




Abstract
Proteoglycans are among the most abundant and structurally complex biomacromolecules and play critical roles in connective tissues. They are composed of a core protein onto which glycosaminoglycan (GAG) side chains are attached via a linker region. Biallelic mutations in B3GALT6, encoding one of the linker region glycosyltransferases, are known to cause either spondyloepimetaphyseal dysplasia (SEMD) or a severe pleiotropic form of Ehlers–Danlos syndromes (EDS). This study provides clinical, molecular and biochemical data on 12 patients with biallelic B3GALT6 mutations. Notably, all patients have features of both EDS and SEMD. In addition, some patients have severe and potential life-threatening complications such as aortic dilatation and aneurysm, cervical spine instability and respiratory insufficiency. Whole-exome sequencing, next generation panel sequencing and direct sequencing identified biallelic B3GALT6 mutations in all patients. We show that these mutations reduce the amount of β3GalT6 protein and lead to a complete loss of galactosyltransferase activity. In turn, this leads to deficient GAG synthesis, and ultrastructural abnormalities in collagen fibril organization. In conclusion, this study redefines the phenotype associated with B3GALT6 mutations on the basis of clinical, molecular and biochemical data in 12 patients, and provides an in-depth assessment of β3GalT6 activity and GAG synthesis to better understand this rare condition.
Introduction
Proteoglycans are structurally complex biomacromolecules and a major component of the extracellular matrix and cell membranes. They support the biomechanical function of connective tissues by organizing the extracellular matrix, and serve as signaling molecules controlling cell–cell interactions, cell growth, morphogenesis and tissue repair (1–3).
Proteoglycans are composed of a core protein and one or more glycosaminoglycan (GAG) chains that consist of repeating disaccharide units. Depending on the composition of these units, the proteoglycan superfamily is subdivided into heparan sulfate (HS) proteoglycans and chondroitin sulfate (CS)/dermatan sulfate (DS) proteoglycans. The biosynthesis of both starts with the formation of a tetrasaccharide linker region that is covalently attached to serine residues on the core protein. This linker region is synthesized in a successive and coordinated effort by specific glycosyltransferases. It starts with the transfer of a xylose residue by xylosyltransferases I/II (encoded by XYLT1/XYLT2), followed by the addition of two galactose residues by galactosyltransferase I (β4GalT7, encoded by B4GALT7) and galactosyltransferase II (β3GalT6, encoded by B3GALT6). It is completed by the transfer of glucuronic acid catalyzed by glucuronosyltransferase I (GlcAT-I, encoded by B3GAT3). The final result is a glucuronic acid-β1,3-galactose-β1,3-galactose-β1,4-xylose-β1-O- (GlcA-β1,3-Gal-β1,3-Gal-β1,4-Xyl-β1-O-) tetrasaccharide that serves as a primer for HS and CS/DS chain polymerization.
Mutations in genes encoding the linker region enzymes cause clinically overlapping autosomal recessive (AR) disorders, called ‘linkeropathies’. XYLT1 mutations cause various skeletal dysplasias, including Desbuquois Dysplasia type 2 (OMIM 615777) (4–8). XYLT2 mutations cause spondylo-ocular syndrome (OMIM 605822) (9–11). B3GAT3 mutations cause a Larsen-like syndrome with joint hypermobility and cardiac defects, and more severe skeletal dysplasias with and without cardiac defects (OMIM 245600) (12–17). Finally, mutations in B4GALT7 and B3GALT6 cause both skeletal dysplasia and an Ehlers–Danlos syndrome (EDS)-like disorder. EDS is an umbrella term for a group of heritable connective tissue disorders hallmarked by joint hypermobility, hyperextensible, fragile skin and generalized connective tissue friability. B4GALT7 mutations cause these EDS features plus short stature, radioulnar synostosis, osteopenia, eye anomalies and a recognizable facial gestalt with some progeroid features (18–21). Because of the latter, this disorder was initially called ‘progeroid EDS’ (OMIM 130070). In addition, a p.(Arg270Cys) mutation causes the so-called Larsen of Reunion Island syndrome (22). B3GALT6 mutations cause both spondyloepimetaphyseal dysplasia (SEMD) with joint hypermobility (SEMDJL1 or SEMDJL Beighton type, OMIM 271640) and a severe pleiotropic EDS-like disorder (OMIM 615349) (23,24). In view of the mixed EDS and skeletal dysplasia phenotype, B4GALT7- and B3GALT6-related EDS have been categorized as ‘spondylodysplastic EDS’ in the recently revised classification on EDS (25,26). The variable phenotypes brought on by mutations in the linker region enzymes that are all presumed to be indispensable in the synthesis of both HS and CS/DS chains, remains largely unexplained.
