




Abstract
The accumulation of abnormal proteins is a common characteristic of neurodegenerative diseases. This accumulation reflects a severe disturbance of cellular homeostasis in pathogenic protein clearance. Here, we demonstrated that the activation of the two major proteolytic machineries, the molecular chaperone–ubiquitin proteasome system (UPS) and the autophagy system, were simultaneously enhanced by paeoniflorin (PF), a major component of Paeonia plants, and exerted therapeutic effects in models of spinal and bulbar muscular atrophy (SBMA). PF significantly increased the expression of nuclear factor-YA (NF-YA), which strongly upregulated the molecules involved in the proteolytic machinery [molecular chaperones, carboxyl terminus of Hsc70-interacting protein and transcription factor EB], which thus mitigated the behavioral and pathological impairments in an SBMA mouse model through the upregulation of pathogenic androgen receptor protein clearance in motor neurons and muscles. These findings demonstrated that PF is able to enhance both the UPS and autophagy systems by upregulating the expression of NF-YA, which promotes therapeutic effects in an SBMA model.
INTRODUCTION
Polyglutamine (polyQ) diseases are inherited neurodegenerative disorders caused by the expansion of a trinucleotide CAG repeat in the causative genes. To date, nine disorders have been identified as polyQ diseases (1). The expanded polyQ stretch is thought to confer a toxic gain of function to the mutant protein. Spinal and bulbar muscular atrophy (SBMA) is a motor neuron disease caused by a polyQ tract within the androgen receptor (AR) (2). SBMA is characterized by motor neuron loss in the spinal cord and brainstem that is accompanied by diffuse nuclear accumulation and nuclear inclusions (NIs) of the mutant AR in motor neurons and specific visceral organs (3,4). SBMA patients also show myogenic changes together with the neurogenic atrophy according to muscle biopsies (5). SBMA patients gradually develop progressive weakness of the bulbar and extremity muscles, and muscle strength and function are inversely correlated with the CAG repeat length, age and duration of weakness (6,7). Nuclear and cytoplasmic inclusions are common pathological features in polyQ diseases. The inclusions are colocalized with many components of molecular chaperones, the ubiquitin proteasome system (UPS) and the autophagy system, raising the possibility that this proteolytic machinery may actively degrade components of these inclusions (8). Molecular chaperones facilitate the refolding and proteolysis of toxic, misfolded proteins and may play a role in protecting neuronal cells against the toxic properties of polyQ expansions (9–11). The carboxyl terminus of the Hsc70-interacting protein (CHIP), a U-box-type E3 ubiquitin ligase, interacts with heat shock protein 90 (Hsp90) or Hsp70 and ubiquitylates and degrades unfolded proteins that are trapped by molecular chaperones. Thus, CHIP acts as a ‘quality-control E3’ (12). Macroautophagy is a set of bulk degradation processes in which cells form double-membrane vesicles, called autophagosomes, around a portion of the cytoplasm. These autophagosomes ultimately fuse with lysosomes, resulting in the degradation of their substrates. The microtubule-associated protein light-chain 3 (LC3)-II specifically associates with autophagosomes, and thus, the levels of LC3-II are correlated with the number of autophagic vacuoles (13). The transcription factor EB (TFEB) regulates autophagy by upregulating genes that belong to the coordinated lysosomal expression and regulation (CLEAR) network (14,15), which thereby controls lysosomal biogenesis.
Paeoniflorin (PF) is the principal active ingredient extracted from the roots of Paeonia plants, including Paeonia alba and Paeonia lactiflora. PF has been shown to have various biological and biomodulating activities, including the improvement of memory, antioxidant activity, antiepileptic activity, anti-stroke properties and anti-neuroinflammation. Several studies have suggested that PF upregulates the expression of molecular chaperones and LC3-II and enhances the degradation of disease-causative proteins via the proteolytic machinery, indicating that PF may have neuroprotective effects (16,17). However, the possible implications for neuronal disease treatment have not been assessed in models of disease, and the mechanisms underlying the activation of the proteolytic machinery are not understood.
Here, we present findings supporting the concept of a proteolytic, machinery-dependent pathophysiology in a neurodegenerative disease. We examined the effects of PF in cultured cells and the transgenic mouse model of SBMA. PF administration inhibited the nuclear accumulation of the mutant AR and significantly ameliorated the motor phenotype of the SBMA mouse model without detectable toxicity. Mutant AR was preferentially degraded over wild-type AR in the presence of PF in both cell culture and mouse models of SBMA. PF significantly induced nuclear factor-YA (NF-YA), also known as CCAAT box-binding factor (CBF) (18,19), resulting in the upregulation of the molecular chaperones CHIP and TFEB. Furthermore, because this proteolytic machinery is considered to be an effect common to other polyQ diseases, our observations suggest that PF is a promising therapeutic candidate for a wide range of polyQ-mediated neurodegenerative diseases, including SBMA. PF enhanced two major proteolysis systems, the molecular chaperone–UPS and the autophagy system. Thus, we demonstrated that PF promoted the degradation of mutant AR to a greater extent than the induction of either the molecular chaperone–UPS or the autophagy system alone.
RESULTS
PF preferentially reduces the amount of polyQ-expanded mutant AR by integrating into the proteolytic machinery
To examine the efficacy of PF in promoting the degradation of mutant polyQ-expanded AR, we treated NSC34 cells stably expressing the wild-type (AR-24Q) or mutant AR (AR-97Q) with the indicated doses of PF or phosphate buffered saline (PBS) for 24 h. Although the anti-AR immunoblot showed a dose-dependent decline in both wild-type and mutant AR expression in response to PF (Fig. 1A), the decrease in monomeric mutant AR (62.8% at 10 µm PF) was significantly greater than that of wild-type AR (31.3%; Fig. 1B). These data suggest that the accumulation of mutant AR is selectively reduced at higher levels in response to PF than wild-type AR (P < 0.01). A similar preferential reduction in mutant AR was observed in another cell model expressing transiently transfected ARs (Supplementary Material, Fig. S1A and B). To reveal the mechanism underlying the PF-induced activation of the proteolytic machinery and the enhancement of the degradation of the mutant AR, we screened the expression of key transcription factors and molecules, which trigger the induction of the molecular chaperone–UPS or autophagy (or both), in the cell culture and transgenic mouse models of SBMA expressing AR-24Q. Among these factors, PF treatment significantly increased the expression level of two transcription factors, NF-YA and TFEB (Supplementary Material, Fig. S2A–D). PF treatment also significantly increased the expression levels of molecular chaperones and CHIP in a dose-dependent manner (Fig. 1A and B), and it also upregulated NF-YA mRNA (Fig. 1C). However, there were no significant differences in the expression levels of these molecules between cells expressing the wild-type or mutant AR (Fig. 1B). There was also no deterioration of cell viability after PF treatment in cells that stably expressed either wild-type or mutant AR (Supplementary Material, Fig. S3). These data indicate that PF preferentially degrades the mutant AR protein without cellular toxicity. Luciferase activity assays revealed that PF increased the transactivation of NF-YA, TFEB, Hsp70 and CHIP in a dose-dependent manner (Fig. 1D). To determine whether the decrease in AR was due to protein degradation or changes in RNA expression, the turnover of wild-type and mutant AR was assessed with a pulse-chase labeling assay. Without treatment, the wild-type and mutant AR were degraded to levels similar to those reported previously (20). However, in the presence of PF, both proteins had shortened half-lives. The wild-type AR decreased from ∼6.5 to 5.5 h, and the mutant AR decreased from 5.5 to 3.5 h (Fig. 1E and F). The mRNA expression levels of both the wild-type and mutant AR were similar (Fig. 1G). The degradation of mutant AR by PF was inhibited both by the proteasome inhibitor lactacystin and bafilomycin A1, a potent inhibitor of autophagy (Fig. 1F and H). This evidence suggests that the PF-induced degradation was dependent on both the proteasome and autophagy systems. These data indicate that PF is non-toxic and preferentially degrades the mutant AR protein without altering mRNA levels.
