



Abstract
Spinocerebellar ataxia type 35 (SCA35) is a rare autosomal-dominant neurodegenerative disease caused by mutations in the TGM6 gene, which codes for transglutaminase 6 (TG6). Mutations in TG6 induce cerebellar degeneration by an unknown mechanism. We identified seven patients bearing new mutations in TGM6. To gain insights into the molecular basis of mutant TG6-induced neurotoxicity, we analyzed all the seven new TG6 mutants and the five TG6 mutants previously linked to SCA35. We found that the wild-type (TG6-WT) protein mainly localized to the nucleus and perinuclear area, whereas five TG6 mutations showed nuclear depletion, increased accumulation in the perinuclear area, insolubility and loss of enzymatic function. Aberrant accumulation of these TG6 mutants in the perinuclear area led to activation of the unfolded protein response (UPR), suggesting that specific TG6 mutants elicit an endoplasmic reticulum stress response. Mutations associated with activation of the UPR caused death of primary neurons and reduced the survival of novel Drosophila melanogaster models of SCA35. These results indicate that mutations differently impacting on TG6 function cause neuronal dysfunction and death through diverse mechanisms and highlight the UPR as a potential therapeutic target for patient treatment.
Introduction
Spinocerebellar ataxia (SCA) type 35 is a rare, autosomal-dominant neurodegenerative disorder characterized by gait and limb ataxia, ocular dysmetria, tremor, hyperreflexia and sometimes dystonia. Disease onset ranges from teenage years to late adulthood with a slow progression. In addition to the typical symptoms associated with cerebellar ataxia, a considerable heterogeneity in the phenotype of patients has been reported with cases of mild mental retardation (1,2). SCA35 was first described in a four-generation Chinese family pedigree, in which nine individuals developed symptoms (3). The disease was later on diagnosed in another three-generation family pedigree with seven affected individuals (2), and in four patients from three Han Chinese families (1). Brain magnetic resonance imaging showed cerebellar atrophy particularly evident in the vermis (3). However, a detailed analysis of the cerebellar neurons undergoing degeneration in SCA35 is still missing. Genome-wide linkage analysis of the first described SCA35 family identified an 8.5-Mb locus on chromosome 20p13-p12.2 associated with the disease (3). Exome sequencing revealed that SCA35 is caused by mutations in the TGM6 gene, which codes for transglutaminase 6 (TG6) (3). This analysis led to identification of two missense mutations, i.e. leucine 517-to-tryptophan (L517W) and aspartic acid 327-to-glycine (D327G) substitutions (3). Subsequently, two additional missense mutations, arginine 111-to-cysteine (R111C) and aspartic acid 510-to-histidine (D510H) substitutions, as well as deletion of glutamate 574 (E574del), were linked to SCA35 (1).
TG6 is a calcium-dependent enzyme that belongs to a family of proteins including eight other members encoded by closely related genes, namely TGM1-7, F13A1 and EPB42 (4). These enzymes are differentially expressed in the organs and tissues, except for TG2 that is ubiquitous. TG6 is specifically expressed in the central nervous system, in particular in neuronal cells of the olfactory lobe, layers II–IV of the cerebral cortex, cerebellum and brainstem (5). Similar to the other members of the TG family, TG6 is composed of four structurally distinct domains, namely an N-terminal β-sandwich domain, a catalytic core, and two β-barrel domains. The catalytic core harbors three critical amino acids (cysteine, histidine and aspartic acid), through which TG6 binds to glutamine residues to form a thioester intermediate. The thioester intermediate is then attacked by the surface amine of a second substrate that can be either a peptide-bound lysine residue or a structurally related primary amine. The end product of the reaction is a stable isopeptide bond between the two substrates, also known as transglutaminase-catalyzed protein crosslinking (5,6). The reaction can be intra- or inter-molecular and typically occurs at high intracellular calcium levels (>100 µm). This enzymatic activity is inhibited by the nucleotide guanosine triphosphate (GTP), which binds to TG6 at the interface between the core and first beta-barrel domain, leading to a conformational change that makes the catalytic site inaccessible for transamidation (5). Mutations in TG6 have been reported to decrease its transamidase activity and increase susceptibility to staurosporine-induced apoptosis in non-neuronal cells (7). However, the molecular mechanism through which mutant TG6 induces cerebellar damage in SCA35 remains elusive. Here, we show that specific TG6 mutations result in a nearly complete loss of protein function, nuclear depletion, enrichment in the perinuclear area, insolubility and activation of the unfolded protein response (UPR). Overexpression of TG6-WT and TG6 mutants with normal subcellular localization caused neuronal death, indicating that dysregulation of TG6 activity is neurotoxic. Notably, overexpression of those TG6 mutants that induce an UPR resulted in enhanced toxicity compared with TG6-WT, suggesting that activation of the UPR is a pathogenic event in SCA35.
Results
Identification of new pathogenetic mutations of TG6 linked to SCA35
In search for TG6 mutations in SCA patients with undefined genetic diagnosis, we sequenced the TGM6 gene in 963 patients affected by hereditary forms of SCA. Analysis was performed in four different centers in Paris, Sheffield, Cardiff and Boston. We found five mutations, i.e. glutamine-181-to-histidine (TG6-Q181H), valine-391-to-methionine (TG6-V391M), tyrosine-441-to-cytosine (TG6-Y441C), arginine-448-to-tryptophan (TG6-R448W), and leucine-502-to-glutamine (TG6-L502Q), which are predicted to be damaging by SIFT and PolyPhen (Table 1). In addition, we found two mutations presenting a duplication, i.e. glutamine-652-duplication (TG6-Q652dup) and missense mutation plus duplication (TG6-L502Q Q652dup) (Table 1). All the newly identified SCA35 patients were heterozygous for the mutation, with an age of onset between late teens and 50 years old, and generally showed spasticity and cerebellar ataxia (Table 2).
