons 2–19 are colored in dark blue and non-coding exons 1 and 20 in purple and light blue, respectively. In the electropherograms of the genomic DNA of the index patient and her parents and of the patient's cDNA, the exonic SNP rs8766 (exon 19) is marked by an arrow.
To further define the intragenic breakpoint and to address the question whether the deletion was maternal or paternal in origin, we performed amplification and direct sequencing of all coding exons and adjacent splice sites of TCF4 in the patient and her parents. We could identify a heterozygous status of SNPs rs1788027 in intron 10 and rs8766 in exon 19 in the patient, demonstrating that at least exons 11–20 were not deleted (Fig. 2 ). With respect to the exonic SNP, the father was shown to be heterozygous A/G, whereas the mother was homozygous A/A (Fig. 2 , bottom), which meant an informative situation for a cDNA analysis. We therefore amplified a part of TCF4 comprising exons 17–20 on cDNA from whole blood of the patient. Direct sequencing of the single polymerase chain reaction (PCR) product revealed hemizygosity for rs8766 with expression of the A allele only (Fig. 2 ), proving that the TCF4 copy on the derivative chromosome 18 is not transcribed. Moreover, we could show that the deletion was of paternal origin.
We also performed a systematic mutation screen of the 18 coding exons and flanking intronic sites of TCF4 in 46 patients with IMR and at least part of our patient's phenotypic spectrum, such as speech delay, ataxia or muscular hypotonia (see Supplementary Material, Table S1). However, none of these patients had a suspected diagnosis of PHS. No pathogenic alterations of TCF4 were detected in any of these patients.
tcf4 expression analysis in Danio rerio (zebrafish)
To study the role of TCF4 in early development, we wanted to analyze its expression pattern in zebrafish. As the TCF4 ortholog has not been described yet, we performed BLAST analyses on EST and genomic data from D. rerio to predict a part of the zebrafish ortholog tcf4 . We could identify an 801 bp spanning fragment predicted to encode 267 amino acids of a central part of tcf4 (Supplementary Material, Fig. S1A). A BLASTP search confirmed that this fragment is most closely related to TCF4 orthologs of different species with an overall identity to the human ortholog of 79% (Supplementary Material, Fig. S1B). A multiple sequence alignment with TCF4 orthologs from Homo sapiens , Mus musculus , and the predicted fragment of D. rerio with the respective homologous bHLH transcription factors TCF3 and TCF12 from all three species confirmed that the predicted fragment represents the zebrafish ortholog of TCF4 (the corresponding dendrogram built by Clustal_X is depicted in Supplementary Material, Fig. S1C).
WISHs were performed with a 468 bp spanning antisense probe based on the zebrafish tcf4 sequence fragment identified. At 1 dpf (late somitogenesis stage), when the patterning of the brain is advanced and neural differentiation commences, tcf4 expression was found in a single domain in the dorsal telencephalon (pallium) across the telencephalic/diencephalic border (Fig. 3 A–C). At this stage, tcf4 is not expressed elsewhere in the brain or in the trunk region. At 2 dpf and 3 dpf, tcf4 continues to be expressed in the dorsal telencephalon (pallium), and starts to be expressed in additional regions in the brain. At 2 dpf, tcf4 is expressed faintly in the hindbrain, and stronger in multiple regions in the diencephalon, including thalamus, ventral thalamus and posterior tuberculum, and in the midbrain tegmentum. tcf4 is not expressed in the diencephalic preoptic region or in the hypothalamus (Fig. 3 D), whereas it is expressed in the retina, as well as the branchial arches (Fig. 3 E–F, arrowheads). At 3 dpf, tcf4 is expressed in the dorsal telencephalon, diencephalon (thalamus, ventral thalamus and posterior tuberculum), midbrain tegmentum, hindbrain (cerebellum and at low level in medulla) and in the inner nuclear layer of the retina (Fig. 3 G–H). No expression is detected in the trunk region at 2 and 3 dpf.