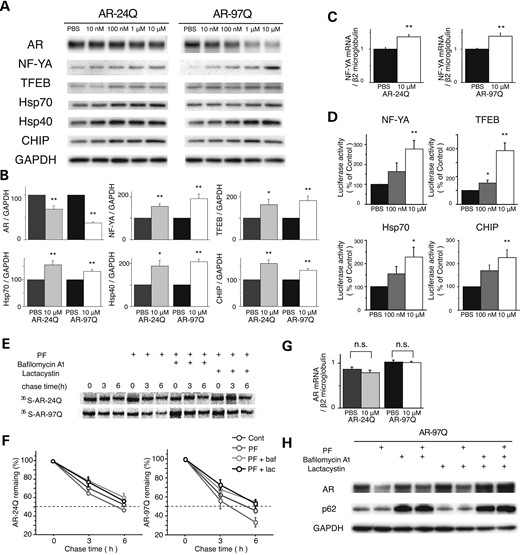
Effects of PF on AR in a cultured cell model of SBMA. (A and B) Western blots and densitometric analyses from NSC34 cells stably expressing wild-type (24Q) or mutant (97Q) AR (*P < 0.05; **P < 0.01, n = 5, unpaired t-test). (C) The expression level of NF-YA mRNA from NSC34 cells stably expressing wild-type (24Q) or mutant (97Q) AR (**P < 0.01, n = 5, unpaired t-test). (D) Constructs containing NF-YA, TFEB, Hsp70 or CHIP and a luciferase reporter gene were transfected into NSC34 cells. Luciferase activity assays were performed in triplicate in five independent experiments. Relative luciferase activity is represented as a ratio against the luciferase activity in cells transfected with an empty promoter vector (**P < 0.01, n = 5, one-way ANOVA with Tukey–Kramer post hoc test). (E) Pulse-chase analysis of two forms of AR. Data from one representative experiment for wild-type and mutant AR. Stably transfected NSC34 cells were treated with PF (10 µm), bafilomycin A1 (baf; 20 nm) and lactacystin (lac; 2.5 μm) for 16 h. (F) Pulse-chase assessment of the half-life of wild-type (left) and mutant AR (right). (G) Real-time RT–polymerase chain reaction (PCR) of wild-type and mutant AR mRNA. Quantities are presented as the ratio to β2 microglobulin mRNA. The wild-type and mutant AR mRNA levels were similar under PF treatments (n = 5, unpaired t-test). (H) Stably transfected NSC34 cells were treated with PF (10 µm), bafilomycin A1 (20 nm) and lactacystin (2.5 μm) for 16 h. Error bars (B, C, D, F, G), SEM.
NF-YA regulates both the proteasomal and autophagic proteolytic machinery
We next addressed whether PF promotes the degradation of mutant, polyQ-expanded AR via the upregulation of transcription factor NF-YA. NSC34 cells stably expressing AR-97Q were transfected with the indicated amount of NF-YA or mock transfected and were analyzed 24 h later (Fig. 2A). Immunoblot analysis demonstrated a dose-dependent decline in mutant AR expression following NF-YA overexpression (Fig. 2A and B). The expression levels of Hsp70, CHIP and TFEB were also significantly increased following NF-YA overexpression (Fig. 2A and B). Moreover, the effects of PF were completely blocked in cells depleted of NF-YA (Fig. 2C and D), which suggests that PF primarily enhances mutant AR degradation through NF-YA upregulation and the downstream transactivation of the proteasomal and autophagic proteolytic machinery.
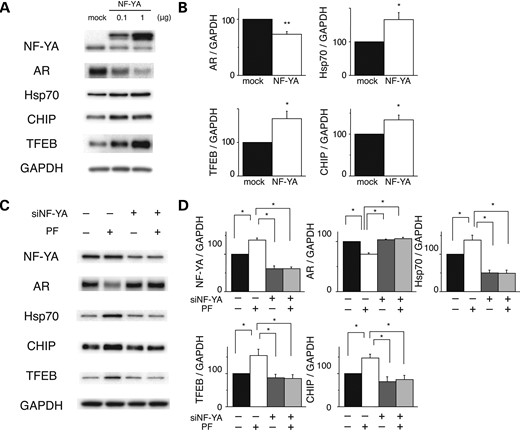
Effects of NF-YA overexpression in cultured cell models of SBMA. (A and B) Western blots and densitometric analyses of NSC34 cells stably expressing mutant AR. Cells were transfected with NF-YA with a V5 tag or the mock vector (*P < 0.05; **P < 0.01, n = 5, unpaired t-test). (C and D) Western blots and densitometric analyses of NSC34 cells stably expressing mutant AR that were transfected with control or NF-YA siRNA and treated with or without PF (*P < 0.05, n = 5, one-way ANOVA with Tukey–Kramer post hoc test). Error bars (B and D), SEM.