List of TG6 variants found in patients affected by hereditary forms of SCAs
| Nucleotide change | Amino acid change | ExAC | gnomAD | Amino acid conservation | SIFT | Score SIFT | PolyPhen | ||
|---|---|---|---|---|---|---|---|---|---|
| Allele count | Ethnicity | Allele count | Ethnicity | ||||||
| c.543G>T | Q181H | 0 | – | 0 | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING | |
| c.1171G>A | V391M | 109 | Asian | 193 | Mixed | Very well conserved | DAMAGING | 0.03 | PROBABLY DAMAGING |
| c.1322A>G | Y441C | 0 | – | 2 | European | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING |
| c.1342C>T | R448W | 1306 | Mixed | 3752 | Mixed | Conserved across Mammals | DAMAGING | 0.02 | PROBABLY DAMAGING |
| c.1505T>A | L502Q | 0 | – | 2 | European | Conserved across Mammals and X.tropicalis | TOLERATED | 0.35 | PROBABLY DAMAGING |
| c.1630G>T | A544S | 6 | European | 16 | Mostly European | Well conserved, except in X.tropicalis | TOLERATED | 0.82 | BENIGN |
| c.1951_1952insAAC | Q652dup | 162 | European | 359 | Mostly European | Well conserved, except in D.rerio | – | – | – |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 0 | 0 | – | – | – | |||
| Nucleotide change | Amino acid change | ExAC | gnomAD | Amino acid conservation | SIFT | Score SIFT | PolyPhen | ||
|---|---|---|---|---|---|---|---|---|---|
| Allele count | Ethnicity | Allele count | Ethnicity | ||||||
| c.543G>T | Q181H | 0 | – | 0 | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING | |
| c.1171G>A | V391M | 109 | Asian | 193 | Mixed | Very well conserved | DAMAGING | 0.03 | PROBABLY DAMAGING |
| c.1322A>G | Y441C | 0 | – | 2 | European | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING |
| c.1342C>T | R448W | 1306 | Mixed | 3752 | Mixed | Conserved across Mammals | DAMAGING | 0.02 | PROBABLY DAMAGING |
| c.1505T>A | L502Q | 0 | – | 2 | European | Conserved across Mammals and X.tropicalis | TOLERATED | 0.35 | PROBABLY DAMAGING |
| c.1630G>T | A544S | 6 | European | 16 | Mostly European | Well conserved, except in X.tropicalis | TOLERATED | 0.82 | BENIGN |
| c.1951_1952insAAC | Q652dup | 162 | European | 359 | Mostly European | Well conserved, except in D.rerio | – | – | – |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 0 | 0 | – | – | – | |||
List of TG6 variants found in patients affected by hereditary forms of SCAs
| Nucleotide change | Amino acid change | ExAC | gnomAD | Amino acid conservation | SIFT | Score SIFT | PolyPhen | ||
|---|---|---|---|---|---|---|---|---|---|
| Allele count | Ethnicity | Allele count | Ethnicity | ||||||
| c.543G>T | Q181H | 0 | – | 0 | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING | |
| c.1171G>A | V391M | 109 | Asian | 193 | Mixed | Very well conserved | DAMAGING | 0.03 | PROBABLY DAMAGING |
| c.1322A>G | Y441C | 0 | – | 2 | European | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING |
| c.1342C>T | R448W | 1306 | Mixed | 3752 | Mixed | Conserved across Mammals | DAMAGING | 0.02 | PROBABLY DAMAGING |
| c.1505T>A | L502Q | 0 | – | 2 | European | Conserved across Mammals and X.tropicalis | TOLERATED | 0.35 | PROBABLY DAMAGING |
| c.1630G>T | A544S | 6 | European | 16 | Mostly European | Well conserved, except in X.tropicalis | TOLERATED | 0.82 | BENIGN |
| c.1951_1952insAAC | Q652dup | 162 | European | 359 | Mostly European | Well conserved, except in D.rerio | – | – | – |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 0 | 0 | – | – | – | |||
| Nucleotide change | Amino acid change | ExAC | gnomAD | Amino acid conservation | SIFT | Score SIFT | PolyPhen | ||
|---|---|---|---|---|---|---|---|---|---|
| Allele count | Ethnicity | Allele count | Ethnicity | ||||||
| c.543G>T | Q181H | 0 | – | 0 | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING | |
| c.1171G>A | V391M | 109 | Asian | 193 | Mixed | Very well conserved | DAMAGING | 0.03 | PROBABLY DAMAGING |
| c.1322A>G | Y441C | 0 | – | 2 | European | Very well conserved | DAMAGING | 0 | PROBABLY DAMAGING |
| c.1342C>T | R448W | 1306 | Mixed | 3752 | Mixed | Conserved across Mammals | DAMAGING | 0.02 | PROBABLY DAMAGING |
| c.1505T>A | L502Q | 0 | – | 2 | European | Conserved across Mammals and X.tropicalis | TOLERATED | 0.35 | PROBABLY DAMAGING |
| c.1630G>T | A544S | 6 | European | 16 | Mostly European | Well conserved, except in X.tropicalis | TOLERATED | 0.82 | BENIGN |
| c.1951_1952insAAC | Q652dup | 162 | European | 359 | Mostly European | Well conserved, except in D.rerio | – | – | – |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 0 | 0 | – | – | – | |||
Clinical features
| Nucleotide change | Amino acid change | Age of onset | Progression | Main symptoms |
|---|---|---|---|---|
| c.543G>T | Q181H | 19 | Slow | Ataxia, myoclonus, epilepsy, mobile without needing a walking aid |
| c.1171G>A | V391M | 36 | Slow | Ataxia with predominant gait disorder requiring a cane, extra-pyramidal signs (hypophonia, rigidity and bradykinesia); dystonic posturing of hands and fee |
| c.1322A>G | Y441C | 54 | Slow/ moderate | Ataxia; spasticity, pyramidal features; dysarthria, swallowing difficulties |
| c.1342C>T | R448W | 20 | Slow | Gait ataxia without any additional features |
| c.1505T>A | L502Q | >50 | Slow | Pure ataxia |
| c.1951_1952insAAC | Q652dup | NA | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 40s | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
| Nucleotide change | Amino acid change | Age of onset | Progression | Main symptoms |
|---|---|---|---|---|
| c.543G>T | Q181H | 19 | Slow | Ataxia, myoclonus, epilepsy, mobile without needing a walking aid |
| c.1171G>A | V391M | 36 | Slow | Ataxia with predominant gait disorder requiring a cane, extra-pyramidal signs (hypophonia, rigidity and bradykinesia); dystonic posturing of hands and fee |
| c.1322A>G | Y441C | 54 | Slow/ moderate | Ataxia; spasticity, pyramidal features; dysarthria, swallowing difficulties |
| c.1342C>T | R448W | 20 | Slow | Gait ataxia without any additional features |
| c.1505T>A | L502Q | >50 | Slow | Pure ataxia |
| c.1951_1952insAAC | Q652dup | NA | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 40s | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
Clinical features
| Nucleotide change | Amino acid change | Age of onset | Progression | Main symptoms |
|---|---|---|---|---|
| c.543G>T | Q181H | 19 | Slow | Ataxia, myoclonus, epilepsy, mobile without needing a walking aid |
| c.1171G>A | V391M | 36 | Slow | Ataxia with predominant gait disorder requiring a cane, extra-pyramidal signs (hypophonia, rigidity and bradykinesia); dystonic posturing of hands and fee |
| c.1322A>G | Y441C | 54 | Slow/ moderate | Ataxia; spasticity, pyramidal features; dysarthria, swallowing difficulties |
| c.1342C>T | R448W | 20 | Slow | Gait ataxia without any additional features |
| c.1505T>A | L502Q | >50 | Slow | Pure ataxia |
| c.1951_1952insAAC | Q652dup | NA | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 40s | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
| Nucleotide change | Amino acid change | Age of onset | Progression | Main symptoms |
|---|---|---|---|---|
| c.543G>T | Q181H | 19 | Slow | Ataxia, myoclonus, epilepsy, mobile without needing a walking aid |
| c.1171G>A | V391M | 36 | Slow | Ataxia with predominant gait disorder requiring a cane, extra-pyramidal signs (hypophonia, rigidity and bradykinesia); dystonic posturing of hands and fee |
| c.1322A>G | Y441C | 54 | Slow/ moderate | Ataxia; spasticity, pyramidal features; dysarthria, swallowing difficulties |
| c.1342C>T | R448W | 20 | Slow | Gait ataxia without any additional features |
| c.1505T>A | L502Q | >50 | Slow | Pure ataxia |
| c.1951_1952insAAC | Q652dup | NA | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
| c.1505T>A; c.1951_1952insAAC | L502Q Q562dup | 40s | Slow | Mainly gait ataxia; myoclonus; uses one stick to walk |
Mutations in TG6 modify its subcellular distribution
To investigate the mechanism through which the newly identified TG6 mutants cause SCA35, we generated vectors to express all these TG6 mutants in mammalian cells and compared their behavior with the five previously identified TG6-mutants (TG6-R111C, TG6-D327G, TG6-D510H, TG6-L517W and TG6-E574del). In addition, we analyzed one variant predicted to be well tolerated and benign (TG6-A544S) by SIFT and Polyphen as internal control of our analysis (Table 1). We asked whether mutations in TG6 alter its subcellular localization. To address this question, we first analyzed the subcellular distribution of endogenous TG6 in the mouse cerebellum, as well as in cultured primary mouse cerebellar and cortical neurons (Fig. 1A and B and Supplementary Material, Fig. S1). TG6 was expressed in the granular layer of the cerebellar cortex and to a lower extent in Purkinje cells (Fig. 1A and inset in Fig. 1). TG6 mostly localized in the nucleus with a marginal localization at the perinuclear area in the cerebellar sections as well as in cerebellar and cortical primary neurons (Fig. 1A and B and Supplementary Material, Fig. S1). Next, we analyzed the subcellular distribution of TG6-WT and all the mutations listed above in COS-7 cells, which do not express endogenous TG6 (Fig. 2A). Similar to what we observed in cultured neurons and in vivo, TG6-WT localized in both the nuclear and perinuclear compartments, with 70% of the protein in the nucleus (Fig. 2A). Among the TG6 mutants analyzed here, TG6-A544S, TG6-D327G, TG6-V391M, TG6-R448W, TG6-L502Q, TG6-L502Q Q652dup, TG6-D510H and TG6-Q652dup showed a pattern of distribution similar to that of TG6-WT. Surprisingly, we found that TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W and TG6-E574del localized almost exclusively in the perinuclear/cytosolic space (P = 0.001). To determine whether the increased perinuclear staining of TG6 mutants matched with the endoplasmic reticulum (ER), we performed co-localization experiments of TG6-WT and TG6-R111C with the ER resident protein, calnexin (Fig. 2B). We found that 13 and 45% of TG6-WT and TG6-R111C, respectively, co-localized with calnexin, indicating that the ER localization of TG6-R111C was increased compared with TG6-WT. Similar results were obtained in primary cerebellar neurons transfected with TG6-WT and TG6-R111C (Fig. 2C), and in cortical neurons transduced with lentiviruses expressing TG6 under the control of the synapsin promoter (Supplementary Material, Fig. S4A). These results indicate that specific mutations in TG6, i.e. TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W and TG6-E574del, result in loss of nuclear TG6 with increased accumulation of the mutant protein at the level of the ER.