In this study, we more accurately define the clinical spectrum associated with B3GALT6 mutations on the basis of clinical and molecular data in 12 novel patients. We combine these data with an in-depth characterization of B3GALT6 expression, β3GalT6 protein level and activity, and GAG synthesis to expand our understanding of this rare condition.
Results
Clinical phenotype
Detailed case reports are available in Supplementary Material, Note. The salient clinical/radiographic findings are summarized in Table 1 and illustrated in Figure 1.
Overview of salient clinical and radiographic findings
| PI:1 | PII:1 | PIII:1 | PIV:1 | PIV:2 | PIV:3 | PV:1 | PVI:1 | PVII:1 | PVIII:1 | All | SEMDJL1 | EDS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Female | Female | Male | Male | Male | Female | Male | Male | NA | NA | NA |
| Ethnicity | USA | USA | Dutch | Congolese— Rwandan | Congolese— Rwandan | Congolese— Rwandan | Indian | Dutch | Iranian | French | NA | NA | NA |
| Consanguinity | − | − | − | − | − | − | − | − | + | − | 1/10 | ||
| Age at last evaluation | 6 years | 8 months | 9 months | 25 years | 31 years | 28 years | 7 years | 3 years | 3 years | 37 years | NA | NA | NA |
| Pregnancy and birth | |||||||||||||
| Prenatal ultrasound abnormalities | + | − | + | ? | ? | ? | − | ? | ? | − | 2/10 | 2/14 | 1/18 |
| Perinatal complications | + | + | + | ? | ? | ? | + | + | ? | + | 6/10 | 1/14 | 3/18 |
| Clinical phenotype | |||||||||||||
| Short stature | + | + | + | + | + | + | + | + | + | + | 10/10 | 8/14 | 10/18 |
| Kyphoscoliosis | +° | + | + | + | + | + | +° | + | + | + | 10/10 | 13/14 | 11/18 |
| Multiple contractures | +° | +° | +° | + | + | + | +° | +° | + | + | 10/10 | 5/14 | 13/18 |
| Joint hypermobility | + | + | + | + | + | + | + | + | + | + | 10/10 | 6/14 | 14/18 |
| Joint dislocations | + | +° | + | + | + | + | + | +° | + | + | 10/10 | 2/14 | 8/18 |
| Congenital hip dysplasia | − | +° | +° | ? | ? | ? | +° | +° | ? | − | 4/10 | 6/14 | 6/18 |
| Cervical spine instability | + | + | + | − | − | − | ? | ? | ? | − | 3/10 | 0/14 | 0/18 |
| Hypotonia | +° | +° | +° | − | − | + | +° | − | ? | − | 5/10 | 1/14 | 11/18 |
| Soft skin | +° | + | + | + | + | + | + | − | + | + | 9/10 | 2/14 | 9/18 |
| Transluscent skin | +° | + | − | − | − | − | − | + | ? | + | 4/10 | 0/14 | 5/18 |
| Hyperextensible skin | + | + | − | + | + | + | + | − | ? | + | 7/10 | 2/14 | 10/18 |
| Cutis laxa | +° | + | + | − | − | − | + | − | + | − | 5/10 | 0/14 | 3/18 |
| Atrophic scarring | + | − | − | + | + | + | − | − | ? | + | 5/10 | 0/14 | 1/18 |
| Aortic root dilatation | + | + | − | − | − | − | − | ? | ? | + | 3/10 | 0/14 | 0/18 |
| Cardiac valve abnormalities | + | − | − | − | + | − | − | ? | ? | − | 2/10 | 1/14 | 5/18 |
| Chronic respiratory insufficiency | + | + | − | − | − | − | − | − | − | − | 2/10 | 1/14 | 3/18 |
| Facial dysmorphologya | +° | +° | +° | + | + | + | + | +° | + | + | 10/10 | 12/14 | 13/18 |