Effect of PF on expression levels of AR via TFEB activation in vitro
The aforementioned results demonstrate that PF increases the expression of TFEB, a master autophagy regulator, via the upregulation of NF-YA. TFEB translocates from the cytoplasm to the nucleus, which results in the activation of its target genes (14). We hypothesized that if PF contributes to the activation of autophagy, then the expression of the mutant polyQ protein would be reduced by TFEB-activated autophagy in addition to the UPS degradation. To test this hypothesis, the nuclear translocation of TFEB was investigated using western blot analysis, which showed that PF increased the amount of TFEB in the nucleus at a dose of 10 µm in NSC34 cells (Fig. 3A and B). Next, we investigated whether TFEB promotes the degradation of mutant AR. NSC34 cells stably expressing AR-24Q or AR-97Q were transfected with the indicated amount of TFEB or mock transfected (Fig. 3C). After 48 h, the expression levels of both AR-24Q and AR-97Q similarly decreased following the overexpression of TFEB (Fig. 3C and D). The expression of the autophagic marker LC3-II was significantly elevated in the cells expressing AR-24Q or AR-97Q (Fig. 3C and D), suggesting that the high expression of TFEB induced autophagosome formation and enhanced the degradation of the AR proteins. However, there were no significant differences in the reduction rate between the wild-type and mutant AR (Fig. 3C and D), which indicates that the activation of UPS is the basis for preferential degradation of the polyQ-expanded mutant AR following PF treatment.
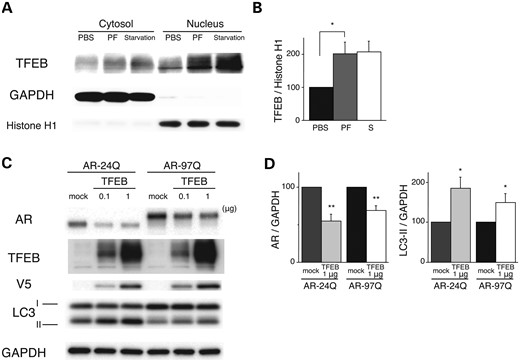
Effects of PF on expression levels of AR via TFEB activation. (A and B) PF facilitates the nuclear translocation of TFEB. Western blots and densitometric analyses from NSC34 cells treated with 10 µm PF or starvation. Cell lysates were subjected to nuclear or cytosolic fractionation and probed with a TFEB antibody. GAPDH and H1 were used as cytosolic and nuclear markers, respectively (*P < 0.01, n = 5, one-way ANOVA with Tukey–Kramer post hoc test). (C and D) Effects of TFEB overexpression on the expression levels of wild-type (24Q) and mutant (97Q) ARs in NCS34 cells. Western blots and densitometric analyses from NSC34 cells stably expressing AR-24Q or AR-97Q. Cells were transfected with the mock or TFEB-V5 vector (*P < 0.01, n = 5, unpaired t-test). Error bars (B and D), SEM.
PF ameliorates the phenotype of male AR-97Q mice
To verify that PF-mediated activation of the proteolytic machinery mitigates polyQ toxicity, we administered PF intraperitoneally every day at doses of 6.7 mg/kg (PF1) or 13.4 mg/kg (PF2) to males of the transgenic mouse model carrying the full-length human AR with either 24Q or 97Q (21). To demonstrate that PF slows the onset and progression of the disease, PF treatments were initiated when the mice reached 5 weeks of age and continued until they were 32 weeks old. The AR-97Q mice displayed progressive muscular atrophy and weakness that was accompanied by diffuse nuclear accumulation and NIs of mutant AR in neuronal and non-neuronal tissues (21). We examined various neurological and behavioral parameters. In the AR-97Q mice treated with 13.4 mg/kg PF (PF2), the phenotype and disease progression were markedly ameliorated, and the progression in mice treated with 6.75 mg/kg PF (PF1) was moderately ameliorated (Fig. 4A–F). The untreated AR-97Q mice (PF0) exhibited motor impairments (assessed by the rotarod task), as early as 9 weeks after birth, whereas the PF2 group showed initial impairments at 13 weeks after birth and had less deterioration than the PF0 mice (Fig. 4A). The PF1 mice exhibited intermediate levels of impairment in their rotarod performance (Fig. 4A). The locomotor cage activity of the PF0 mice was also markedly decreased at 15 weeks compared with the PF1 and PF2 groups, which exhibited decreases in activity at 24 and 26 weeks of age, respectively (Fig. 4B). No mouse lines were distinguishable in terms of body weight at birth; however, the PF0 mice lost weight significantly earlier (by 16 weeks) and to a greater extent than the PF1 and PF2 mice (Fig. 4C). PF treatment also significantly prolonged the lifespan of the PF1 and PF2 mice compared with the PF0 mice (Fig. 4D). Additionally, the PF0 mice exhibited motor weakness, as demonstrated by the dragging of their legs and shortened steps, whereas the PF2 mice had nearly normal ambulation (Fig. 4E and F). AR-24Q mice treated with PF did not have altered phenotypes (Supplementary Material, Fig. S4A–D). To evaluate the possible toxic effects of PF, we examined blood samples from 16-week-old AR-97Q mice treated with 13.4 mg/kg PF for 11 weeks. Measurements of aspartate aminotransferase, alanine aminotransferase, blood urea nitrogen and serum creatinine demonstrated that PF resulted in neither infertility nor liver or renal dysfunction in the AR-97Q male mice (Supplementary Material, Fig. S5A–D). The serum testosterone levels were unchanged in the PF2 mice (1.029 ± 0.369 ng/ml, n = 15) versus the PF0 mice (0.857 ± 0.406 ng/ml, n = 15).
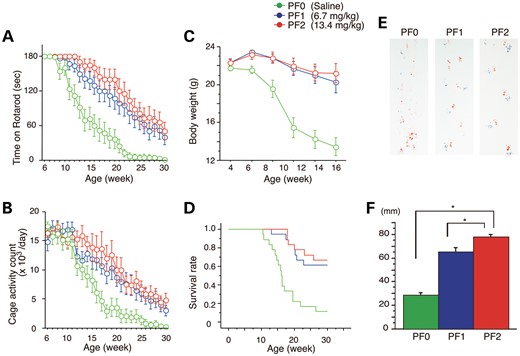
Effects of PF on behavioral phenotypes in AR-97Q mice. Untreated AR-97Q mice (PF0) or mice administered 6.7 mg/kg (PF1) or 13.4 mg/kg (PF2) PF intraperitoneally every day from ages 5 to 30 weeks were tested for rotarod performance (A), cage activity (B) and body weight (C). PF1 and PF2 mice remained on the rotarod longer and exhibited greater cage activity than PF0 mice. PF0 mice lost weight earlier than PF1 and PF2 mice. All parameters were significantly different in PF2 mice compared with PF0 mice: P < 0.01 at 25 weeks (rotarod and cage activity) and P < 0.01 at 10 weeks (body weight) (n = 18, two-way ANOVA with Tukey–Kramer post hoc test). (D) A Kaplan–Meier plot shows the prolonged survival of PF1 and PF2 mice compared with PF0 mice (P = 0.0002 and 0.0001, respectively; n = 18, log-rank test). (E) Footprints of representative 16-week-old AR-97Q mice treated with or without PF. Front paws are indicated in red, and hind paws are in blue. PF0 mice exhibited motor weakness, demonstrated by the dragging of the legs. PF1 mice walked with somewhat longer steps, and PF2 mice walked almost normally. (F) The length of the steps was measured in 16-week-old AR-97Q mice. Each column shows the average length of the steps of the hind paw. PF1 and PF2 mice walked with significantly longer steps than PF0 mice (*P < 0.01, n = 6, one-way ANOVA with Tukey–Kramer post hoc test). These data suggest that PF ameliorates the phenotypic expression of SBMA in AR-97Q mice in a dose-dependent manner. Error bars (F), SEM.