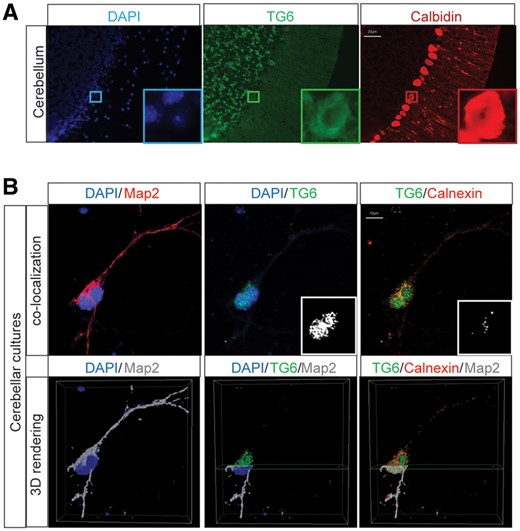
TG6 is expressed in cerebellar neurons and Purkinje cells. (A) Immunohistochemical analysis of TG6 in cerebellar sections of 2-month-old mice revealed that TG6 localizes to the soma of cells present in the granular layer and calbindin-positive cells (Purkinje neurons). Bar, 20 µm. Inset, a Purkinje neuron expressing TG6 in the cell body. (B) Immunocytochemical analysis of TG6 and Map2 in DIV7 primary cerebellar neurons showed that TG6 is mostly expressed in the nucleus and perinuclear area. Bottom panel, 3D rendering of a neuron showing that the majority of endogenous TG6 localizes in the nucleus. Bar, 10 µm. Shown are representative images from three mice and three cerebellar cultures. TG6, calbindin, calnexin and Map2 were detected with specific antibodies, nuclei were detected with DAPI.
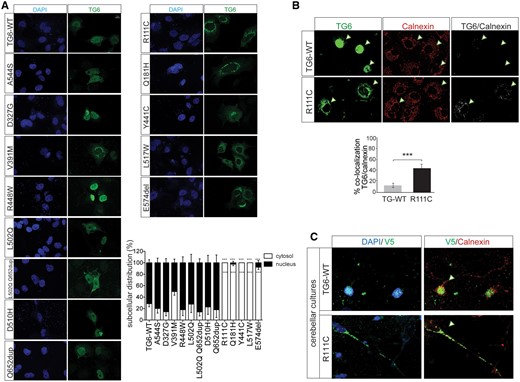
Mutations in TG6 alter its subcellular distribution. (A) Immunocytochemical analysis of TG6 subcellular distribution in COS-7 cells showed decreased nuclear localization of TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W and TG6-E574del. Shown are representative images from three independent experiments. Bar, 10 µm. Quantification of the subcellular distribution of the indicated TG6 mutants is shown at the bottom. Graph, mean±SEM, n = 100 cells from three independent experiments; ***P <0.001, 2-way ANOVA with Bonferroni’s post hoc test. (B) Representative confocal images of TG6 subcellular localization in COS-7 cells expressing TG6-WT and TG6-R111C showed TG6 co-localization with calnexin. Arrowheads indicate co-localization of TG6 and calnexin. Graph, mean±SEM, Student’s t test, ***P<0.001. (C) Representative confocal images of TG6 subcellular localization in DIV7 cerebellar neurons transfected with vectors expressing V5-tagged TG6-WT and TG6-R111C. Nuclei were stained with DAPI, and ER was revealed with calnexin. Arrowheads highlight the different subcellular localization of TG6-WT and TG6-R111C.
Mutations in TG6 affect transamidase function
Mutations in TG6 have previously been reported to reduce its enzymatic activity (1,8). We investigated whether the newly identified TG6 mutants show defects in transamidase activity. We measured TG6 transamidase activity using a biotinylated peptide that is irreversibly cross-linked to proteins in the presence of high calcium concentration (5 mm) and can be detected in streptavidin blotting (Fig. 3) (9). TG6 enzymatic activity was normalized to the amount of TG6 present in each cell lysate. In agreement with previous data, the transamidase activity of TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W, TG6-E574del, TG6-D327G and TG6-V391M was significantly decreased by 86, 60, 77, 90, 80, 54 and 77%, respectively, compared with TG6-WT. The activity of all the other TG6 mutants was similar or slightly reduced compared with TG6-WT. These observations indicate that, except for TG6-D327G and TG6-V391M, specific mutations in TG6 that alter its subcellular localization result in a substantial loss of protein function, with nearly 10–40% residual activity left.
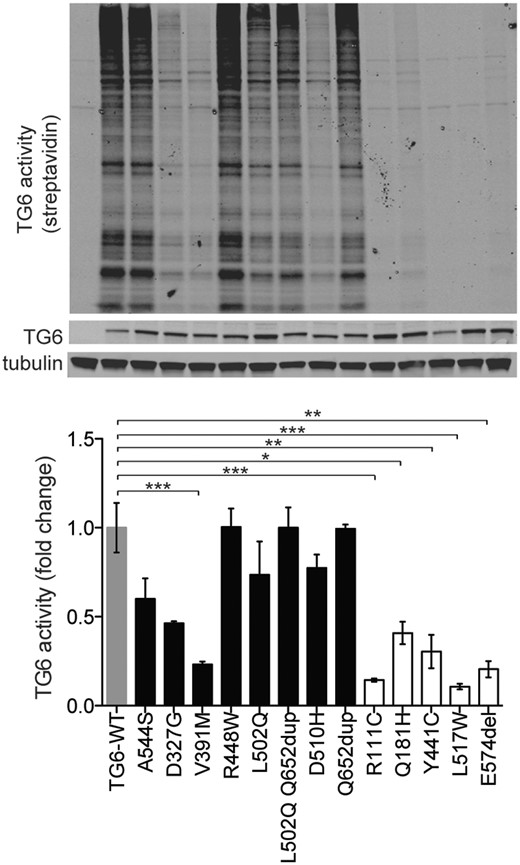
Mutations in TG6 alter its enzymatic activity. Immunoblot (top) and analysis of enzymatic activity (bottom) in transiently transfected HEK293T cells showed a significant reduction of transamidase activity of TG6-V391M, TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W and TG6-E574del. TG6 activity was revealed with streptavidin conjugated to a fluorophore with emission at 800 nm and it is shown as fold change where TG6-WT activity was set as 1. Graph, mean±SEM, *P <0.05; **P <0.01; ***P <0.001, 1-way ANOVA with Tukey’s post hoc test.
