




Abstract
We sought to verify whether variation in the promoter of the gene encoding placental anticoagulant protein annexin A5 (ANXA5) represents a risk factor for recurrent pregnancy loss (RPL). Sequence analysis of 70 German RPL patients, all known to carry neither factor V Leiden nor a prothrombin mutation, revealed four consecutive nucleotide substitutions in the ANXA5 promoter, which were transmitted as a joint haplotype (M2). Reporter gene assays revealed that M2 reduces the in vitro activity of the ANXA5 promoter to 37–42% of the normal level. The possible relationship between M2 and RPL was evaluated by comparing RPL patients with two independent control groups recruited from the registry of the Institut für Humangenetik in Münster and the PopGen biobank in Kiel, respectively. Carriers of M2 were found to exhibit a > 2-fold higher RPL risk than non-carriers (odds ratio, 2.42; 95% confidence interval, 1.27–4.58) when using unselected controls (PopGen) and an almost 4-fold higher risk when using the Münster ‘super-controls’, i.e. women with successful pregnancies and no previous history of pregnancy losses (odds ratio, 3.88; 95% confidence interval, 1.98–7.54). This statistically significant association should facilitate the development of improved prognostic algorithms for RPL, involving a more precise assessment of individual disease risks, and provide a guide to offering adequate therapies where relevant.
INTRODUCTION
Recurrent pregnancy loss (RPL) is a complex, multifactorial condition with a polygenic background. Prime candidates for the molecular basis of RPL are various inherited hypercoagulation disorders that promote thrombosis, collectively termed ‘thrombophilias’. Among these, the most significant defects (associated with a 3–6-fold risk of miscarriage) are carriership of either the factor V Leiden mutation or the factor II (prothrombin) 20210G → A (PTm) mutation. For both lesions, an association with RPL has been proved by statistical meta-analyses (1).
The PTm mutation is a G to A transition at position 20 210 in the 3′-untranslated region (3′-UTR) of the factor II (F2) gene, which causes gain of function via an increased recognition of the 3' cleavage signal and concomitantly enhanced 3' end processing of the mRNA. The mutation eventually results in elevated mRNA production and an increased synthesis of prothrombin (2).
Factor V Leiden is a mutation that leads to an R to Q substitution at position 506 of the factor V amino acid sequence, resulting in a 10-fold slower cleavage of factor V by activated protein C and, hence, an increased level of thrombin generation and a pro-coagulant state (3).
Another major risk factor for RPL is the presence of circulating maternal antiphospholipid antibodies (aPL). A higher incidence of RPL has been documented in both low-risk and high-risk pregnancies when aPL were present (4). aPL are thought to lead to fetal loss by causing thrombosis of the placental vessels, although the observed variability in placental pathology may argue against such a direct involvement (5,6). Annexin A5 (placental anticoagulant protein) occurs on normal placental villi and appears to be reduced in the presence of aPL (7). Reduced annexin A5 expression on placental trophoblasts has also been documented immunohistochemically in patients with pre-eclampsia (8). On the basis of these observations and the reported anticoagulation activity of annexin A5 (9), it has been suggested that annexin A5 molecules form an antithrombotic shield on the apical surface of placental syncytiotrophoblasts, which may then be disrupted by aPL (10). This hypothesis has recently received additional support from in vitro studies employing atomic force microscopy and functional assays (11).
Annexin A5 is a typical member of the chordate annexin family. It shows the essential tetrad structure and calcium-dependent phospholipid binding and is one of the few annexins that can be found extracellularly (12). Annexin A5 is thought to function as an inhibitor of coagulation owing to its ability to bind to anionic phospholipids exposed on the surface of, for example, platelets, thereby inhibiting aggregation (13) and/or down-regulating the cell surface presentation of tissue factor (14). The annexin A5 (ANXA5) gene covers ∼9 kb of human chromosome 4q27 and consists of one non-translated exon and 12 coding exons (15). To date, little is known about the regulation of ANXA5 gene expression. Annexin A5 is an abundantly and ubiquitously expressed protein showing the highest levels of concentration in kidney, liver and placenta (16). The human ANXA5 gene produces several transcripts and possesses a complex promoter that is subject to intricate regulation (17). To date, mutations of ANXA5 have not been associated with disease phenotypes, with the exception of a − 1C → T variant that is proposed to protect young individuals against myocardial infarction by being associated with higher plasma levels of the protein in T allele carriers (18). However, the validity of this finding has recently been questioned (19,20).