PF reduces diffuse nuclear staining of mutant AR and degrades mutant AR protein in the mouse model of SBMA
Having demonstrated that PF promotes mutant AR degradation in vitro, we examined the levels of AR in the AR-24Q and AR-97Q mice. We performed immunohistochemical staining for mutant AR using the 1C2 antibody in the spinal cord and skeletal muscle of 16-week-old AR-97Q mice. In the PF0 mice, intense staining was frequently observed in the nuclei, whereas staining was infrequent in the PF1 mice and much less frequent in the PF2 mice (Fig. 5A). There were significantly more 1C2-positive cells in the spinal cord and muscle tissue of the PF0 mice than in the PF1 and PF2 mice (Fig. 5B). The 1C2-positive cell populations were not significantly different between the PF1 and PF2 mice (Fig. 5B). There were no NIs in the spinal cord and skeletal muscle of the AR-24Q mice (Supplementary Material, Fig. S4E and F).
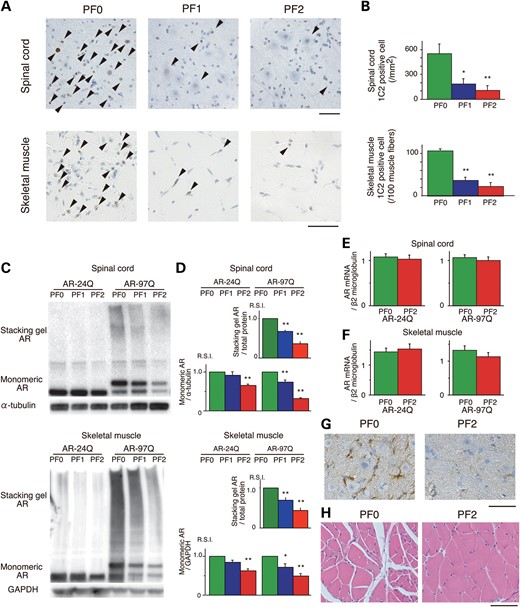
Effects of PF on the nuclear accumulation of abnormal AR in 16-week-old AR-24Q and AR-97Q mice. (A) Immunohistochemical staining for mutant AR using the 1C2 antibody showed a marked reduction in diffuse nuclear staining and NIs (arrow head) in the spinal cord and skeletal muscle of the treated mice compared with the PF0 mice. (B) Quantification of the number of 1C2-positive cells in the spinal cord and skeletal muscle of AR-97Q mice treated with or without PF (*P < 0.05; **P < 0.01, n = 6, one-way ANOVA with Tukey–Kramer post hoc test). (C and D) Western blot analysis of total tissue homogenates from the spinal cord and muscle of AR-24Q and AR-97Q mice probed with anti-AR (*P < 0.05; **P < 0.01, n = 5, one-way ANOVA with Tukey–Kramer post hoc test). (E and F) Real-time RT–PCR of wild-type and mutant AR mRNA in the spinal cord (E) and skeletal muscle (F). Quantities are presented as the ratio to β2 microglobulin mRNA (n = 5, unpaired t-test). (G) Immunohistochemical staining with anti-GFAP antibody in the spinal anterior horn of AR-97Q mice. (H) Hematoxylin and eosin staining of the muscles of AR-97Q mice. Error bars (B, D, E and F), SEM. Scale bars: 50 μm.
Treatment with PF markedly diminished both the high-molecular-weight complex and monomeric mutant AR in the spinal cord and muscle of AR-97Q mice (P < 0.01). PF significantly diminished the wild-type monomeric AR in AR-24Q mice (Fig. 5C and D). In the PF2 mice, monomeric AR decreased by 68.5 and 34.5% in the spinal cord and by 50.3 and 37.2% in the skeletal muscle for the AR-97Q and AR-24Q mice, respectively (Fig. 5C and D). Thus, the rate of reduction of the monomeric mutant AR was significantly higher than for wild-type AR in both the spinal cord (P < 0.01) and skeletal muscle (P < 0.01). Reverse transcription (RT)–polymerase chain reaction (PCR) of both the AR-24Q and AR-97Q mice showed that the levels of AR mRNA were similar in the spinal cord and muscle of the treated (PF2) and untreated (PF0) mice (Fig. 5E and F); thus, the decreased protein level was due to degradation and not to lower mRNA levels. These observations indicate that PF markedly degrades not only the high-molecular-weight mutant AR complex but also the monomeric mutant AR protein compared with the wild-type AR, which reflects the preferential degradation of the mutant AR through the UPS.
Anti-glial fibrillary acidic protein (GFAP) staining showed an apparent reduction in reactive astrogliosis in the PF2 mice in the spinal anterior horn (Fig. 5G). Muscle histology also demonstrated a marked amelioration of muscle atrophy in the PF2 mice (Fig. 5H).