TG6 mutants interact with and affect the function of TG6-WT
Since SCA35 patients are all heterozygous for the mutations, we asked whether mutations in TG6 exert dominant-negative effects on the WT counterpart. To address this question, we generated V5- and HA-tagged TG6-WT, TG6-R111C, which shows altered subcellular distribution and almost complete loss of function, and TG6-D327G, which shows normal subcellular distribution and partial loss of function. The enzymatic activity of the tagged proteins was similar to that of untagged TG6, indicating that fusion of these tags does not alter TG6 function (Supplementary Material, Fig. S2A and B). By immunoprecipitation assay, we found that TG6-WT forms a complex with TG6-R111C and TG6-D327G (Fig. 4A). To determine in which subcellular compartment TG6-WT and the TG6 mutants interact with each other, we used the proximity ligation assay (PLA), which is based on the employment of oligonucleotide-conjugated secondary antibodies to detect in situ protein–protein interactions (see Materials and Methods) (10). By expressing HA-tagged TG6-WT together with V5-tagged TG6-WT and TG6 mutants, we found that TG6-WT forms a complex with TG6-WT and TG6-D327G in the nucleus, and with TG6-R111C in the perinuclear space (Fig. 4B). Next, we tested whether TG6-R111C and TG6-D327G affect the function of TG6-WT (Fig. 4C). To achieve this aim, we expressed HA-tagged TG6-WT and V5-tagged TG6-WT alone and HA-tagged TG6-WT together with V5-tagged TG6-WT and TG6-mutants. The activity detected by expressing HA-tagged TG6-WT and V5-tagged TG6-WT alone was similar to that detected by co-expressing HA-tagged TG6-WT and V5-tagged TG6-WT together, indicating that this assay measures maximal TG6 activity in both conditions. Surprisingly, TG6-R111C significantly reduced the net enzymatic activity by 85%. On the other hand, TG6-D327G decreased TG6 activity by 58%, and this effect was not significant. These results indicate that the TG6 mutants tested here retain their ability to bind TG6-WT. However, TG6-R111C specifically altered the enzymatic activity of the WT counterpart, suggesting a dominant negative loss of function mechanism in SCA35 linked to the mutations that present an altered subcellular localization.
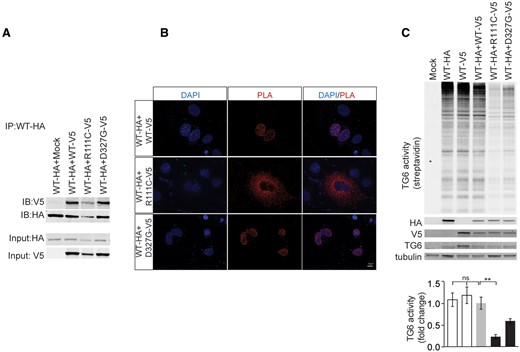
TG6 mutants interact with and affect the function of TG6-WT. (A) Immunoprecipitation assay in HEK293T cells transiently transfected with vectors expressing HA- and V5-tagged TG6-WT, TG6-R111C, TG6-D327G revealed that TG6 mutants form a complex with TG6-WT. Shown is one experiment representative of three. IP, immunoprecipitation with HA antibody-conjugated beads. IB, immunoblot with anti-HA and V5 antibodies. (B) The interaction between HA-tagged TG6-WT with V5-tagged TG6-WT or V5-tagged mutants was analyzed in intact cells by the PLA (red). The red dots represents areas of close proximity of HA and V5 epitopes. Nuclei were revealed with DAPI. Shown are representative images from three independent experiments. Bar, 10 µm. (C) Functional analysis of HA-tagged TG6-WT co-expressed with either V5-tagged TG6-WT or V5-tagged TG6 mutants in HEK293T cells revealed that TG6-R111C significantly reduce TG6-WT activity. The activity of co-expressed HA-tagged TG6-WT and V5-tagged TG6-WT was set as 1. Graph, mean±SEM, n =3; **P<0.01, 1-way ANOVA with Tukey’s post hoc test.
Specific mutations in TG6 enhance its degradation via the ubiquitin-proteasome system (UPS) and decrease its solubility
Mutations in TG6 linked to SCA35 have been previously shown to reduce protein stability (3). Therefore, we asked whether the reduction in mutant TG6 enzymatic activity was associated with an effect on protein turnover. To measure protein turnover, we expressed TG6-WT, TG6-R111C, TG6-D327G in HEK293T cells and examined TG6 levels 6 h after treatment with cycloheximide, an inhibitor of protein translation (Fig. 5A). Accumulation of TG6-WT was decreased by 23% in the presence of cycloheximide. Accumulation of TG6-D327G was decreased by about 27%, yet this difference was not significant compared with TG6-WT. On the other hand, accumulation of TG6-R111C was significantly decreased by 53%, indicating increased protein turnover. By real-time PCR, we verified that this effect on TG6-R111C accumulation was not due to decreased TG6 mRNA transcript level (Supplementary Material, Fig. S3). To establish whether the increased turnover of TG6 mutants was due to enhanced protein degradation through either the UPS or autophagy, we incubated the cells expressing TG6-R111C with cycloheximide together with the proteasome inhibitor, MG132, and the autophagy inhibitor, bafilomycin (BA) (Fig. 5B). The degradation of TG6-R111C was specifically inhibited by MG132, indicating that this TG6 mutant is mainly degraded via the UPS. We wondered whether the enhancement in turnover and degradation via proteasome of TG6-R111C was linked to increased protein misfolding. Misfolded proteins are prone to aggregate into oligomers and higher-order structures that are insoluble in buffers containing mild non-ionic detergents, such as NP40 (11). We lysed cells overexpressing TG6-WT, TG6-R111C and TG6-D327G in presence and absence of MG132, and we analyzed TG6 levels in the soluble and insoluble fractions (Fig. 5C). TG6-WT and TG6-D327G were recovered mostly in the soluble fraction. In absence of MG132, TG6-R111C was equally distributed in soluble and insoluble fractions. Notably, in the presence of MG132, TG6-R111C appeared to accumulate in the insoluble fraction (Fig. 5C). These observations indicate that TG6-R111C has decreased solubility with aggregation-prone forms readily degraded by the proteasome.
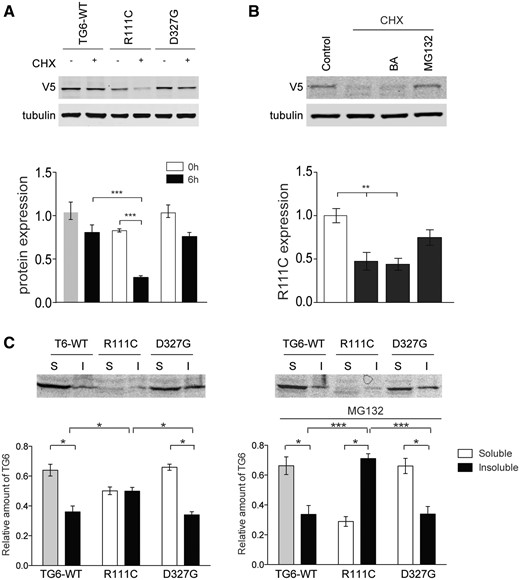
Increased turnover and decreased solubility of TG6-R111C. (A) Western blotting analysis of TG6-WT, TG6-R111C and TG6-D327G expression levels in HEK293T cells treated with cycloheximide (CHX, 0.1 mg/ml, 6 h) revealed that the turnover of TG6-R111C is increased compared with TG6-WT. Graph, mean±SEM, n = 3; ***P <0.001, 2-way ANOVA with Bonferroni’s post hoc test. (B) Western blotting analysis of TG6-R111C expression levels in HEK293T cells treated with cycloheximide (CHX, 0.1 mg/ml), the autopohagy inhibitor bafilomycin (BA, 200 nm) and the proteasome inhibitor MG132 (10 μm), revealed that TG6-R111C is degraded by the UPS. Graph, mean±SEM, n = 3, **P <0.01, 1-way ANOVA with Tukey’s post hoc test. (C) Western blotting analysis of TG6-WT, TG6-R111C and TG6-D327G recovery in soluble (S) and insoluble (I) fractions in HEK293T cells treated with and without MG132 (10 µm) showed increased accumulation of TG6-R111C in the insoluble fraction compared with TG6-WT and TG6-D327G. Graph, mean±SEM, n=3, *P <0.05; ***P <0.001; 2-way ANOVA with Bonferroni’s post hoc test.