To test the hypothesis of whether ANXA5 gene variant(s) could influence the risk for RPL, we examined the coding region of the ANXA5 gene and its core promoter (17) in 70 RPL patients, pre-screened for the absence of the PTm and factor V Leiden mutations. We found two new genetic variants, in the form of common haplotypes (M1 and M2), in the ANXA5 gene promoter and investigated the influence of these haplotypes on the expression of the ANXA5 gene and on the risk for RPL.
RESULTS
Structural and functional characterization of ANXA5 promoter haplotypes
Upon systematic screening of all exons, exon–intron boundaries and some 270 bp of the 5′-UTR, a set of four consecutive nucleotide substitutions in the ANXA5 gene promoter, − 19G → A, 1A → C, 27T → C and 76G → A (Fig. 1), was identified in 18 of the 70 patients analyzed. Nucleotide numbering refers to the first transcription start point of the gene, tsp1 (17). In seven additional patients, only two of the four variants were present, namely 1A → C and 27T → C. We further analyzed whether the observed changes were in linkage disequilibrium with each other by cloning and sequencing the relevant promoter region in patients who carried the changes in heterozygous state. This revealed the presence of only two variant ANXA5 promoter haplotypes in addition to the wild-type: M1, comprising 1A → C and 27T → C (six heterozygotes) and M2, comprising − 19G → A, 1A → C, 27T → C and 76G → A (16 heterozygotes). All substitutions changed a transcription factor consensus site or affected a nucleotide adjacent to it. The − 19 G → A substitution (Fig. 1) abuts a gGCCc consensus for the MTF-1 transcription factor at the point where zinc finger binding occurs (21). The transcription start point, tsp1, which is changed by the 1A → C mutation in both variant haplotypes, lies in close proximity to an HNF-3 consensus (17). The 27T → C substitution disrupts an Sp1 consensus and 76G → A destroys a BamHI restriction site in the immediate vicinity of an AP4/MED-1 consensus (motif B), which has been shown to be indispensable for the ANXA5 promoter activity (17). Reporter gene assays on both M1 and M2 were performed in triplicate and showed a drastic reduction of the ANXA5 promoter activity when all four nucleotide substitutions were present (haplotype M2, 37–42% activity compared with N; Fig. 2). The decrease in activity was less pronounced for M1, which contains only the two ‘core’ replacements (57–62% activity).

Structure of the ANXA5 gene core promoter region. The boundaries are marked by vertical bars and are numbered according to the position of the first transcription start point (tsp1). Non-translated exon 1 is shaded in gray. Transcription factor consensus motifs are in small print, and abbreviations of the corresponding transcription factors are displayed in italics above the sequence information. NotI and BamHI restriction sites are underlined and the sequence of the Z-DNA stretch in the promoter is given in italics. Nucleotides marking transcription start points (tsp) are underlined. Regions important for promoter function (motifs A and B) cover nucleotide positions 295–311 and 328–337. Nucleotides changed in the M2 ANXA5 promoter haplotype are printed in bold and substituting nucleotides are given in bold capital letters on top of the respective positions.
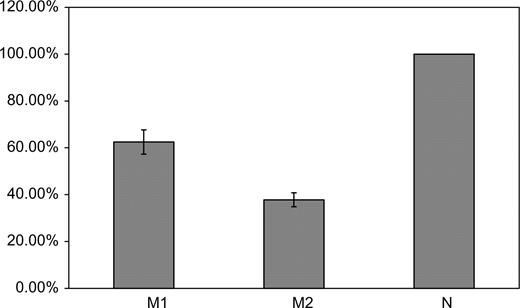
Activity of ANXA5 gene promoter variants in luciferase reporter gene assays. N denotes the wild-type promoter sequence, the activity of which was normalized to 100%. M1 contains nucleotide changes 1A → C and 27T → C; M2 contains all four substitutions, i.e. − 19G → A, 1A → C, 27T → C and 76G → A.