| Dental abnormalities | + | + | ? | + | + | + | − | + | + | + | 8/10 | 0/14 | 4/18 |
| Delayed motor development | + | + | + | − | − | − | + | + | ? | − | 5/10 | 2/14 | 10/18 |
| Delayed cognitive development | + | + | ? | − | − | − | + | − | ? | − | 3/10 | 3/14 | 7/18 |
| Radiological features | |||||||||||||
| Metaphyseal widening | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 4/18 |
| Narrowing and bowing long bones | + | + | + | + | + | + | + | + | + | + | 10/10 | 0/14 | 10/18 |
| Hypoplastic ilia | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 7/18 |
| Acetabular/femoral head dysplasia | + | + | + | + | + | − | + | + | + | + | 9/10 | 8/14 | 4/18 |
| Vertebral body changes | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 8/18 |
| Osteopenia | + | +° | + | + | + | + | + | ? | ? | + | 8/10 | 3/14 | 9/18 |
| Fractures | + | +° | + | + | + | + | + | − | ? | + | 8/10 | 0/14 | 9/18 |
| PI:1 | PII:1 | PIII:1 | PIV:1 | PIV:2 | PIV:3 | PV:1 | PVI:1 | PVII:1 | PVIII:1 | All | SEMDJL1 | EDS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Female | Female | Male | Male | Male | Female | Male | Male | NA | NA | NA |
| Ethnicity | USA | USA | Dutch | Congolese— Rwandan | Congolese— Rwandan | Congolese— Rwandan | Indian | Dutch | Iranian | French | NA | NA | NA |
| Consanguinity | − | − | − | − | − | − | − | − | + | − | 1/10 | ||
| Age at last evaluation | 6 years | 8 months | 9 months | 25 years | 31 years | 28 years | 7 years | 3 years | 3 years | 37 years | NA | NA | NA |
| Pregnancy and birth | |||||||||||||
| Prenatal ultrasound abnormalities | + | − | + | ? | ? | ? | − | ? | ? | − | 2/10 | 2/14 | 1/18 |
| Perinatal complications | + | + | + | ? | ? | ? | + | + | ? | + | 6/10 | 1/14 | 3/18 |
| Clinical phenotype | |||||||||||||
| Short stature | + | + | + | + | + | + | + | + | + | + | 10/10 | 8/14 | 10/18 |
| Kyphoscoliosis | +° | + | + | + | + | + | +° | + | + | + | 10/10 | 13/14 | 11/18 |
| Multiple contractures | +° | +° | +° | + | + | + | +° | +° | + | + | 10/10 | 5/14 | 13/18 |
| Joint hypermobility | + | + | + | + | + | + | + | + | + | + | 10/10 | 6/14 | 14/18 |
| Joint dislocations | + | +° | + | + | + | + | + | +° | + | + | 10/10 | 2/14 | 8/18 |
| Congenital hip dysplasia | − | +° | +° | ? | ? | ? | +° | +° | ? | − | 4/10 | 6/14 | 6/18 |
| Cervical spine instability | + | + | + | − | − | − | ? | ? | ? | − | 3/10 | 0/14 | 0/18 |
| Hypotonia | +° | +° | +° | − | − | + | +° | − | ? | − | 5/10 | 1/14 | 11/18 |
| Soft skin | +° | + | + | + | + | + | + | − | + | + | 9/10 | 2/14 | 9/18 |
| Transluscent skin | +° | + | − | − | − | − | − | + | ? | + | 4/10 | 0/14 | 5/18 |
| Hyperextensible skin | + | + | − | + | + | + | + | − | ? | + | 7/10 | 2/14 | 10/18 |
| Cutis laxa | +° | + | + | − | − | − | + | − | + | − | 5/10 | 0/14 | 3/18 |
| Atrophic scarring | + | − | − | + | + | + | − | − | ? | + | 5/10 | 0/14 | 1/18 |
| Aortic root dilatation | + | + | − | − | − | − | − | ? | ? | + | 3/10 | 0/14 | 0/18 |