Mutations in TG6 induce the UPR
Based on our observations that TG6-R111C show increased co-localization with the ER-resident protein, calnexin, we asked whether this specific mutation in TG6 leads to activation of the UPR. The UPR is initiated by three ER transmembrane proteins: Inositol requiring 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6). In normal conditions, the ER chaperone, immunoglobin binding protein (BiP), binds to the luminal domains of these UPR master regulators keeping them inactive (12). Upon ER stress, BiP dissociates from these sensors resulting in their activation. Moreover, BiP is induced at the transcription level. We transiently transfected TG6-WT, TG6-R111C and TG6-D327G in HEK293T cells and measured BiP levels. BiP mRNA transcript and protein levels were significantly upregulated by 2- and 1.4-fold, respectively, in cells expressing TG6-R111C compared with cells expressing TG6-WT or cells mock-transfected (Fig. 6A and B). No changes in BiP mRNA or protein levels were observed in cells expressing TG6-D327G. We also analyzed the expression levels of two additional markers of UPR activation: spliced XBP1 (sXBP1) and C/EBP homologous protein (CHOP). XBP1 is spliced upon IRE1 activation and autophosphorylation and controls the transcription of genes that increase the folding capacity of ER (13), while CHOP is a transcription factor induced by the PERK-eIF2α-ATF4 pathway with a role in ER stress-mediated apoptosis (13). sXBP1 mRNA was significantly increased by 2-fold in cells expressing TG6-R111C compared with cells transfected with TG6-WT and empty vector, while it was increased by 1.6-fold in cells expressing TG6-D327G without statistical significance (Fig. 6C). CHOP mRNA levels were significantly increased by 2.1-fold in cells expressing TG6-R111C compared to TG6-WT and cells mock-transfected, while no difference was observed in cells expressing TG6-D327G (Fig. 6D). These results indicate that a TG6 mutant with enriched perinuclear localization induces the UPR.
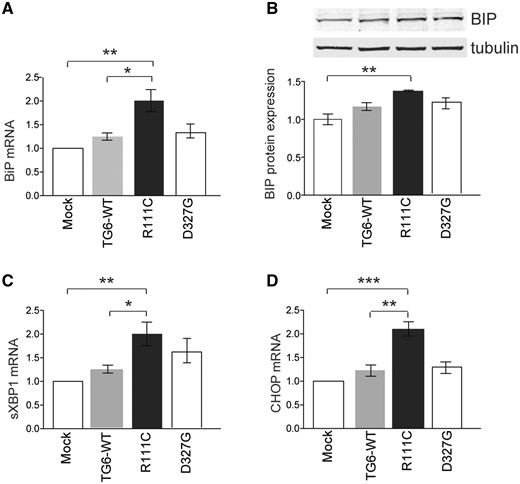
Specific TG6 mutants elicit the UPR. (A–B) Real-time PCR (A) and western blotting (B) analyses of BiP mRNA transcript and protein levels in HEK293T cells transfected with either empty vector (Mock) or vectors expressing TG6-WT, TG6- R111C and TG6-D327G, showed that BiP is induced in cells expressing TG6-R111C. (C–D) Real-time PCR analysis of spliced XBP1 (sXBP1) (C) and CHOP (D) in HEK293T cells transfected as in (A) showed that sXBP1 and CHOP are induced in cells expressing TG6-R111C. Graph, mean±SEM, n = 3, *P <0.05; **P <0.01; ***P <0.001, 1-way ANOVA with Tukey’s post hoc test.
TG6 mutants exert toxicity in vitro and in vivo
To determine whether accumulation in the perinuclear area and activation of the UPR by TG6-R111C was associated with an effect on neuronal degeneration, we transduced primary cortical neurons with lentiviruses expressing TG6 and the GFP through independent promoters and measured cell viability (Fig. 7A). We quantified the number of viable neurons expressing GFP at DIV7 with the Operetta High Content Imaging System. We found that overexpression of TG6-WT reduces neuronal viability by 50%, indicating that dysregulation of TG6 function is neurotoxic per se. Overexpression of TG6-D327G did not further modify toxicity. Notably, overexpression of TG6-R111C enhanced neuronal toxicity by 76.5% compared with TG6-WT (Fig. 7B), although the expression levels of TG6-R111C were significantly lower compared with TG6-WT (Supplementary Material, Fig. S4B). To validate our results in vivo, we generated Drosophila melanogaster models of SCA35 by expressing either soluble mCherry or mCherry-tagged human TG6-WT and TG6-R111C. We first verified whether fusion of mCherry to TG6 alters its enzymatic activity (Supplementary Material, Fig. S5A). We cloned mCherry either at the N-terminus or at the C-terminus of TG6. Similar to HA- and V5-tagged TG6, the enzymatic activity of the N-terminal mCherry-tagged TG6-WT was similar to untagged TG6, whereas that of the TG6-WT construct bearing mCherry at the C-terminal tail was decrease by 40%. Similarly to the untagged constructs, the enzymatic activity of mCherry-TG6-R111C was significantly decreased by 52% (Supplementary Material, Fig. S5B). Notably, mCherry-TG6-R111C expression in the fly head was 70% less than that of mCherry-TG6-WT (Fig. 7C). However, flies expressing mCherry-TG6-R111C were more susceptible to death compared with flies expressing TG6-WT at early stages of life, with 25% of the flies dying as early as 17 days compared with 31 days of the control flies (mCherry) and 18 days of mCherry-TG6-WT flies. Kaplan–Meier analysis of survival showed that mCherry-TG6-WT and mCherry-TG6-R111C-expressing flies have significantly lower lifespan compared with control flies (Log-rank test, P < 0.0001) (Fig. 7D). Collectively, these results indicate that dysregulation of TG6 function is neurotoxic, and mutations affecting TG6 subcellular localization, function, folding and turnover exacerbate TG6-mediated neurotoxicity in vitro and in vivo.
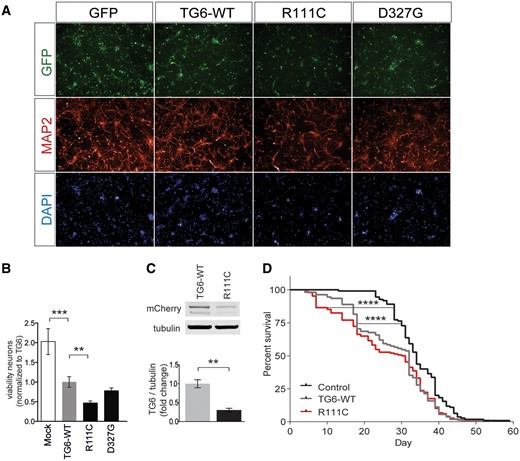
TG6 mutants exert toxicity in vitro and in vivo. (A–B) Analysis of cell viability in mouse primary cortical neurons expressing TG6-WT, TG6-R111C and TG6-D327G mutants showed that overexpression of TG6-WT is toxic in neurons, and overexpression of TG6-R111C enhanced toxicity compared with TG6-WT. Graph, mean±SEM, n = 3, **P <0.01; ***P <0.001, 1-way ANOVA with Tukey’s post hoc test. (C) Western blotting analysis of fly heads expressing mCherry-tagged TG6-WT and TG6-R111C showed that expression of mutant TG6 is significantly lower compared with that of TG6-WT. Graph, mean±SEM, n=3, **P <0.01, Student’s t-test. (D) Kaplan–Meier analysis of survival of flies ubiquitously expressing mCherry (control) and mCherry-tagged TG6-WT and TG6-R111C. ****P <0.0001, Log-Rank test.
Discussion
In this study, we identified seven new TG6 mutations linked to SCA35. We showed that five out of thirteen TG6 mutants are depleted from nucleus, are enriched in the perinuclear area and induce the UPR in cultured cells. Overexpression of TG6 was toxic in cultured neurons and in vivo, yet toxicity was exacerbated by mutations associated with activation of the UPR. Our results imply that different mutations in TG6 may cause neuronal death through distinct mechanisms. Remarkably, among the new mutants, Q181H and Y441C are either not or rarely present in the ExAC database and in gnomAD, in which 60 706 and 123 136 exome sequences from unrelated individuals are collected (Table 1). Notably, these are the only TG6 mutants that show at the same time a decrease in TG activity, a change in the subcellular localization with an increase in the perinuclear area, and an increased activation of the UPR. Our findings suggest that analysis of the levels of expression of specific UPR markers may represent a means to identify the TG6 variants that lead to SCA35 by a toxic gain of TG6 function through chronic ER stress. Other TG6 mutations, such as TG6-V391M, showed a loss of activity with no changes in subcellular distribution, thereby implying different pathogenetic pathways to neuronal dysfunction and death in these SCA35 patients.