Effect of PF on the regulation of proteolytic machinery in vivo
Both low and high doses of PF treatment of the SBMA transgenic mice were accompanied by marked increases in NF-YA mRNA levels and protein synthesis in the spinal cord and muscle tissue (Fig. 6A–F). These results were well correlated with those of anti-NF-YA immunohistochemical-stained tissue sections (Supplementary Material, Fig. S6A and B). In contrast, PF treatment in the cell and mouse models of SBMA had no effect on the expression levels of hyper-phosphorylated heat shock factor-1 (10,22), p53 (23) or transcription factors regulating Hsp expression, namely, TBP and SP1 (Supplementary Material, Fig. S2A). AR-97Q transgene expression decreased the expression level of NF-YA in both the spinal cord and muscle (Fig. 6C and D). This effect might be due to a decrease in mRNA levels (Fig. 6A and B) and sequestration of endogenous NF-YA in the inclusions in the spinal anterior horn cells and muscle tissue of AR-97Q mice; this finding was also observed in the spinal anterior horn cells of SBMA patients (Supplementary Material, Fig. S6A and B). To ensure that PF activated the proteolytic machinery in vivo, the expression levels of the molecular chaperones, CHIP and TFEB, were determined using western blotting. In the AR-24Q and AR-97Q mice, the levels of Hsp70, Hsp40, CHIP and TFEB were significantly increased (Fig. 6C–F), and PF facilitated the nuclear translocation of TFEB in the spinal cord and muscle of the AR-24Q and AR-97Q mice (Supplementary Material, Fig. S7). Furthermore, western blotting showed that PF treatment resulted in increased expression levels of lysosomal proteases and the autophagic marker LC3-II in the spinal cord and muscle of the AR-24Q and AR-97Q mice (Fig. 7A–D). The expression level of protease mRNA also increased in the AR-24Q and AR-97Q mice treated with 13.4 mg/kg PF as well as in the cell culture model treated with 10 µm PF (Supplementary Material, Fig. S8A–C). These observations suggest that an increase in the levels of the molecular chaperones CHIP and TFEB promotes the downregulation of ARs via the UPS and autophagic degradation. Collectively, these data suggest that the expression of molecules related to the proteolytic machinery is enhanced by PF treatment, which results in a heightened pharmacological effect. The expression of mutant AR is reduced, and the polyQ-dependent phenotype is mitigated by PF in the mouse model of SBMA.
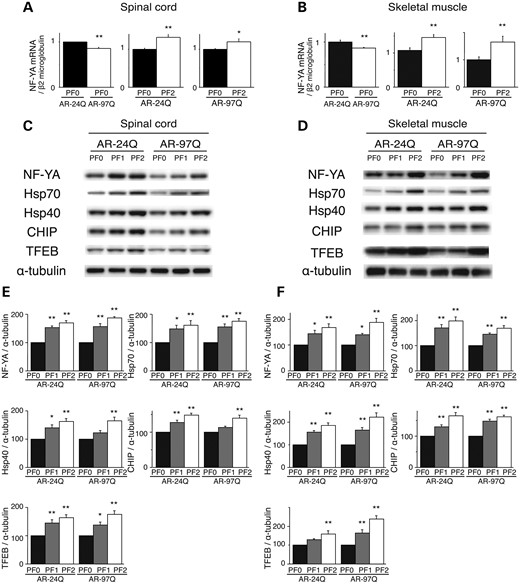
PF increased the expression of molecular chaperones, CHIP and transcription factors in AR-24Q mice and AR-97Q mice. The expression levels of NF-YA mRNA in the spinal cord (A) and skeletal muscle (B) of 16-week-old AR-24Q mice and AR-97Q mice (**P < 0.01; ***P < 0.001, n = 5, unpaired t-test). Western blots and densitometric analyses of NF-YA, molecular chaperones, CHIP and TFEB in the spinal cord (C and E) and skeletal muscle (D and F) of 16-week-old AR-24Q mice and AR-97Q mice (*P < 0.05; **P < 0.01, n = 5, one-way ANOVA with Tukey–Kramer post hoc test). Error bars (A, B, E, F), SEM.
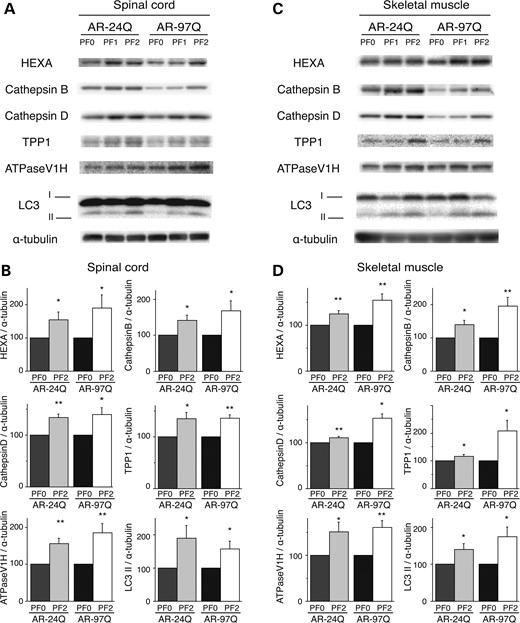
PF increased the expression of lysosomal proteases and LC3-II in AR-24Q and AR-97Q mice. Western blots and densitometric analyses for lysosomal proteases in the spinal cord (A and B) and skeletal muscle (C and D) of 16-week-old AR-24Q mice and AR-97Q mice (*P < 0.05; **P < 0.01, n = 5, unpaired t-test). Error bars (B, D), SEM.
DISCUSSION
Our results demonstrated that mutant AR is sensitive to PF treatment and that the PF-mediated downregulation of mutant AR can ameliorate motor abnormalities in an SBMA mouse model without detectable toxicity. PF treatment reduced the levels of monomeric and aggregated forms of mutant AR. In chronic neurodegenerative disorders, such as polyQ diseases, Parkinson's disease, Alzheimer disease and amyotrophic lateral sclerosis, commonly observed phenotypes include the abnormal accumulation of disease-causing proteins and the formation of nuclear and cytoplasmic inclusions. Under pathologic conditions, the accumulated levels of misfolded and toxic proteins may exceed the protective ability of the proteolytic machinery; the inability to either maintain misfolded proteins in a soluble form or degrade them results in their accumulation and the formation of inclusions. In this study, we showed that PF potently induced a novel, NF-YA-mediated mechanism involving both the UPS and autophagy systems, which downregulated mutant AR accumulation and thereby ameliorated SBMA phenotypes in a mouse model. Motor neuron degeneration induced by mutant AR involves several distinct mechanisms, including transcriptional dysregulation, aggregate formation, impaired axonal transport, DNA binding and mitochondrial dysfunction. Although it remains unclear which of these mechanisms has a pivotal impact on SBMA pathogenesis, the accumulation of mutant AR is a well-established upstream molecular event of neuro- and myodegeneration (3). The successful anti-androgen therapy in SBMA mouse models has translated to clinical trials, which showed that swallowing function improved in patients with a disease duration of <10 years (24,25). Many studies have sought to elucidate the pathogenesis of polyQ diseases and develop treatment methods. Among these treatments, the pharmacologic induction of molecular chaperones, the UPS and autophagy may ameliorate these disorders (26). The mutant polyQ AR adopts an altered conformation that results in protein aggregation and inclusion formation; however, recent studies have suggested that soluble forms of the causative protein, including oligomeric protein, aggregate. Mutant monomers in the nuclear space may also be toxic (rather than protein inclusions). Thus, soluble protein aggregates, or possibly mutant monomers, represent targets in the treatment of neurodegenerative disorders (27–29). Indeed, we have demonstrated that reducing the levels of the pathogenic AR protein in SBMA mice attenuates the expression of the disease (30–35), which suggests that the clearance of mutant AR may therapeutically benefit SBMA. Our present data also confirm that PF passes through the blood–brain barrier and that long-term treatment with PF exerts significant pharmacological effects in the central nervous system.