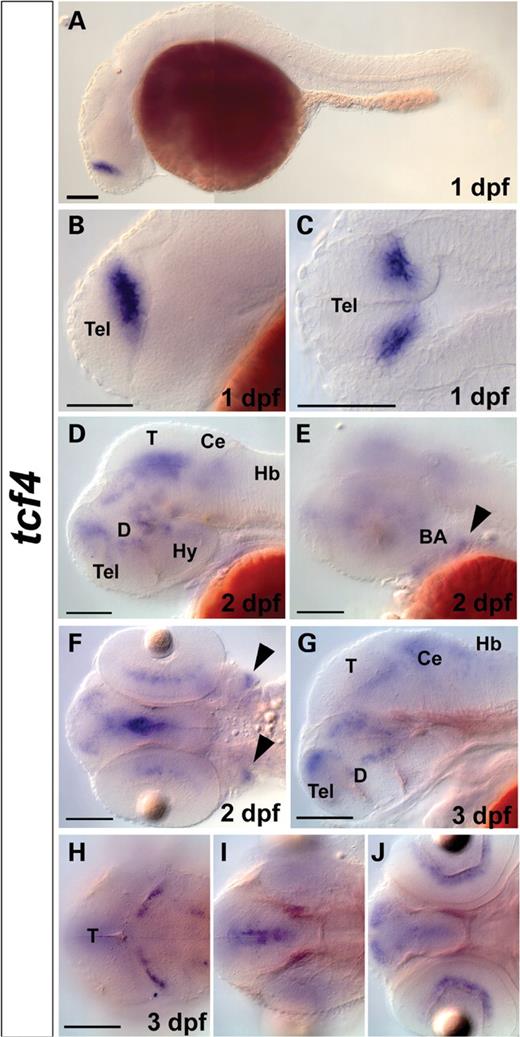
Whole-mount in situ hybridization (WISH) of tcf4 in zebrafish embryos. ( A – C ) At 1 day post-fertilization (dpf), tcf4 is expressed in a single domain in dorsal telencephalon. At 2 dpf ( D – F ) and 3 dpf ( G – H ), tcf4 is expressed in the retina, telencephalon, hindbrain and multiple regions in the midbrain and the diencephalon. At 2 dpf, tcf4 is expressed in branchial arches (black arrowheads in E and F). (A, B, D, E, G) Lateral views. (C) Flat-mounted dorsal view. (F, H – J ) Dorsal views. (H–J) Dorsal views at different planes with (H) representing most dorsal and (J) most ventral plane of tcf4 expression. Scale bars represent 100 µm. BA, branchial arches; Ce, cerebellum; D, diencephalon; Hb, hindbrain; Hy, hypothalamus; T, tectum; Tel, telencephalon.
DISCUSSION
Using array-based CGH, we identified a novel microdeletion on the long arm of chromosome 18 in a 7-year-old girl with severe MR and absence of speech, muscular hypotonia, ataxia, breathing abnormalities and a coarse face, thus suspected to have PHS. The microdeletion at 18q21.2 was confirmed by FISH studies and found to be de novo making a causal involvement likely, particularly as there is no known copy number variation in this chromosomal region. The extent of the deletion was further characterized by FISH, shown to be 0.5 Mb in size and to include parts of the TCF4 gene. This transcription factor is an excellent candidate gene for the MR and abnormalities of the autonomic nervous system, which were observed in our patient and are characteristic for PHS, for two reasons: (i) TCF4 is the only gene in the small deleted region and (ii) it is expressed in the brain and interacts with the tyrosine hydroxylase enhancer ( 10 ). As TCF4 expression is found mostly in the brain in embryonic human tissues ( 13 ) and is abundant in the adult human brain ( 14 ), it seems to play an important role in neural development and differentiation.
By cDNA analysis, we could then show that the TCF4 copy on the derivative chromosome 18 is not transcribed in our patient. That in fact TCF4 is the gene responsible for PHS has independently been found by two other groups ( 11 , 12 ), who identified larger genomic rearrangements, nonsense and missense mutations in a total of 10 cases with suspected PHS. The combined data of our report and the two other studies confirm that haploinsufficiency is the most probable mechanism of disease development. Interestingly, homozygous tcf4 knockout mice show early lethality because of unspecified reasons, whereas heterozygous tcf4+/− mice were not reported to have any obvious phenotype ( 15 ), a finding which should be re-evaluated in the light of the novel molecular data on PHS.