Association of the ANXA5 M2 haplotype with RPL
An association between RPL and haplotypes M1 and M2 was initially tested by comparing the patients with the 500 Münster ‘super-controls’, who all had successful pregnancies and lacked a recorded history of pregnancy loss (Table 1). The frequency of M2 was found to be substantially higher among patients (0.143) than among controls (0.051). Consequently, carriers of M2 appeared to face a four times higher risk of RPL than non-carriers (odds ratio, 3.88; 95% confidence interval, 1.98–7.54). In contrast, carriership of M1, in either homozygous or heterozygous state, was not associated with a higher RPL risk. In the Münster control group, we also observed a significant deviation from the Hardy–Weinberg equilibrium [Monte-Carlo Markov Chain (MCMC) P < 10−4], which was mainly due to an excess of M2 homozygotes (10 observed versus 1.3 expected).
Genotype frequencies of ANXA5 gene promoter haplotypes in German RPL patients and two different control groups
| Genotype | Patients | Münster controls | PopGen controls | |||
|---|---|---|---|---|---|---|
| Observed, n (%) | Expected | Observed, n (%) | Expected | Observed, n (%) | Expected | |
| N/N | 45 (64.3) | 44.8 | 356 (71.2) | 343.6 | 415 (77.9) | 413.3 |
| N/M1 | 6 (8.6) | 6.4 | 87 (17.4) | 99.5 | 35 (6.6) | 47.8 |
| M1/M1 | 1 (1.4) | 0.2 | 16 (3.2) | 7.2 | 1 (0.2) | 1.5 |
| N/M2, M1/M2a | 16 (22.9) | 17.2 | 31 (6.2) | 48.4 | 77 (14.4) | 69.0 |
| M2/M2 | 2 (2.9) | 1.4 | 10 (2) | 1.4 | 5 (0.9) | 1.4 |
| Total | 70 | 70 | 500 | 500 | 533 | 533 |
| Genotype | Patients | Münster controls | PopGen controls | |||
|---|---|---|---|---|---|---|
| Observed, n (%) | Expected | Observed, n (%) | Expected | Observed, n (%) | Expected | |
| N/N | 45 (64.3) | 44.8 | 356 (71.2) | 343.6 | 415 (77.9) | 413.3 |
| N/M1 | 6 (8.6) | 6.4 | 87 (17.4) | 99.5 | 35 (6.6) | 47.8 |
| M1/M1 | 1 (1.4) | 0.2 | 16 (3.2) | 7.2 | 1 (0.2) | 1.5 |
| N/M2, M1/M2a | 16 (22.9) | 17.2 | 31 (6.2) | 48.4 | 77 (14.4) | 69.0 |
| M2/M2 | 2 (2.9) | 1.4 | 10 (2) | 1.4 | 5 (0.9) | 1.4 |
| Total | 70 | 70 | 500 | 500 | 533 | 533 |
Expected, genotype frequency expected at the Hardy–Weinberg equilibrium.
aGenotype M1/M2 was only observed in one Münster and five PopGen controls.
Genotype frequencies of ANXA5 gene promoter haplotypes in German RPL patients and two different control groups
| Genotype | Patients | Münster controls | PopGen controls | |||
|---|---|---|---|---|---|---|
| Observed, n (%) | Expected | Observed, n (%) | Expected | Observed, n (%) | Expected | |
| N/N | 45 (64.3) | 44.8 | 356 (71.2) | 343.6 | 415 (77.9) | 413.3 |
| N/M1 | 6 (8.6) | 6.4 | 87 (17.4) | 99.5 | 35 (6.6) | 47.8 |
| M1/M1 | 1 (1.4) | 0.2 | 16 (3.2) | 7.2 | 1 (0.2) | 1.5 |
| N/M2, M1/M2a | 16 (22.9) | 17.2 | 31 (6.2) | 48.4 | 77 (14.4) | 69.0 |
| M2/M2 | 2 (2.9) | 1.4 | 10 (2) | 1.4 | 5 (0.9) | 1.4 |
| Total | 70 | 70 | 500 | 500 | 533 | 533 |
| Genotype | Patients | Münster controls | PopGen controls | |||
|---|---|---|---|---|---|---|
| Observed, n (%) | Expected | Observed, n (%) | Expected | Observed, n (%) | Expected | |
| N/N | 45 (64.3) | 44.8 | 356 (71.2) | 343.6 | 415 (77.9) | 413.3 |
| N/M1 | 6 (8.6) | 6.4 | 87 (17.4) | 99.5 | 35 (6.6) | 47.8 |
| M1/M1 | 1 (1.4) | 0.2 | 16 (3.2) | 7.2 | 1 (0.2) | 1.5 |
| N/M2, M1/M2a | 16 (22.9) | 17.2 | 31 (6.2) | 48.4 | 77 (14.4) | 69.0 |
| M2/M2 | 2 (2.9) | 1.4 | 10 (2) | 1.4 | 5 (0.9) | 1.4 |
| Total | 70 | 70 | 500 | 500 | 533 | 533 |
Expected, genotype frequency expected at the Hardy–Weinberg equilibrium.