| Cardiac valve abnormalities | + | − | − | − | + | − | − | ? | ? | − | 2/10 | 1/14 | 5/18 |
| Chronic respiratory insufficiency | + | + | − | − | − | − | − | − | − | − | 2/10 | 1/14 | 3/18 |
| Facial dysmorphologya | +° | +° | +° | + | + | + | + | +° | + | + | 10/10 | 12/14 | 13/18 |
| Dental abnormalities | + | + | ? | + | + | + | − | + | + | + | 8/10 | 0/14 | 4/18 |
| Delayed motor development | + | + | + | − | − | − | + | + | ? | − | 5/10 | 2/14 | 10/18 |
| Delayed cognitive development | + | + | ? | − | − | − | + | − | ? | − | 3/10 | 3/14 | 7/18 |
| Radiological features | |||||||||||||
| Metaphyseal widening | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 4/18 |
| Narrowing and bowing long bones | + | + | + | + | + | + | + | + | + | + | 10/10 | 0/14 | 10/18 |
| Hypoplastic ilia | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 7/18 |
| Acetabular/femoral head dysplasia | + | + | + | + | + | − | + | + | + | + | 9/10 | 8/14 | 4/18 |
| Vertebral body changes | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 8/18 |
| Osteopenia | + | +° | + | + | + | + | + | ? | ? | + | 8/10 | 3/14 | 9/18 |
| Fractures | + | +° | + | + | + | + | + | − | ? | + | 8/10 | 0/14 | 9/18 |
aFacial dysmorphology is characterized by frontal bossing, mid-facial hypoplasia, low−set and posteriorly rotated ears, prominent eyes and blue sclerae, a long philtrum and a short nose with anteverted nares. +, present; −, absent; ?, unknown; °, present at birth; NA, not applicable; SEMDJL1, spondyloepimetaphyseal dysplasia with joint hypermobility; EDS, Ehlers–Danlos syndrome. The columns ‘SEMDJL1’ and ‘EDS’ summarize the clinical and radiographic findings of all hitherto reported patients for whom this data was available (16, 23, 24, 27, 29–32).
Overview of salient clinical and radiographic findings
| PI:1 | PII:1 | PIII:1 | PIV:1 | PIV:2 | PIV:3 | PV:1 | PVI:1 | PVII:1 | PVIII:1 | All | SEMDJL1 | EDS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Female | Female | Male | Male | Male | Female | Male | Male | NA | NA | NA |
| Ethnicity | USA | USA | Dutch | Congolese— Rwandan | Congolese— Rwandan | Congolese— Rwandan | Indian | Dutch | Iranian | French | NA | NA | NA |
| Consanguinity | − | − | − | − | − | − | − | − | + | − | 1/10 | ||
| Age at last evaluation | 6 years | 8 months | 9 months | 25 years | 31 years | 28 years | 7 years | 3 years | 3 years | 37 years | NA | NA | NA |
| Pregnancy and birth | |||||||||||||
| Prenatal ultrasound abnormalities | + | − | + | ? | ? | ? | − | ? | ? | − | 2/10 | 2/14 | 1/18 |
| Perinatal complications | + | + | + | ? | ? | ? | + | + | ? | + | 6/10 | 1/14 | 3/18 |
| Clinical phenotype | |||||||||||||
| Short stature | + | + | + | + | + | + | + | + | + | + | 10/10 | 8/14 | 10/18 |
| Kyphoscoliosis | +° | + | + | + | + | + | +° | + | + | + | 10/10 | 13/14 | 11/18 |
| Multiple contractures | +° | +° | +° | + | + | + | +° | +° | + | + | 10/10 | 5/14 | 13/18 |