Concerning the clinical presentation, it is now clear that virtually all PHS patients share the severe MR with absent speech, a typical and coarse facial gestalt and the breathing abnormalities, whereas other possible features are more variable between different patients (see Table 1 ). However, because the breathing abnormalities may not be present in young children, which was also true in our patient, the diagnosis of PHS may be delayed, since this symptom is often crucial for the clinical diagnosis. After gene identification, it will now be possible to prove or exclude this differential diagnosis by genetic testing in suspected cases even before the onset of hyperventilation, which will be of great value for genetic counseling and further medical management of PHS patients. As larger genomic rearrangements do not seem to be uncommon, the genetic testing strategy will have to include (i) techniques to identify larger deletions like multiplex ligation-dependent probe amplification (MLPA) technology and (ii) direct sequencing especially of the highly conserved C-terminal part of the gene. Nevertheless, our study and the study of Zweier et al . ( 11 ) indicate that TCF4 mutations are not a common cause for IMR or other syndromes with overlapping clinical features, so that a more general screening of TCF4 in severe MR is currently not warranted.
| Clinical features | PHS patient with deletion in TCF4 (this study) | PHS patients with TCF4 mutations [Zweier et al . ( 11 ) and Amiel et al . ( 12 )] ( n = 10) |
|---|---|---|
| Postnatal growth | ||
| Short stature | − | 4/10 |
| Dystrophy | − | 2/8 |
| Microcephaly | − | 8/10 |
| PHS facial gestalt | + | 10/10 |
| Severe mental retardation | + | 10/10 |
| Severe motor retardation | + | 6/6 |
| Speech delay/absence | + | 6/6 |
| Muscular hypotonia | + | 6/6 |
| Ataxia | + | 2/6 |
| Intestinal anomalies | − | 7/10 |
| Breathing anomalies (age of onset) | + (7 years) | 8/10 (2–8 years) |
| Epilepsy | − | 5/10 |
| Simian crease | + | 8/10 |
| Myopia | + | Not analyzed |
| Brain MRI anomalies | − | 7/7 |
| Clinical features | PHS patient with deletion in TCF4 (this study) | PHS patients with TCF4 mutations [Zweier et al . ( 11 ) and Amiel et al . ( 12 )] ( n = 10) |
|---|---|---|
| Postnatal growth | ||
| Short stature | − | 4/10 |
| Dystrophy | − | 2/8 |
| Microcephaly | − | 8/10 |
| PHS facial gestalt | + | 10/10 |
| Severe mental retardation | + | 10/10 |
| Severe motor retardation | + | 6/6 |
| Speech delay/absence | + | 6/6 |
| Muscular hypotonia | + | 6/6 |
| Ataxia | + | 2/6 |
| Intestinal anomalies | − | 7/10 |
| Breathing anomalies (age of onset) | + (7 years) | 8/10 (2–8 years) |
| Epilepsy | − | 5/10 |
| Simian crease | + | 8/10 |
| Myopia | + | Not analyzed |
| Brain MRI anomalies | − | 7/7 |
n , number of patients; MRI, magnetic resonance imaging.