NF-Y is a heterotrimeric complex that consists of three subunits, NF-YA, NF-YB and NF-YC, and binds to CCAAT motifs in the promoter regions of a variety of genes (18). NF-YA was reported to upregulate the expression of molecular chaperones. Our results demonstrate that NF-YA is also able to induce the expression of CHIP and TFEB, which suggests that NF-YA plays a pivotal role in the clearance of pathogenic proteins and the maintenance of proteostasis. The upregulation of diverse molecules involved in the proteolytic machinery must contribute to the marked beneficial effects of PF treatment in the AR-97Q mice. AR-97Q transgene expression decreased the expression of NF-YA, due to the decrease in mRNA levels (Fig. 6A and B) and sequestration of endogenous NF-YA in the inclusions (Supplemental Material, Fig. S6A and B). Consequently, the expression levels of molecular chaperones, CHIP and TFEB decreased in the spinal cord and muscle (Fig. 6C and D). The reduction of these molecules can impair the function of the proteolytic machinery and enhance polyQ toxicity. NF-YA also colocalized with mutant AR NIs that were present in the motor neurons and muscles of SBMA patients (Supplementary Material, Fig. S6A and B). NIs that were positive for NF-YA have been identified in models of SBMA (36), Huntington's disease (HD) (37) and spinocerebellar ataxia 17 (SCA17) (38), which suggests that NF-YA is a protein targeted for sequestration in polyQ diseases. Collectively, these data suggest that PF can increase the expression of NF-YA and restore the proteolytic machinery. It is likely that the proteolytic machinery is inhibited by misfolded proteins in the affected tissues of diverse polyQ diseases, such as SBMA, HD and SCA17 (36–38).
Mutant AR was preferentially degraded compared with wild-type AR in the presence of PF in both the cell culture and mouse models of SBMA. Mutant AR elimination was mediated by the upregulation of molecular chaperones and CHIP in which it was then prone to proteasomal degradation (39). Hsp70 and Hsp40 suppress aggregate formation and cellular toxicity in a wide range of polyQ disease models (30,40–43). CHIP interacts with Hsp90 or Hsp70 and ubiquitinates and degrades unfolded proteins trapped by molecular chaperones (44). A wide variety of proteins have been identified as CHIP substrates, including polyQ disease-causative proteins (45). Unfolded mutant proteins are more efficiently ubiquitinated by CHIP in the presence of molecular chaperones (12). Furthermore, mutant AR with a longer polyQ length is more efficiently ubiquitinated by CHIP (46). Based on these findings, the upregulation of CHIP expression by PF treatment may facilitate the preferential degradation of mutant AR in close cooperation with upregulated molecular chaperones.
Our experiments demonstrated that PF elevated the expression of TFEB and promoted the degradation of ARs via the activation of the autophagy system. AR has been demonstrated to be a direct target of autophagy (47,48). TFEB is a master regulator of lysosomal biogenesis and function (14) and upregulates lysosomal pathways and autophagy by modulating the expression of lysosomal enzymes involved in protein degradation (15). TFEB also regulates lysosomal proteostasis (49) and exocytosis (50). Importantly, TFEB regulates the clearance of disease-causing proteins, including huntingtin (14) and Aβ (51), and its overexpression reduces the expression level of polyQ-expanded huntingtin protein (52). However, chronically enhanced autophagy contributed to the detrimental consequences in the skeletal muscle of an SBMA mouse model (53). Although future studies should weigh the detrimental effects of chronically inducing autophagy to address proteinopathies against our observation of beneficially elevated autophagy in SBMA, sufficient inhibition of AR accumulation may overcome the deleterious effects of autophagy induction on muscle atrophy. Overall, based on our observations and taking the results from the previous studies into account, PF treatment promoted the degradation of both wild-type and mutant ARs by activating lysosomal biogenesis via the TFEB pathway and thereby restored the phenotypic defects in the SBMA mouse model. Collectively, PF treatment could provide a two-pronged cooperative therapeutic effect against SBMA pathogenesis by activating the molecular chaperone–UPS system and the autophagy system to degrade the mutant AR.
In conclusion, our present study demonstrated the safety and efficacy of PF in a mouse model of SBMA. PF potently induced the proteolytic machinery of UPS and autophagy, which lead to mutant AR degradation. In SBMA patients, the diffuse nuclear accumulation of mutant AR is frequent and extensive and occurs both in regions of the central nervous system and in muscles. The accumulation of pathogenic proteins causes many age-related neurodegenerative diseases. Inducers of the proteolytic machinery appear to be potent agents for disease-modifying therapies that inhibit the pathogenic process of neuron degeneration (54); thus, the pharmacological induction of the proteolytic machinery through PF treatment appears to be an applicable therapeutic strategy for multiple neurodegenerative disorders, including SBMA.
MATERIALS AND METHODS
Plasmid constructs and siRNA
pcDNA3.1/V5-His-NF-YA plasmid (37) was kindly provided by Dr Nobuyuki Nukina in the Department of Neuroscience for Neurodegenerative Disorders, Juntendo University Graduate School of Medicine. The plasmids pCR3.1-AR-24Q and pCR3.1-AR-97Q (32) were also used. The human TFEB cDNA was cloned into pcDNA 6.2/V5-GW/D-TOPO (Invitrogen) from the RNA of SH-SY5Y human neuroblastoma cells (ATCC No. CRL-2266). All of the constructs were verified by DNA sequencing. For the knock-down of NF-YA, 10 nm siRNA was used with the following sequence: 5′-CAGUAUCACACCGCAUCCUUATT-3′ (sense) and 5′-UAAGGAUGCGGUGAUACUGTT-3′ (anti-sense). We used MISSION siRNA Universal Negative Control (Sigma) as the control siRNA.
Cell culture and DNA transfection
NSC34 cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) with plasmids encoding ARs containing normal (24 CAGs) or expanded (97 CAGs) polyQ repeats (32). Stable clones expressing these normal and mutant ARs were established by selection with the antibiotic G418 (0.4 mg/ml final concentration). The AR is not expressed in non-transfected NSC34 cells. All cell cultures were propagated in the absence of androgen. In western blots from these cultures, we detected a band of monomeric mutant AR in the separating gel but could hardly detect the high-molecular-weight mutant AR protein complex, which was retained in the stacking gel. Therefore, this cultured cell model is better suited for estimating the change in monomeric mutant AR expression.