| Joint hypermobility | + | + | + | + | + | + | + | + | + | + | 10/10 | 6/14 | 14/18 |
| Joint dislocations | + | +° | + | + | + | + | + | +° | + | + | 10/10 | 2/14 | 8/18 |
| Congenital hip dysplasia | − | +° | +° | ? | ? | ? | +° | +° | ? | − | 4/10 | 6/14 | 6/18 |
| Cervical spine instability | + | + | + | − | − | − | ? | ? | ? | − | 3/10 | 0/14 | 0/18 |
| Hypotonia | +° | +° | +° | − | − | + | +° | − | ? | − | 5/10 | 1/14 | 11/18 |
| Soft skin | +° | + | + | + | + | + | + | − | + | + | 9/10 | 2/14 | 9/18 |
| Transluscent skin | +° | + | − | − | − | − | − | + | ? | + | 4/10 | 0/14 | 5/18 |
| Hyperextensible skin | + | + | − | + | + | + | + | − | ? | + | 7/10 | 2/14 | 10/18 |
| Cutis laxa | +° | + | + | − | − | − | + | − | + | − | 5/10 | 0/14 | 3/18 |
| Atrophic scarring | + | − | − | + | + | + | − | − | ? | + | 5/10 | 0/14 | 1/18 |
| Aortic root dilatation | + | + | − | − | − | − | − | ? | ? | + | 3/10 | 0/14 | 0/18 |
| Cardiac valve abnormalities | + | − | − | − | + | − | − | ? | ? | − | 2/10 | 1/14 | 5/18 |
| Chronic respiratory insufficiency | + | + | − | − | − | − | − | − | − | − | 2/10 | 1/14 | 3/18 |
| Facial dysmorphologya | +° | +° | +° | + | + | + | + | +° | + | + | 10/10 | 12/14 | 13/18 |
| Dental abnormalities | + | + | ? | + | + | + | − | + | + | + | 8/10 | 0/14 | 4/18 |
| Delayed motor development | + | + | + | − | − | − | + | + | ? | − | 5/10 | 2/14 | 10/18 |
| Delayed cognitive development | + | + | ? | − | − | − | + | − | ? | − | 3/10 | 3/14 | 7/18 |
| Radiological features | |||||||||||||
| Metaphyseal widening | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 4/18 |
| Narrowing and bowing long bones | + | + | + | + | + | + | + | + | + | + | 10/10 | 0/14 | 10/18 |
| Hypoplastic ilia | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 7/18 |
| Acetabular/femoral head dysplasia | + | + | + | + | + | − | + | + | + | + | 9/10 | 8/14 | 4/18 |
| Vertebral body changes | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 8/18 |
| Osteopenia | + | +° | + | + | + | + | + | ? | ? | + | 8/10 | 3/14 | 9/18 |
| Fractures | + | +° | + | + | + | + | + | − | ? | + | 8/10 | 0/14 | 9/18 |
| PI:1 | PII:1 | PIII:1 | PIV:1 | PIV:2 | PIV:3 | PV:1 | PVI:1 | PVII:1 | PVIII:1 | All | SEMDJL1 | EDS | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Female | Female | Male | Male | Male | Female | Male | Male | NA | NA | NA |
| Ethnicity | USA | USA | Dutch | Congolese— Rwandan | Congolese— Rwandan | Congolese— Rwandan | Indian | Dutch | Iranian | French | NA | NA | NA |
| Consanguinity | − | − | − | − | − | − | − | − | + | − | 1/10 | ||
| Age at last evaluation | 6 years | 8 months | 9 months | 25 years | 31 years | 28 years | 7 years | 3 years | 3 years | 37 years | NA | NA | NA |
| Pregnancy and birth | |||||||||||||
| Prenatal ultrasound abnormalities | + | − | + | ? | ? | ? | − | ? | ? | − | 2/10 | 2/14 | 1/18 |
| Perinatal complications | + | + | + | ? | ? | ? | + | + | ? | + | 6/10 | 1/14 | 3/18 |
| Clinical phenotype | |||||||||||||