Loss of TG6 function in SCA35
Transglutaminases are a complex enzyme family that comprises eight different isoforms all encoded by independent genes (4). Each protein is preferentially expressed in selected tissues with TG6 being the neuronal-specific isoform (5). Genetic mutations in the TG family have been linked to loss of protein function, with damage in the tissue where they are predominantly expressed. Mutations in Factor XIIIA are associated with bleeding disorders (14), and mutations in TG1, which is mostly expressed in skin, cause lamellar ichthyosis, a chronic skin condition (15). At the subcellular level, TG6 localized to the nucleus and perinuclear area in both neurons and non-neuronal cells. Interestingly, TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W and TG6-E574del were almost completely depleted from the nucleus. Emerging evidence supports a key role for TGs in the nucleus (16–19). We have previously shown that TG2 modulates gene transcription in immortalized striatal cells expressing normal or polyglutamine expanded huntingtin (20). TG2 acts as a transcription co-repressor of genes involved in mitochondrial biogenesis (e.g. PGC-1α) and antioxidant response (e.g. cytochrome c). Although further analysis will establish whether mutations in TG6 cause SCA35 through a loss of function mechanism, it is possible that similar to TG2, loss of nuclear TG6 causes neuronal damage through a loss of function mechanism that involves dysregulation of gene expression.
Consistent with previous studies (1,3), we confirmed that several mutations in TGM6 result in loss of TG6 enzymatic activity. It is noteworthy that there was a correlation between transamidase activity and subcellular localization. TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W and TG6-E574del were depleted from nucleus and enriched in the perinuclear area and showed the lowest activity compared with mutations that retained a normal subcellular distribution. It is possible that mutations that hamper TG6 nuclear import result in loss of interaction with specific cofactors required for full enzyme activation. Interestingly, both TG6-R111C and TG6-D327G, two representative TG6 mutants with different behavior, retained the ability to interact and form a complex with TG6-WT. Importantly, we found that expression of TG6-R111C reduces the activity of TG6-WT, suggesting that specific mutations in SCA35 are associated with a dominant negative loss of TG6 function. In the TG family, Factor XIIIA, which is implicated in clotting, works as homodimer (21). We found that TG6-WT and TG6 mutants form a complex. Therefore, we speculate that TG6 also works as a dimer or in a bigger complex and that specific TG6 mutants affect the enzymatic activity of the WT protein, leading to TG6 loss of function.
TG6-R111C showed an increased rate of protein turnover via the UPS, which may at least in part explain why some TG6 mutants showed lower activity, compared to other TG6 mutants with normal protein turnover. The inhibition of UPS through MG132 halted TG6-R111C degradation. Interestingly, TG6-R111C was mostly recovered in a mild detergent-insoluble fraction, suggesting that TG6-R111C is prone to misfold and aggregate, or form large complexes. Misfolded proteins impact the cellular homeostasis inducing the Protein Quality Control System that when overloaded can activate other chaperone-mediated response such as the UPR (22).
ER-induced toxic gain of function in SCA35
We obtained evidence suggesting that mutations in TG6 may cause neuronal damage through a toxic gain of function arising from increased insolubility, aberrant subcellular enrichment in proximity of the ER and activation of the UPR. We found that mutations in TG6 increase co-localization with calnexin. TG2 has been reported to co-localize with calnexin in catecholaminergic neurons modeling Parkinson’s disease in vitro (22). Biochemical analysis revealed that TG2 is mostly associated with the cytoplasmic face of the ER, and only a small portion is retrieved in the ER lumen (23,24). Similar to TG2, also mutations in TG6 (TG6-R111C, TG6-Q181H, TG6-Y441C, TG6-L517W and TG6-E574del) may alter protein folding, leading to accumulation of the mutant protein on the cytosolic side of the rough ER membranes in close proximity to calnexin, and over time induce the UPR. Genetic diseases associated with the accumulation of misfolded proteins affect the ER structure, function and integrity, inducing the ER stress response independently of whether the misfolded proteins localize within or outside the ER (25). Excessive accumulation of mutant TG6 in the perinuclear area may alter the cellular proteostasis, thereby resulting in a toxic gain of function that leads to neuronal damage. UPR is mediated by proteins that initially halt protein translation and increase protein folding and degradation capacity of the ER, to finally reduce the protein load (26). Of interest, induction of the UPR by mutations in SIL1, a nucleotide exchange factor for BiP, is associated with a recessive form of cerebellar ataxia, namely Marinesco–Sjogren syndrome (27,28). Furthermore, mutations in eukaryotic elongation factor 2 lead to an imbalance in protein homeostasis and are linked to SCA26 (29). These observations hint toward the susceptibility of cerebellar neurons to UPR, and this may be the case also for SCA35. Therapies aimed at lowering ER stress may be beneficial for patients with forms of ataxias associated with activation of the UPR.
Alteration of TG6 function causes neuronal death
We found that gain of normal TG6 function is neurotoxic since overexpression of normal TG6 caused neural death in cultured neurons and in flies. At the same time, we identified TG6 mutants with reduced activity and with dominant negative loss of function activity over the WT counterpart. The most studied member of the TG family, TG2, has been defined as a double-edge sword enzyme because both its aberrant activity and its inhibition correlate with cellular toxicity (30,31). In acute and chronic neurodegenerative diseases, such as Huntington’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, stroke and traumatic brain injury, levels and activity of TG2 along with formation of insoluble cross-linked GGEL peptides were reported to be augmented (32,33). At the same time, genetic removal of TG2 worsened neuronal survival in brain ischemia (16). Our results support a model whereby both loss and toxic gain of TG6 function contributes to disease. It remains to be elucidated why cerebellar neurons are specifically vulnerable to the expression of mutant TG6. Nonetheless, our results support a model whereby specific TG6 mutations cause neuronal dysfunction and death through a mechanism associated with chronic depletion of the protein from nucleus, loss of enzymatic activity, reduced solubility, increased turnover and activation of the UPR. Such analysis of basic disease mechanisms that takes advantage of a specific mutation like TG6-R111C, which reproduces toxicity in vivo and in vitro, has therapeutic relevance because it helps to further understand the etiology of SCA35 and suggest looking closely at aberrant ERUPR activation as a plausible mechanism inducing Purkinje cell degeneration. Finally, we cannot exclude that different mutations in TG6 exert toxicity through distinct mechanisms, by affecting diverse properties of the enzyme.
Materials and Methods
Cell lines
HEK293T cells were used for expression and activity studies for TG6-WT and mutant proteins. COS-7 cells were used for immunocytochemistry experiments to study the subcellular distribution of TG6-WT and mutant proteins. These cells were maintained in DMEM supplemented with 10% fetal bovine serum, l-Glutamine (2 mm) and PenStrep (1%).
Animals
Animal care and experimental procedures were conducted in accordance with the University of Trento ethics committee and were approved by the Italian Ministry of Health. Primary mouse cortical and cerebellar neurons were obtained from C57BL/6J mated to generate male and female WT embryos at day 15 (E15.5).
Primary neurons
Primary cortical neurons were cultured from embryonic E15.5 C57BL/6J mice as previously described (9). In brief, the cortices were dissected out, digested in Papain solution (20 U Papain, 5-mm EDTA and 30 mm Cysteine in 1× Eagles’ Balanced Salt Solution (EBSS)) for 20 min. This was followed by a DNase I treatment for 3 min. The dissociated cells were centrifuged at 1000g for 5 min. Supernatant was discarded and the digestion was blocked with a solution containing Soybean Trypsin inhibitor and bovine serum albumin in EBSS. Following a centrifugation of 1000g for 10 min the cells were plated in MEM media supplemented with 10% fetal bovine serum, L-Glutamine (2mM) and PenStrep (1%). The day after half the media was replaced with neurobasal medium supplemented with B27, PenStrep (100 units), l-Glutamine (2 mm) and AraC (100 µm). Half of the media was replaced with fresh media every 3 days. For dissociated cerebellar cultures, cerebellum was dissected out from neonatal mice (P0-1) of C57BL/6 strain (34). For digestion the same procedure was used as for cortical neurons (9). The dissociated cells were centrifuged at 100g for 5 min. Supernatant was discarded and the digestion was blocked with a solution containing Trypsin inhibitor and bovine serum albumin in EBSS. Following a centrifugation of 100g for 10 min the cells were plated in Neurobasal medium supplemented with B27, PenStrep (100 units), l-Glutamine (2 mm), KCl (3 mm), Horse serum (1%), Glucose (2.1 mg/mL) and NGF (50 ng/mL). Half of the media was replaced with fresh media every 3 days (34).