| Clinical features | PHS patient with deletion in TCF4 (this study) | PHS patients with TCF4 mutations [Zweier et al . ( 11 ) and Amiel et al . ( 12 )] ( n = 10) |
|---|---|---|
| Postnatal growth | ||
| Short stature | − | 4/10 |
| Dystrophy | − | 2/8 |
| Microcephaly | − | 8/10 |
| PHS facial gestalt | + | 10/10 |
| Severe mental retardation | + | 10/10 |
| Severe motor retardation | + | 6/6 |
| Speech delay/absence | + | 6/6 |
| Muscular hypotonia | + | 6/6 |
| Ataxia | + | 2/6 |
| Intestinal anomalies | − | 7/10 |
| Breathing anomalies (age of onset) | + (7 years) | 8/10 (2–8 years) |
| Epilepsy | − | 5/10 |
| Simian crease | + | 8/10 |
| Myopia | + | Not analyzed |
| Brain MRI anomalies | − | 7/7 |
| Clinical features | PHS patient with deletion in TCF4 (this study) | PHS patients with TCF4 mutations [Zweier et al . ( 11 ) and Amiel et al . ( 12 )] ( n = 10) |
|---|---|---|
| Postnatal growth | ||
| Short stature | − | 4/10 |
| Dystrophy | − | 2/8 |
| Microcephaly | − | 8/10 |
| PHS facial gestalt | + | 10/10 |
| Severe mental retardation | + | 10/10 |
| Severe motor retardation | + | 6/6 |
| Speech delay/absence | + | 6/6 |
| Muscular hypotonia | + | 6/6 |
| Ataxia | + | 2/6 |
| Intestinal anomalies | − | 7/10 |
| Breathing anomalies (age of onset) | + (7 years) | 8/10 (2–8 years) |
| Epilepsy | − | 5/10 |
| Simian crease | + | 8/10 |
| Myopia | + | Not analyzed |
| Brain MRI anomalies | − | 7/7 |
n , number of patients; MRI, magnetic resonance imaging.
To elicit the expression pattern in embryonal development, we chose D. rerio as a model organism, which has not been analyzed concerning tcf4 expression or function. Since the orthologous zebrafish gene had not been identified previously, we predicted a part of the cDNA sequence by BLAST analyses on expressed sequence tag (EST) and genomic data from D. rerio to generate a probe for WISH of tcf4 in zebrafish embryos. Interestingly, already early in development, during late somitogenesis stages (1 dpf), tcf4 was solely expressed in the pallium, which represents the fish embryonic equivalent of the mammalian cortex. In mammals, the cortex later subdivides into the paleo-, archi- and neocortex. These domains affect crucial tasks, like sensomotor functions, perhaps giving rise to an MR phenotype and to hypotonia or ataxia in case of a misdevelopment. In later zebrafish development, tcf4 is also expressed in the branchial arches, which form parts of the facial musculature, thus contributing to craniofacial shape in mammals, which may correlate to the characteristic facial anomalies described in PHS. Notably, tcf4 expression emerges at a time when neuronal differentiation commences in the zebrafish brain, indicating that tcf4 may be involved in differentiation of specific neuronal populations. Together with the known interaction of TCF4 with the tyrosine hydroxylase enhancer ( 10 ) and the functional interaction of TCF4 with achaete-scute complex, drosophila, homolog of, 1 (ASCL1) ( 11 ), a regulator of sympathetic and enteric precursor cells, a crucial role of TCF4 for the autonomic nervous system seems reasonable, which again is well compatible with the phenotype seen in PHS patients. Whether the early expression in the retina found in zebrafish may mean a risk of a retinal disease in PHS patients is currently unknown, however, a respective clinical examination in patients with this syndrome may be warranted based on the expression data.
In conclusion, our study identifies TCF4 haploinsufficiency as the cause of PHS and shows that D. rerio may be a valuable model to study the pathogenesis of this syndrome and the role of TCF4 in brain development.
MATERIALS AND METHODS
Clinical data
The index patient was born spontaneously after an uneventful pregnancy in the 41st gestational week with a birth weight of 4330 g (97th percentile), length of 58 cm (90th percentile) and head circumference of 36.5 cm (90th percentile). She showed a severe delay of motor development with no crawling and only assisted walking at the age of 5 years. Upon examination at the age of 7 years, she had severe MR with absent speech. She suffered from pronounced muscular hypotonia, an ataxia mainly of the trunk, and could merely walk with assistance. The facial features include a coarse face with a broad and slightly depressed nasal bridge, a wide mouth with a bow-shaped upper lip and a short philtrum (Fig. 1 A). The ears were slightly dysplastic with anteverted earlobes. She had a short neck and low frontal and nuchal hairlines. Her nipples were widely spaced. She had long, slightly tapering fingers with finger pads, simian creases and a proximal-inserted thumb on both hands. Her feet were flat with superimposed toes and a moderate syndactyly of toes II and III. Her body measurements were: weight 26 kg (75–90th percentile), length 116 cm (10th percentile) and head circumference 52 cm (75th percentile). She was not affected by gastrointestinal problems. Her disposition was happy and friendly. Hyperventilation attacks commenced at age 7.5 years. Magnetic resonance imaging (MRI) revealed no obvious brain anomalies. Initially suspected epilepsy was excluded at the age of 7 years, and EEG findings were reported normal. Furthermore, the patient was found to have a pronounced myopia (−6.5/−7 dpt). Conventional cytogenetic analyses and a subtelomere screening by FISH gave normal results. Rett and Angelman syndromes were excluded and comprehensive metabolic studies revealed no pathogenic results.