| Short stature | + | + | + | + | + | + | + | + | + | + | 10/10 | 8/14 | 10/18 |
| Kyphoscoliosis | +° | + | + | + | + | + | +° | + | + | + | 10/10 | 13/14 | 11/18 |
| Multiple contractures | +° | +° | +° | + | + | + | +° | +° | + | + | 10/10 | 5/14 | 13/18 |
| Joint hypermobility | + | + | + | + | + | + | + | + | + | + | 10/10 | 6/14 | 14/18 |
| Joint dislocations | + | +° | + | + | + | + | + | +° | + | + | 10/10 | 2/14 | 8/18 |
| Congenital hip dysplasia | − | +° | +° | ? | ? | ? | +° | +° | ? | − | 4/10 | 6/14 | 6/18 |
| Cervical spine instability | + | + | + | − | − | − | ? | ? | ? | − | 3/10 | 0/14 | 0/18 |
| Hypotonia | +° | +° | +° | − | − | + | +° | − | ? | − | 5/10 | 1/14 | 11/18 |
| Soft skin | +° | + | + | + | + | + | + | − | + | + | 9/10 | 2/14 | 9/18 |
| Transluscent skin | +° | + | − | − | − | − | − | + | ? | + | 4/10 | 0/14 | 5/18 |
| Hyperextensible skin | + | + | − | + | + | + | + | − | ? | + | 7/10 | 2/14 | 10/18 |
| Cutis laxa | +° | + | + | − | − | − | + | − | + | − | 5/10 | 0/14 | 3/18 |
| Atrophic scarring | + | − | − | + | + | + | − | − | ? | + | 5/10 | 0/14 | 1/18 |
| Aortic root dilatation | + | + | − | − | − | − | − | ? | ? | + | 3/10 | 0/14 | 0/18 |
| Cardiac valve abnormalities | + | − | − | − | + | − | − | ? | ? | − | 2/10 | 1/14 | 5/18 |
| Chronic respiratory insufficiency | + | + | − | − | − | − | − | − | − | − | 2/10 | 1/14 | 3/18 |
| Facial dysmorphologya | +° | +° | +° | + | + | + | + | +° | + | + | 10/10 | 12/14 | 13/18 |
| Dental abnormalities | + | + | ? | + | + | + | − | + | + | + | 8/10 | 0/14 | 4/18 |
| Delayed motor development | + | + | + | − | − | − | + | + | ? | − | 5/10 | 2/14 | 10/18 |
| Delayed cognitive development | + | + | ? | − | − | − | + | − | ? | − | 3/10 | 3/14 | 7/18 |
| Radiological features | |||||||||||||
| Metaphyseal widening | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 4/18 |
| Narrowing and bowing long bones | + | + | + | + | + | + | + | + | + | + | 10/10 | 0/14 | 10/18 |
| Hypoplastic ilia | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 7/18 |
| Acetabular/femoral head dysplasia | + | + | + | + | + | − | + | + | + | + | 9/10 | 8/14 | 4/18 |
| Vertebral body changes | + | + | + | + | + | + | + | + | + | + | 10/10 | 9/14 | 8/18 |
| Osteopenia | + | +° | + | + | + | + | + | ? | ? | + | 8/10 | 3/14 | 9/18 |
| Fractures | + | +° | + | + | + | + | + | − | ? | + | 8/10 | 0/14 | 9/18 |
aFacial dysmorphology is characterized by frontal bossing, mid-facial hypoplasia, low−set and posteriorly rotated ears, prominent eyes and blue sclerae, a long philtrum and a short nose with anteverted nares. +, present; −, absent; ?, unknown; °, present at birth; NA, not applicable; SEMDJL1, spondyloepimetaphyseal dysplasia with joint hypermobility; EDS, Ehlers–Danlos syndrome. The columns ‘SEMDJL1’ and ‘EDS’ summarize the clinical and radiographic findings of all hitherto reported patients for whom this data was available (16, 23, 24, 27, 29–32).