aGenotype M1/M2 was only observed in one Münster and five PopGen controls.
Next, we re-tested the apparent association between the M2 haplotype and RPL by comparing the patients with the PopGen controls. Analysis of a genome-wide set of 67 intragenic SNPs did not indicate any systematic genetic differences between the two groups, and the PopGen control group was in the Hardy–Weinberg equilibrium for the ANXA5 haplotypes (MCMC P = 0.409). The abundance of M2 among patients was also apparent in comparison to the PopGen controls (0.143 versus 0.082). For M2 carriership, the odds ratio equals 2.42, with a 95% confidence interval of 1.27 − 4.58. Thus, the role of M2 as a risk factor for RPL could be confirmed, although the population relative risk faced by M2 carriers is likely to be closer to 2 than to 4. The association between M1 and RPL was of borderline statistical significance so that, given the current data, we cannot conclude that M1 represents a risk factor for RPL. The case sample was also too small to allow formal statistical testing of any interaction between haplotypes or of differences between homozygosity and heterozygosity. A trend was nevertheless seen for M2, where homozygosity M2/M2 was found to entail a higher risk (OR = 3.11) than heterozygosity N/M2 or M1/M2 (OR = 1.76) when the PopGen controls were taken into consideration. However, the 95% confidence interval for the M2/M2 odds ratio was rather wide (0.41–18.47), indicating that such a conclusion would not yet be formally justified.
DISCUSSION
In the present study, we have tested the hypothesis that variant(s) in the placental anticoagulant protein annexin A5 (ANXA5) gene could influence the risk for RPL through prothrombotic effects on the relevant organ (i.e. the placenta). Three independent lines of evidence support a pathological role of ANXA5 in RPL. First, our mutation screen has identified a new variant promoter haplotype of ANXA5 in a sample of German RPL patients that had been pre-screened for the absence of other genetic risk factors. Secondly, using a reporter gene assay, we demonstrated that this haplotype strongly reduces the activity of the ANXA5 promoter in vitro (37–42% of the wild-type level). Thirdly, we compared our patient sample with two different control samples with the same (proven) genetic background, namely (i) women with successful pregnancies and no recorded history of previous pregnancy losses and (ii) unselected female controls from the PopGen study. Haplotype M2 was found to be consistently rarer in these two samples, and the frequency differences were too large to be attributable solely to the fact that M2 was first detected in the RPL patient sample.
Taken together, these findings imply that carriership of the M2 promoter haplotype of the ANXA5 gene is a risk factor for RPL in German females. The relative risk was found to be 2, when comparing patients with unselected controls, but may be as high as 4 in terms of successful pregnancies and a lack of recorded pregnancy losses (Table 1). Along these lines, we surmise that the deviation from the Hardy–Weinberg equilibrium observed in the Münster control group reflects positive ascertainment bias, bearing in mind that some 10–15% of women worldwide suffer pregnancy loss. Since the PopGen control group was a representative sample from the population of Northwest Germany, we consider the respective increase in the M2 haplotype frequency in this sample to be closer to the true, overall population frequency.
Reduction of the ANXA5 expression in the placenta seems to be a relevant etiological factor, as it most likely favors thrombophilic environments that are causative for pregnancy loss. Annexin A5 (placental anticoagulant protein) has been shown to be critical for maintaining murine placental integrity (22). When mice were infused with anti-annexin A5 antibodies, various degrees of fetal absorption, together with thrombosis and necrosis of absorbed embryos, were observed. As it is enriched along the apical surface of trophoblasts, it has been proposed that annexin A5 builds an anticoagulant shield on the phospholipid membrane surface (10). Recently, evidence for the disruptive action of human monoclonal aPL on such annexin A5 lattices has been obtained using atomic force microscopy and a functional assay for the inhibition of the annexin A5 anticoagulant effect through aPL action (11).