Wild-type Neuro2A cells or NSC34 cells and NSC34 cells stably expressing the human AR24Q or AR97Q were plated in six-well dishes in 5 ml of DMEM/10% fetal bovine serum (FBS) (Dulbecco's modified Eagle's medium supplemented with 10% FBS) with penicillin and streptomycin. Each dish was transfected with 4 μg of DNA plasmids and siRNA or control (mock) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. NSC34 cells were cultured in differentiation medium (DMEM/2% FBS) containing dihydrotestosterone (DHT; 1 nm) after transfection. The transfection efficiency was 60–70%. Neuro2A cells were cultured in differentiation medium (DMEM/2% FBS) containing retinoic acid (20 μm) and DHT (1 nm) after transfection. The transfection efficiency was 60–70%.
Promoter assay
GoClone and the pGL4 Luciferase Reporter Vector encoding the NF-YA, TFEB, Hsp70 and CHIP promoter (SwitchGear Genomics) were transfected into NSC34 cells with Lipofectamine 2000 (Invitrogen). The Steady-Glo Luciferase Assay System (Promega) was used to measure expression with POWERSCAN 4 (DS Pharma Biomedical).
Generation and maintenance of transgenic mice
We used AR-24Q and AR-97Q male mice in this study (21). The AR-97Q (Line #7–8) male mice showed progressive muscular atrophy and weakness accompanied by diffuse nuclear staining and NIs of the mutant AR. Similar to SBMA patients, these phenotypes were very pronounced in the male transgenic mice. The littermates were also used for phenotypic analyses. All of the animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and under the approval of the Nagoya University Animal Experiment Committee.
Therapeutic agents and protocol for administration
PF, purchased from Wako Pure Chemical Industries (Japan) (molecular weight: 480.45, 10 mg), was dissolved in 1 ml of PBS, diluted for use in culture medium and stored in the freezer until use. For the SBMA mouse model, we stored 10 mg/ml stock solutions of PF dissolved in a 0.9% NaCl solution at −20°C. To demonstrate that PF slows the onset and progression of the disease, PF treatments were initiated when the mice reached 5 weeks of age and continued until they were 32 weeks old. The stock solution was diluted to 1 mg/ml with saline solution just before the experiment, and 6.7 or 13.4 mg/kg of PF was administered intraperitoneally every day. The control mice received saline.
Neurological and behavioral assessments of the SBMA mouse model
Mice of varying ages were assessed with two different behavioral tests by investigators blind to the genotype and treatment. The rotarod task (Ugo Basile) was performed on a weekly basis, and cage activity was measured weekly with the AB system (Neuroscience) (21). Spontaneous motor activity was monitored for periods of 24 h; all spontaneous movements, both vertical and horizontal, including locomotion, rearing and head movements, were counted and automatically totaled.
Immunohistochemistry and histopathology
Mice were deeply anesthetized with pentobarbital sodium. The spinal cord and skeletal muscle tissues were removed, fixed overnight in 10% phosphate-buffered formalin and processed for paraffin embedding. Sections (6 μm thick) of tissue were deparaffinized, dehydrated with alcohol and treated in formic acid for 5 min at room temperature. For the immunohistochemical studies, the paraffin sections were preheated in a microwave oven for 10 min and blocked with normal animal serum (1:20). The sections were then incubated with anti-expanded polyQ antibody (1:10 000; 1C2; Millipore), anti-NF-YA antibody (1:1000; H-209; Santa Cruz), anti-TFEB antibody (1:1000; Medical and Biological Laboratories) and anti-GFAP antibody (1:1000; Roche Diagnostics). The primary antibodies were probed with a biotinylated anti-species-specific IgG (Vector), and the immune complexes were visualized using streptavidin–horseradish peroxidase (HRP) (Dako) and 3,3-diaminobenzidine (Dojindo) as a substrate. The sections were counterstained with Mayer's hematoxylin. Paraffin-embedded sections (6 μm thick) of the gastrocnemius muscles were air-dried and stained with hematoxylin and eosin. For double-immunofluorescence staining, the sections were blocked with TSA Blocking Reagent (PerkinElmer) and then sequentially incubated with anti-NF-YA antibody (1:1000; H-209; Santa Cruz) and 1C2 antibody (1:10000; Millipore) at 4°C overnight. The sections were then incubated with Alexa 488-conjugated goat anti-rabbit IgG (1:1000; Invitrogen) and Alexa 546-conjugated goat anti-mouse IgG (1:1300; Invitrogen) for 8 h at 4°C. The stained sections were examined and imaged with a confocal laser-scanning microscope (LSM 710; Carl Zeiss).
Patients
Tissue was obtained from three patients with clinicopathologically and genetically confirmed SBMA (51–77 years of age; mean, 65.7 years) and three non-neurological controls (51–78 years of age; mean, 64.0 years). The patients were hospitalized and evaluated at Nagoya University Hospital and affiliated hospitals. The collection of human tissues and their use for this study were approved by the Ethics Committee of Nagoya University Graduate School of Medicine. Informed consent was obtained to use the tissues for research purposes. Paraffin-embedded sections of the spinal cord were processed and examined in the same manner as those of the transgenic mice.
Quantification of 1C2-positive cells
To assess 1C2-positive cells in the ventral horn of the spinal cord, 50 consecutive transverse sections of the thoracic spinal cord were prepared from each individual mouse. 1C2-positive cells within the ventral horn of every fifth section were counted (55). Populations of 1C2-positive cells were expressed as the number/mm2 (Fig. 5B). To examine 1C2-positive cells in the muscle, the number of 1C2-positive cells was calculated from counts of 500 fibers in randomly selected areas and expressed as the number per 100 muscle fibers (Fig. 5B). The quantitative data of six individual mice were expressed as the mean ± SEM.