Plasmids
TG6-WT (5) was subcloned into the pcDNA3.1 vector under the CMV promoter. Plasmids for the reported mutants were generated by site-directed mutagenesis. In order to generate TG6-V5 tagged constructs, TG6 lacking the stop codon and harboring a C-terminal Acc65I site was generated by amplification with 5′-TGC TAG CGG AGT CCA ACT GGC CTT CAC ATG G and 5′-C GGT ACC CTT GGC AGT AGC GAC ATG GAC GAT C. Complementary oligonucleotides were synthesized (MWG), annealed and ligated into the Acc65I site to generate the C-terminally V5-tagged variant (adding GTGKPIPNPLLGLDSTS to native sequence). Through molecular cloning, Human influenza hemagglutinin (HA)-tagged constructs for TG6-WT and mutants were generated. TG6-WT and mutants were also cloned into the LentiLox 3.7 backbone under the control of the synapsin I promoter for expression/toxicity studies in primary neurons (35).
Viral production and titration
Plasmids of TG6-WT or mutants (cloned into the LentiLox 3.7 backbone under the control of the synapsin I promoter) together with pCMV-dR8.91 (Delta 8.9) plasmid containing gag, pol and rev genes and VSV-G envelope plasmid were expressed in HEK293T cells by calcium phosphate transfection. 16 h post-transfection the medium was discarded and replaced with fresh medium. 24 h later the medium was collected, centrifuged at 1000g for 5 min (to pellet down any cellular debris), filtered (0.45-μm pore size filters) and stored at −80°C in aliquots, until use. Before infection the viruses were quantified using the SG-PERT reverse transcription assay (36). In brief, viral particles were lysed by adding an equal volume of Lysis buffer [0.25% Triton X-100, 50 mm KCl, 100 mm Tris–HCl pH 7.4, and 0.4 U/μL RNase inhibitor (RiboLock, MBI Fermentas)]. Lysates were then added to a single-step, RT-PCR assay with 35 nm MS2 RNA (Roche) as template, 500 nm of each primer (5′-TCCTGCTCAACTTCCTGTCGAG-3′ and 5′-CACAGGTCAAACCTCCTAGGAATG-3′), and hot-start Taq (Promega), all in 20 mm Tris–Cl pH 8.3, 5 mm (NH4)2SO4, 20 mm KCl, 5 mm MgCl2, 0.1 mg/ml BSA, 1/20 000 SYBR Green I (Sigma), and 200 μm dNTPs. The program for the thermocycler was 42°C for 20 min, 95°C for 2 min for enzyme activation, followed by 40 cycles of denaturation at 95°C for 5 s, annealing 60°C for 5 s, extension at 72°C for 15 s and acquisition at 80°C for 5 s. A standard curve was obtained using known concentrations of recombinant viruses (a gift from Dr. Massimo Pizzato).
Immunoblotting
Cells were lysed 48-h post-transfection using 1% Triton lysis buffer (25 mm Tris–HCl pH 7.4, 100 mm NaCl, 1 mm EGTA, 1% Triton X-100, protease and phosphatase inhibitors). 25–50 µg of proteins were used for SDS-PAGE. InfraRed dye-conjugated secondary antibody (1:10 000 in the blocking solution) was used and proteins were detected by an Odyssey infrared imaging system (LI-COR Biosciences). For co-immunoprecipitation experiment, anti HA-agarose beads (Thermo scientific, Pierce #26180) were used. The following primary antibodies were used in the experiments: TG6 (Zedira #A017, 1:1500), BiP (Abcam #ab21685, 1:1000), V5 epitope Tag (Thermo, 1:1500), HA epitope tag (Santa Cruz #sc-805, 1:500), mCherry (Novus Biologicals #NBP2-25157, 1:2000).
In vitro transamidase activity
For the transamidase activity, 25 µg of proteins from cell lysates (cells overexpressing TG6-WT or mutants) was incubated with a biotinylated peptide substrate (Biotin-Thr-Val-Gln-Gln-Glu-Leu-OH; 0.5 mm) in presence of 5 mm DTT and 5 mm CaCl2 for 15 min at 30°C. The reaction mix was then electrophoresed and blotted onto nitrocellulose membranes. After blocking with 5% skimmed milk, membranes were incubated with Anti-Streptavidin (IRDye 800CW #926-32230 1:10 000) antibody, prepared in Blocking Solution, for 1 h at room temperature (in dark). The membranes were then scanned at the Odyssey infrared imaging system.
Immunocytochemistry
Cells were grown on Poly-d-Lysine coated coverslips. 48 h after transfection, the cells were fixed with 4% paraformaldehyde for 15–20 min, at room temperature. For immunocytochemistry, cells were permeabilized with 0.1% Triton X-100 (in phosphate buffered saline, PBS), blocked with PBS containing 10% FBS and 0.05% Triton X-100 for 45 min at room temperature. This was followed by incubation with the primary antibody at 4°C overnight with the following dilutions: TG6 (Zedira #A017, 1:250), Map2 (Merck-Millipore #AB5622, 1:500), calnexin (Abcam #ab31290, 1:200), V5 epitope Tag (Thermo Scientific #PA1-993, 1:500), HA epitope tag (Invitrogen #32-6700, 1:200), GFP (Merck-Millipore #MAB2510, 1:2000). The next day, after three washes with PBS (5 min each), Alexa-fluor conjugated secondary antibody (1:1000) was added for 1 h at room temperature in the dark. The coverslips were then washed with PBS and mounted on glass slides with DAPI containing Prolong Gold antifade mounting media (Life technologies #P36961). Slides were imaged with Zeiss AxioObserver.Z1 microscope.
Co-localization
Confocal imaging was performed using a laser-scanning motorized confocal system (Nikon A1) equipped with an Eclipse Ti-E inverted microscope and four laser lines (405, 488, 561 and 638 nm). Z-series images were taken with an inter-stack interval of 0.5 μm using 60× oil objective. Laser intensities and camera settings were maintained identically within the same experiment to allow comparison of different experimental groups. Quantification of TG6/calnexin co-localization was performed using Mander’s overlap calculated with Nis-element software (Nikon).
In situ PLA
Duolink starter kit (Sigma–Aldrich; DUO92101) was used to study the interaction of TG6-WT and mutant in COS-7 cells co-transfected with the WT and mutant protein. The assay was performed following manufacturer’s instructions.
Immunofluorescence analysis
Mice were perfused transcardially with a 4% paraformaldehyde solution and brains were collected, embedded in paraffin and sections were cut with a microtome at 8 µm (Leica Biosystems RM2255). Brain sections were subjected to antigen retrieval for 45 min at 95°C in a solution of 10 mm sodium citrate (pH 6), followed by blocking in a solution containing 10% normal goat serum, 5% bovine serum albumin and 0.5% fish gelatin in TBS–Tween 0.01%, overnight at 4°C. The incubation with the primary antibodies for detection of calbindin (Sigma-Aldrich #C9848, 1:250) and TG6 (Zedira #A017, 1:250) was performed overnight and the incubation with secondary antibodies for 1 h at room temperature (Alexa Fluor 546 goat anti-rabbit IgG #A-11035 and Alexa Fluor 488 goat anti-mouse IgG #A-11029, Invitrogen). The slices were mounted in mounting medium with DAPI (Abcam) and the images were acquired with the inverted Zeiss Observer Z1 microscope.
Protein turnover
Protein turnover was studied by performing a cycloheximide (CHX) chase experiment. Cells transfected with TG6-WT or mutants were treated with CHX (0.1 mg/mL). As a preliminary experiment (data not shown) protein expression was studied at three different time points (3, 6 and 9 h) and finally 6 h time point was chosen for further experiments. After 6 h of CHX treatment with MG132 (10 µm; proteasome inhibitor) or Bafilomycin a1 (200 nm; autophagy inhibitor), the levels of protein expression were analyzed for TG6-WT or mutants.