A direct link between reduced annexin A5 expression levels and a prothrombotic placental environment, leading to fetal growth restriction, has been established immunohistochemically in pre-eclamptic patients (8). Markedly reduced expression of annexin A5 has also been observed in women with aPL and who suffer repeated pregnancy loss (7). These observations suggest that the reduced expression of annexin A5 could be responsible for immunological and hemostatic phenomena that, together, lead to fetal loss. The lower abundance of the protein on the trophoblast apical surface could result, for example, in inefficient phospholipid shielding and hence in a potential enrichment of antigenic determinants, leading to aPL generation. On the other hand, even in the absence of aPL, markedly reduced annexin A5 expression could possibly cause a hypercoagulable state in the intervillous placental space. No matter what the mechanisms are that lead to RPL via annexin A5 depletion, it is clear that genetic factors mediating such insufficiency could be responsible for the observed pathology. The M2 haplotype of the ANXA5 gene promoter, which we have shown to be over-represented among women who had been diagnosed with RPL and who had been pre-screened for the absence of other major thrombophilic genetic factors, might be the cause of reduced annexin A5 expression. Our functional reporter gene assay shows that this promoter variant leads to a significant reduction in expression (37–42% of the wild-type level). In heterozygous condition, the expression of the ANXA5 gene should be reduced to 60–80%. Whether this reduction is generally representative of annexin A5 plasma levels, or whether it varies within a particular organ (e.g. placenta), and whether the lower expression level is sufficient for a normal function, remains to be clarified. There are reports of reduced circulating annexin A5 levels in women with recurrent spontaneous pregnancy loss (23). Annexin A5 is an intracellular protein (12), and its release into the blood plasma is not governed by any known quantitative mechanism. Moreover, since the ANXA5 gene promoter has a complex structure (17), it is reasonable to propose that a variety of interacting elements, activation strategies and adequate expression levels would be observed in different tissues. Consequently, one would not expect equal annexin A5 expression in placental trophoblasts and in blood plasma, although the possibility of passive annexin A5 recruitment from the bloodstream on to the trophoblast surface cannot be excluded.
Obviously, the distribution of ANXA5 promoter haplotypes in other populations and their impact on RPL and other prothrombotic pathological conditions require further evaluation. Furthermore, it will also be worth investigating whether and to what extent ANXA5 promoter mutations statistically interact, in terms of RPL risk, with other thrombophilic genetic factors, including PTm and factor V Leiden. The latter had been deliberately ruled out in our patients, with the hope that the exclusion of known risk factors should increase the power to detect new ones, but this restriction came at the expense of a limited patient base for recruitment. We therefore strongly urge other groups not only to try to replicate our findings but also to expand them into a multifactorial context. In the same vein, analyses of the presence of RPL-associated aPL in conjunction with ANXA5 promoter haplotypes would also be an important subject for future investigation. Nevertheless, our study adds to the growing list of successful identifications of genetic markers for complex disease, and the diagnostic application of our findings is likely to facilitate the development of paradigms for an early and preventive treatment of RPL.
MATERIALS AND METHODS
Study populations
The present study complied with the ethical guidelines of all the institutions involved. Informed consent was obtained from all subjects examined. We analyzed 70 patients of northern German origin with RPL (defined as having experienced more than two fetal losses), who had been referred to the Institut für Humangenetik, University Clinic Münster, for genetic counseling. With 56 women of this group, pregnancy loss had occurred in the first or in the second trimester, and the remaining 14 subjects had had at least one stillbirth. There were no cases with pre-eclampsia or abruptio placentae. All patients were screened for the PTm and factor V Leiden mutations and were found to be non-carriers. Five hundred anonymized control individuals of northern German origin, all with successful pregnancies and no documented history of RPL, were ascertained from the Institute's registry (Münster controls). Another 533 anonymized female control samples of northern German extraction were obtained from the PopGen biobank (24), University Hospital Schleswig-Holstein, Kiel (PopGen controls). No evidence for population stratification was found when patients were compared with the PopGen controls, using 67 intragenic SNPs uniformly distributed over the human genome (25).