Protein expression analysis
Forty-eight hours after transfection, the cells were lysed in CelLytic-M Mammalian Cell Lysis/Extraction Reagent (Sigma), supplemented with 1 mm phenylmethylsulfonyl fluoride (PMSF) and 6 µg/ml aprotinin and centrifuged at 1000g for 15 min at 4°C. NE-PER Nuclear Cytoplasmic Reagents (Thermo Scientific) were used for the analysis of the nuclear/cytoplasmic translocation of TFEB. Sixteen-week-old mice were exsanguinated under pentobarbital sodium anesthesia, and the tissues were snap-frozen with powdered CO2 in acetone. The tissues were homogenized in CelLytic-M Mammalian Cell Lysis/Extraction Reagent with 1 mm PMSF and 6 μg/ml aprotinin and centrifuged at 2500g for 15 min at 4°C. The supernatant protein concentrations were determined using the DC protein assay (Bio-Rad). Aliquots of the supernatant fractions were loaded on 5–20% SDS–PAGE gels, where each lane contained 7 μg of cell protein and 40 μg of neural and muscle tissue. The gels were then transferred to Hybond-P membranes (GE Healthcare) using transfer buffer (25 mm Tris, 192 mm glycine, 0.1% SDS, 10% methanol). The following antibodies were used in our studies: anti-AR (1:1000; N20; Santa Cruz), anti-AR (1:1000; H280; Santa Cruz), anti-NF-YA (1:1000; H-209; Santa Cruz), anti-TFEB (1:5000; ab2636; Abcam), anti-HSP70 (1:10 000; SPA-810; Enzo Life Sciences), anti-HSP70 (1:1000; #4872; Cell Signaling Technology), anti-HSP40 (1:10 000; SPA-400; Enzo Life Sciences), anti-HSP40 (1:1000; #4868; Cell Signaling Technology), anti-CHIP (1:1000; #C3B6; Cell Signaling Technology), mouse anti-GAPDH (1:10000; MAB374; Millipore), mouse anti-α-tubulin (1:5000, T9026; Sigma), anti-LC3 (1:1000; #D11; Cell Signaling Technology), anti-V5 (1:5000; PM003; Medical & Biological Laboratories), anti-hexosaminidase A (1:1000; N-19; Santa Cruz), anti-Cathepsin B (1:2000; ab33538; Abcam), anti-Cathepsin D (1:2000; ab6313; Abcam), anti-tripeptidyl peptidase 1 (1:1000; G-16; Santa Cruz), anti-V-ATPase H (1:2000; H-300; Santa Cruz), anti-mTOR (1:2000; #2983; Cell Signaling Technology), anti-Phospho-mTOR (1:2000; #2974; Cell Signaling Technology), anti-p70 S6 Kinase (1:2000; #2708; Cell Signaling Technology), anti-Phospho-p70 S6 Kinase (1:2000; #9234; Cell Signaling Technology), anti-eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) (1:2000; #9644; Cell Signaling Technology), anti-Phospho-4E-BP1 (Thr37/46; 1:2000; #2855; Cell Signaling Technology) and anti-Beclin-1 (1:2000; #3495; Cell Signaling Technology). The primary antibodies were probed using HRP-conjugated anti-rabbit IgG F(ab′)2 and anti-mouse IgG F(ab′)2 (1:5000; GE Healthcare) secondary antibodies and were detected with the ECL Prime kit (GE Healthcare). An LAS-3000 imaging system was used to produce digital images and to quantify the band intensities, which were then analyzed with the Image Gauge software version 4.22 (Fujifilm). Densitometric values of ARs were normalized to endogenous GAPDH.
Pulse-chase labeling assay
NSC34 cells stably expressing the human AR24Q or AR97Q were plated in 6-cm dishes in 5 ml of DMEM/10% FBS containing DHT (1 nm) supplemented with penicillin and streptomycin. Each dish was treated with 10 μm of PF or PBS as a control. The cells were starved for 1 h in methionine- and cysteine-free DMEM/10% FBS and then labeled for 1 h with 150 Ci/ml of Redivue Pro-Mix L-[35S] in vitro cell-labeling mix (GE Healthcare). After washing in PBS, the cells were chased for the indicated time intervals in complete medium containing 10 μm of PF or saline. PF-treated cells were also treated with 20 nm bafilomycin A1 (11707; Sigma) or 2.5 μm lactacystin (PEPTIDE INSTITUTE, Inc.). Immunoprecipitation was performed using equivalent amounts of protein lysates, 50 μl of protein G Dynabeads (Invitrogen) and 5 μl of anti-AR antibody (N20; Santa Cruz) for 10 min at room temperature. Each sample was separated by 5–20% SDS–PAGE and analyzed by phosphorimaging (Typhoon LFA 9000 PhosphorImager; GE Healthcare) and Image Gauge software version 4.22 (Fujifilm).
Quantitative real-time reverse transcription–PCR
Total RNA was isolated from the NSC34 cells using the RNeasy Mini kit (Qiagen) and from mouse tissues using the PureLink RNA Mini kit (Invitrogen) according to the manufacturer's instructions. Total RNA from cells (3 μg) and mouse tissues (2.5 μg) was transcribed using the SuperScript VILO cDNA Synthesis Kit (Invitrogen). Real-time RT–PCR was performed in a total volume of 25 μl containing 12.5 μl of 2× QuantiFast SYBR Green PCR Master Mix (Qiagen) and 1 μm of each primer. The PCR products were detected by the iCycler system (Bio-Rad). The reaction conditions were 95°C for 5 min, 40 cycles of 10 s at 95°C and 30 s at 60°C. As an internal standard control, the expression level of β2 microglobulin was simultaneously quantified. The primer sequences are listed in Supplementary Material, Table S1.
Cell viability assay
The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS)-based cell proliferation assay was performed in triplicate in NSC34 cells stably expressing the human AR24Q or AR97Q using the Cell Proliferation Reagent WST-1 (Roche Applied Science) and the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) 48 h after incubation with PF.
Serum testosterone assay
Mice receiving PF or saline were deeply anesthetized with pentobarbital sodium at 16 weeks of age, 1 ml of blood was collected by cardiocentesis, and serum testosterone was assayed with the Coat-A-Count Total Testosterone radioimmunoassay (Roche) according to the manufacturer's instructions.
Statistical analysis
The data were analyzed using unpaired t-tests and post hoc tests (Tukey–Kramer post hoc tests) for multiple comparisons. The Kaplan–Meier and log-rank tests were used to assess the survival rate using StatView software version 5 (Hulinks).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by a Center-of-Excellence (COE) grant, KAKENHI (20390243) and additional grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
ACKNOWLEDGEMENTS
We thank Miwa Ito and Kazuko Matsuba in the Department of Neurology and Yasutaka Ohya and Kumiko Yano in the Division for Research of Laboratory Animals at the Center for Research of Laboratory Animals and Medical Research Engineering for their technical assistance. We also thank Dr Nobuyuki Nukina in the Department of Neuroscience for Neurodegenerative Disorders, Juntendo University Graduate School of Medicine, for kindly providing the vectors containing NF-YA.
Conflict of Interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}