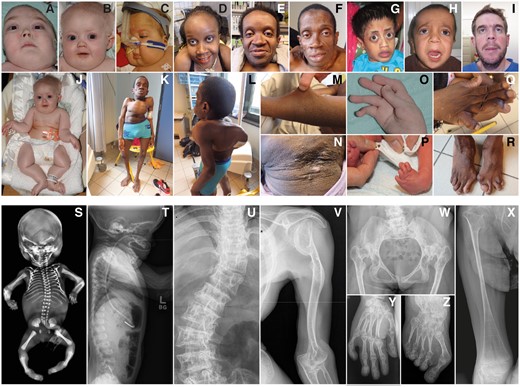
Clinical spectrum of the B3GALT6 spondylodysplastic type of the EDS. Clinical pictures and radiographs of PI:1 at age 4 years and 11 months (A, O), PII:1 at age 10 years (B, J) and 7 months (T), PIII:1 at age 8 months (C), PIV:1 at age 26 (D, Q) and age 24 (U, W, Y, Z) years, PIV:2 at age 28 years (E, M, N), PIV:3 at age 30 (F, K, L, R) and age 28 (V) years, PV:1 at age 7 years (G, X), PVI:1 at birth (P), PVII:1 at age 5 years (H), PVIII:1 at age 5 years (I), and PIX:2 (S) at 22 weeks of gestation.
The complexity of the phenotype is illustrated by the presumed diagnoses at first presentation: they were referred for genetic testing of osteogenesis imperfecta (Families I, II, IV), cutis laxa (Families I, V), Loeys–Dietz syndromes (Families I, II), AR EDS (Family I), congenital contractural arachnodactyly (Family II), Shprintzen–Goldberg syndrome (Family II), Larsen syndrome (Family III), vascular EDS (Family VIII) and mesomelic dysplasia (Family IX).
Sex ratio was seven males to five females. One patient was born to consanguineous parents (PVII:1). In the live born patients, the molecular diagnosis was made at a mean age of 14 years and 9 months (range: 5 months to 38 years). Clinical data on adulthood are limited to the siblings from Family IV and PVIII: 1.
Prenatal ultrasound abnormalities were reported in four pregnancies. PI:1 displayed bilateral lower limb anomalies, a small cerebellum and oligohydramnios. PIII:1 had shortening of the tubular bones and multiple contractures. In Family IX, two pregnancies were terminated at 22 weeks of gestation because of severe skeletal dysplasia.
From this point forward, the clinical description will be limited to the remaining 10 patients. Five patients were born at term; gestational age was not reported in the others. Anthropometric measurements were usually within normal limits, but post-natal growth restriction was noted in all patients. It was reported as early as a few months after birth and was severe in all patients (length below third percentile). Contributing to the short stature was the progressive and severe kyphoscoliosis, a consistent finding in all patients. It was congenital in only two, but progressive and severe in all but PIII:1. In all patients, multiple contractures of small (i.e. adducted thumbs) and large joints (i.e. talipes equinovarus) were present either at birth or developed at a later age. Joint hypermobility with multiple dislocations of small and large joints were reported in all patients. Congenital hip dysplasia was reported in four. Cervical spine instability was reported in three patients. At the age of 12 months, PIII:1 had severe torticollis, secondary to posterior displacement of the vertebral column with atlanto-occipital and atlanto-axial dislocation. Likewise, invagination of the atlas into the foramen magnum and anterior atlanto-axial subluxation were reported in PI:1 and PII:1, respectively. In PI:1 and PIII:1, this cervical instability was associated with hydrocephalus. Hypotonia was reported in five patients and was congenital in all but one. PV:1 presented with pectus carinatum, PVII:1 with pectus excavatum and PI:1 had an asymmetrical chest. Other musculoskeletal features included arachnodactyly (n = 3/10) and hallux valgus (n = 3/10).
The skin was hyperextensible (n = 7/10), and soft with a doughy texture in the majority of patients (n = 9/10). A thin and translucent skin was reported in four. A cutis laxa-like aspect was described in five patients. Atrophic scarring was reported in five patients. Easy bruising was reported in only two.