Genotyping and haplotype determination
Genomic DNA was extracted from peripheral blood lymphocytes using a salting procedure (26). A mutation search was performed in the 70 RPL patients, covering the entire coding sequence of ANXA5 together with 60–80 bp of flanking introns and the ‘core’ promoter region (i.e. the first non-translated exon and ∼270 bp of the 5′-UTR). Following PCR amplification, amplicons were sequenced directly. PCR reactions were performed on 100 ng of genomic DNA in a total volume of 25 ml, and the reaction mix contained the following: 50 mm Tris–HCl, pH 9.5; 20 mm (NH4)2SO4; 1 mm DTT, 0.005% NP-40; 1.5 mm MgCl2; 1 M betaine; 5% DMSO; 20 pm of each (forward and reverse) primer; 300 mm dNTP; 100–200 ng DNA and 1.25 U Taq polymerase from a local source (Institute of Experimental Pathology, ZMBE, University of Münster, Münster, Germany). The primers used are listed in panel 1. Cycling conditions were as follows: denaturation at 94°C for 45 s; annealing at 60°C for 30 s; extension at 68°C for 1 min in 25 cycles. PCR products were purified using MultiScreen™ PCR plates (Millipore, Eschborn, Germany) according to the manufacturer's instructions. Purified amplicons were directly sequenced in 96-well plates using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (ABI/Perkin-Elmer, Weiterstadt, Germany); sequencing reactions were analyzed on an ABI PRISM® 3700 DNA Analyzer (ABI/Perkin-Elmer, Weiterstadt, Germany). The 67 intragenic SNPs were genotyped using Taqman MGB technology (ABI/Perkin-Elmer).
Amplicons containing 436 bp of the ANXA5 gene promoter region were obtained from carriers and non-carriers of the M1 and M2 haplotypes using primers with added restriction sites, as listed in panel 1. The amplicons were purified and the ends were hydrolyzed with MluI and XhoI before cloning into the pGL3-Basic vector (Promega, Freiburg, Germany) to generate luciferase reporter constructs. Ten insert-carrying clones were selected at random and plasmid DNA was sequenced in both directions using the amplification primers.
Reporter gene assays
pGL3-Basic plasmids containing haplotypes M1, M2 and normal ANXA5 promoter sequence (N) were transfected into HeLa cells in equimolar quantities with a pCMV-beta(gal) plasmid (BD Biosciences Clontech, Heidelberg, Germany). Transfections were performed in triplicate for each promoter construct. A total of 0.6 µg DNA was used on 3 × 105 cells in 6-well culture plates, following the Effectene (Qiagen, Hilden, Germany) transfection protocol. Cells were lysed 24 h after transfection and the lysate was analyzed in parallel reactions for β-galactosidase and luciferase activities using the luminescent β-galactosidase Gene Reporter Assay and the luciferase Gene Reporter Assay (constant light) kits (Roche, Mannheim, Germany) following the manufacturers’ instructions. Luminescence was measured in 96-well non-transparent plates and documented using the LumiImager (Roche). Luciferase activity was scored by its ratio to measured β-gal activity for each transfection (internal standard), and these values were taken to reflect the comparative promoter activity of the assayed constructs.
Statistical analysis
Odds ratios with 95% confidence intervals were calculated using procedure FREQ of the SAS statistical software package V8 (SAS Institute, Cary, NC, USA). The statistical significance of departures from the Hardy–Weinberg equilibrium was assessed using an MCMC implementation of an exact test, with 1 million proposals following a burn-in period of 100 000 proposals (27). Computer program STRUCTURE (28,29) was used to examine possible population genetic differences between the patient and PopGen control samples.
ACKNOWLEDGEMENTS
The authors would like to thank Tanja Wesse for the excellent technical assistance and Alexandre Todorov for the initial statistical evaluation of data. Grant support was received from the Deutsche Forschungsgemeinschaft (DFG) and Interdisziplinäres Zentrum für Klinische Forschung (IZKF) to J.H. and V.G. and from the German National Genome Research Network (NGFN-2) to S.S. and M.K. A.T. was a Humboldt Stiftung fellow.
Conflict of Interest statement. None declared.

{kind=link}
{kind=link}