In addition to the index patient, 46 patients (29 males, 17 females) with IMR, at least part of our patient's phenotypic spectrum, but no suspected diagnosis of PHS were tested for mutations in TCF4 (for clinical characteristics of this patient sample see Supplementary Material, Table S1).
All participating families gave their written informed consent and the molecular study was approved by the local Ethics Committee of the Medical Faculty, Rheinische Friedrich-Wilhelms-University, Bonn, Germany.
Molecular cytogenetic analyses
We performed array-based CGH on a genomic DNA microarray comprising 8000 large insert clones (8k array). Hybridization and data analysis were carried out as described previously ( 16 ). FISH with BAC/PAC DNA was performed as described previously ( 16 ) to verify the array-CGH results, to analyze whether the deletion occurred de novo and to characterize the size of the chromosomal aberration.
Molecular genetic investigations
We elucidated the genomic structure of TCF4 using a human cDNA sequence from GenBank (NM_003199.1). All coding exons and adjacent splice sites were amplified by standard PCR on genomic DNA and directly sequenced in our index patient, her parents and 46 individuals with IMR. The sequences were compared with the GenBank entry NM_003199 and with additional genomic data (primer sequences and PCR conditions are available upon request). RNA from the index patient's and her parents' blood lymphocytes was isolated using the PAXgene blood RNA kit (Qiagen, Hilden, Germany) and cDNA was synthesized using the OneStep RT–PCR kit (Qiagen) according to manufacturer's instructions. Primers located in exons 17 and 20 were used to amplify a 497 bp cDNA fragment harboring rs8766 (located in exon 19) on the patient's RNA (forward primer: 5′-tgcaagacacgaaatcttcgg-3′, reverse primer: 5′-atacagctgtt aaggaagtgg-3′).
D. rerio in situ hybridizations
Using public sequence databases ( http://www.genome.ucsc.edu/ ), we predicted a part of the formerly unknown cDNA sequence of tcf4 in D. rerio from genomic and EST data which allowed to define a segment of 801 bp from the central part of the gene. Very recently, a novel, predicted zebrafish gene (XM_692142) including the fragment identified by us was deposited in GenBank. In addition to the homology to TCF4 , this much longer sequence, predicted from genomic data but not verified experimentally, also has strong homology to an ADAM-type metallopeptidase in a different part of the sequence. Thus, this GenBank entry most probably represents a bioinformatical artifact erroneously combining exons of tcf4 and adamts3 , which—on the genomic level—seem to be adjacent genes transcribed in the same direction. PCR amplification with deduced primer pairs on whole zebrafish embryo cDNA and direct sequencing of a 468 bp fragment confirmed our partial prediction of tcf4 and allowed subcloning in an expression vector [TOPO TA cloning system (Invitrogen)]. WISH for fish embryos with alkaline phosphatase-based color reaction was performed as described ( 17 ). Digoxigenin-labeled antisense RNA probes were prepared using RNA-labeling reagents (Roche).
SUPPLEMENTARY MATERIAL
Supplementary material is available at HMG Online.
ACKNOWLEDGEMENTS
The authors thank the affected individuals and their families, especially the index patient and her parents, for participation. We also thank Marion Ehrler and Sabrina Wolf for excellent technical assistance. This work was supported by the BONFOR program of the Medical Faculty, Rheinische Friedrich-Wilhelms University, Bonn (grant no. O-149.0081), the Doktor Robert Pfleger-Stiftung, the Deutsche Forschungsgemeinschaft (grant no. PR 131/19-1), the Bundesministerium für Bildung und Forschung (NGFN SMP-DNA 01GR0417) and the EU-IP ZF-MODELS.
Conflict of Interest statement . None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}