Assessment of protein solubility
The protocol for extraction of the soluble and insoluble protein fractions was performed as previously reported (11). In brief, cells were collected in PBS and centrifuged at 500g for 5 min, 4°C. The cell pellet was then lysed in RIPA buffer (1% NP-40, 0.1% SDS, 50 mm Tris–HCL pH 7.4, 150 mm NaCl, 0.5% sodium deoxycholate, 1 mm EDTA) and sonicated. The lysates were then centrifuged at 5000g for 10 min, 4°C, to remove the cellular debris. The supernatant was collected and further centrifuged at 100 000g for 30 min. The supernatant was collected as the soluble fraction and the pellet was washed in RIPA lysis buffer and centrifuged again at 100 000g for 30 min. After this final step of centrifugation, the supernatant was discarded and the pellet was resuspended in Urea buffer (8 m Urea, 4% CHAPS, 50 mm DTT, 40 mm Tris HCL pH 7.4, 2.5 mm EDTA) and considered as the insoluble fraction. For experiments with proteasome inhibitor, cells were treated with 10 µm of MG132 and lysed 6 h after the treatment.
Quantitative PCR
Total RNA was obtained from cells following the manufacturer instructions using TRIzol (Invitrogen). 1 μg of RNA was reverse transcribed using iScript Reverse Transcription Supermix (Bio-Rad). The cDNA was diluted 10 times and then 1 μl was used for qPCR using the iTaq Universal SYBR Green Supermix (Bio-Rad). The primer efficiency was calculated for each set of primers and the primers with efficiency approximately 1 were used for the study. Primer used: h_TG6 Fwd: TGTCACCAGGATCATCAGTGCCAT, Rev: TGGTACCACCTCCATATTTGCCCT; h_BiP Fwd: TGTTCAACCAATTATCAGCAAACTC, Rev: TTCTGCTGTATCCTCTTCACCAGT; h_CHOP Fwd: AGAACCAGGAAACGGAAACAGA, Rev: TCTCCTTCATGCGCTGCTTT; h_splicedXBP1 Fwd: CTGAGTCCGAATCAGGTGCAG, Rev: ATCCATGGGGAGATGTTCTGG; h_ACTIN Fwd: GACAGGATGCAGAAGGAGATTACTG, Rev: CTCAGGAGGAGCAATGATCTTGAT.
Toxicity assay
Primary cortical neurons were seeded on 96-well tissue culture plates at a concentration of 2.5×104 cells/well. At DIV1 the immature neurons were transduced with viral particles (at MOI 3) expressing only GFP (Mock) or GFP along with TG6-WT or mutants. The average efficiency of transduction was 47.4 ± 4%. At DIV5 the neurons were fixed and processed for immunocytochemistry for GFP and Map2. The plate was then imaged with Operetta and analyzed for the total number of GFP positive neurons in each condition. The experiments were carried out in triplicates and repeated at least three times independently.
Fly experiments
Transgenic flies, with GAL4-activatable TG6, were generated using P-element transgenic insertion by BestGene. Survival analysis was done by crossing the TG6 lines with a ubiquitous driver tubulin-gal4 (Bloomington Stock Center #5138) at 28°C. Day 1 adult males (n = 100) from each group were separated and maintained on standard food at 28°C and deaths were recorded every day. Kaplan–Meier survival curve analysis was performed using Log-Rank test on Prism software.
For assessment of protein expression, TG6 lines were crossed with GMR-gal4 driver line (Bloomington Stock Center #8605) at 29°C. Day 1 fly heads were used for protein extraction and subsequent immunoblotting. TG6 was detected with an antibody against mCherry (Novus Biologicals, NBP2-25158) at 1:2000. Anti-phospho(p)eIF2α (Ser51) (Cell Signaling Technologies, #9721) was revealed by using a dilution of 1:1000. Goat anti-mouse Dylight 800 (ThermoFisher Scientific) was used as secondary antibody at 1:10 000 concentration. Scanning of western blots was performed using the LI-COR Odyssey Infrared Imaging System.
Patient recruitment
For the French center, 731 index patients were recruited as part of the SPATAX cohort of patients with Spastic Paraplegia and Ataxia (https://spatax.wordpress.com/). They were clinically assessed by at least one member of the SPATAX network, with a standardized evaluation form (https://spatax.files.wordpress.com/2013/09/fichecliniquespatax-eurospa-2011.pdf). Patients gave informed consent to participate in the study, and blood samples were collected in accordance with local French regulations (Paris Necker ethics committee approval [RBM 01-29 and RBM 03-48] to AD). DNA was extracted with classical procedures. For the UK patient cohort, archived DNA samples from 231 ataxia patients collected by Cardiff and Sheffield NHS Trusts were investigated, with informed consent and in accordance with UK regulations (REC13/SW/0235) and the Sheffield NHS diagnostic genetics service. For the US center, only one patient was screened at GeneDx.
TGM6 sequencing
For 412 index patients from the French center with a history of dominant disease transmission, TGM6 was sequenced as part of an amplicon-based gene panel including 34 known cerebellar ataxia genes (37). DNA was amplified using the Fluidigm Access Array system, according to the manufacturer protocol (Fluidigm). Barcoded, pooled and purified library was sequenced on a MiSeq Illumina Sequencer as 2×300 bp reads using standard protocols (Illumina). For 319 index patients from the French center without history of dominant disease transmission, whole exome was sequenced on the Illumina HiSeq 2000 (2 × 100 bp paired end reads) after enrichment with the Extended Nextera Rapid-Capture Exome kit (Illumina, USA). Bioinformatic processing followed current recommendations [alignment against the reference human genome (UCSC hg19) using the Burrows-Wheeler Alignment tool (BWA) (38); local realignment, recalibration and variant calling with the Broad Institute’s Genome Analysis Toolkit (GATK) (39); variants annotation with Annovar (40)]. Further variant processing was performed as previously described (37). For the US center, the exome sequencing was performed commercially by Gene Dx. UK samples were analyzed for polymorphisms using a combination approach employing high resolution melting analysis of amplified exon sequences with subsequent direct sequencing using standard Sanger dideoxy sequencing or using a hereditary ataxia NGS panel (www.sheffieldchildrens.nhs.uk/our-services/laboratory-medicine/laboratory-services.htm#ngs) (41).
Statistics
Statistical analysis was conducted by one-way ANOVA followed by Tukey’s post hoc test, two-way ANOVA followed by Bonferroni’s post hoc test and Student’s t-test according to the specific experiment. Statistically significant results were defined as follows: *P < 0.05; **P < 0.01; ***P < 0.001 and ****P < 0.0001. All the experiments presented here were repeated at least three times.
Supplementary Material
Supplementary Material is available at HMG online.
Acknowledgements
We would like to thank Dr. Massimo Pizzato’s lab for help with the production and titration of lentiviruses; Michael Pancher at the High Throughput Screening Facility for assistance with the Operetta experiments; Marta Tarter and Sergio Robbiati at the Model Organism Facility; the Advanced Imaging Facility at CIBIO. We would like to thank Alessandro Roncador and Alessia Moruzzi for technical assistance, Enrico Domenici for fruitful discussion on genetic variants and Rajiv R. Ratan for valuable comments on the manuscript. We really appreciate the help of Dr. Mark Wardle, Consultant Neurologist at Cardiff and Vale University Health Board for the access to patients’ samples. We are grateful to all the patients and patients’ families for their cooperation in this study.
Conflict of Interest statement. None declared.
Funding
This study was supported by the University of Trento and CIBIO (start-up funding to M.B.), the Alzheimer Trento Onlus with the Legato Baldrachi (to M.B.), Bando Progetti Strategici di Ateneo-University of Trento to M.P. and M.B.), Telethon-Italy and Provincia Autonoma di Trento-Italy (TCP12013 to M.P.). Support from the Ryder Briggs Trust (to D.A. and M.H.) and Cardiff University/Zedira (to D.A. and S.T.) is also gratefully acknowledged.
References
Author notes
Present address: Department of Pharmaceutic and Health Science. Faculty of Pharmacy, Universidad San Pablo CEU, Campus Montepríncipe, Alcorcòn, Madrid, Spain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}