Most patients also had a recognizable facial gestalt characterized by mid-facial hypoplasia (n = 8/10), frontal bossing (n = 8/10), low-set and posteriorly rotated ears (n = 9/10), a depressed nasal bridge (n = 5/10), prominent eyes (n = 7/10), blue sclerae (n = 6/10), a short nose with anteverted nares and a long philtrum (n = 6/10). Less frequent findings included micrognathia (n = 3/10), plagiocephaly (n = 2/10) and a high arched palate (n = 3/10). The siblings from Family IV presented with dentinogenesis imperfecta. PVI:1 had severe caries. Dental hypoplasia/agenesis was reported in four patients. One patient, PI:1, was born with a laryngopharyngeal cleft.
There were no consistent ophthalmic features: PI:1 had optic nerve atrophy due to glaucoma, PIII:1 had moderate myopia with peripapillary atrophy at the age of 4 months and PIV:1 had microcornea. Bilateral mixed sensorineural and conductive hearing loss was documented in PIII:1.
Delayed motor development was reported in five patients and ranged from mild (PVI:1) to severe (PI:1, PII:1, PIII:1, PV:1). Speech development was delayed in two patients. At the age of 6 years, PI:1 babbled and used an iPad to communicate non-verbally using pictures. PV:1 did not speak at the age of 7 years and showed some autistic traits at the age of 7 years.
The phenotype, and more specifically the cardiovascular and respiratory phenotype, of some patients was strikingly more severe.
PI:1 developed seizures shortly after birth. These were caused by an intracranial hemorrhage that presumably occurred during vaginal delivery. He was intubated and ventilated for the first month of life. At the age of 16 months, a dilatation of the aortic root and a mild prolapse of the mitral valve were noted. At the age of 4 years he underwent chemical pleurodesis after recurrent spontaneous pneumothoraces. He still needed non-invasive ventilation at night and oxygen via nasal cannula while awake because of muscular hypotonia, tracheomalacia and restrictive lung disease at the age of 6 years.
PII:1 was born as a floppy infant, received routine resuscitation, and was subsequently transferred to the neonatal intensive care unit with multiple skeletal anomalies and mild respiratory insufficiency. She was also diagnosed with an aneurysm of the ascending aorta.
PIII:1 developed apneas with desaturation 4 days after birth, for which she was treated with non-invasive ventilation for 7 days and caffeine therapy for several weeks. At the age of 2 months, she developed respiratory distress with tachypnea, caused by a combination of mild lung hypoplasia, atelectasis, mild distal tracheomalacia and compression of the proximal trachea due to the scoliosis. She passed away at the age of 18 months due to sputum aspiration and hypoxia.
PVI:1 was diagnosed with a Wilms tumor of the left kidney at the age of 3 years, and she underwent a nephrectomy and received chemotherapy.
Finally, PVIII:1 suffered from a perforated diverticulitis at the age of 33 years, which was complicated by difficult cicatrization and necessity of prolonged ileostomy. He suffered from a cerebral vascular accident at the age of 35 years and was diagnosed with a dilatation of the thoracic aorta (TAA) at the age of 37 years.
Radiological findings
After interruption at 22 weeks of gestation, babygrams of PIX:1–2 were taken. These showed severe mesomelic skeletal dysplasia with bowing of the radius and ulna, hypoplasia of the iliac bones, platyspondyly and ovoid vertebrae, and rocker bottom feet.
Post-natal radiographs were performed in all live born patients. All patients had moderate to severe SEMD. They all showed widening of the metaphyses of long bones and relative narrowing and bowing of the diaphysis of long bones. Another consistent finding was hypoplasia of the iliac bones. In addition, moderate to severe acetabular dysplasia and femoral head dysplasia were present in all patients except PIV:3. All patients showed changes in the shape of the vertebral bodies, including anterior beaking (n = 4/10), flattening (n = 2/10) or ovoid vertebrae (n = 3/10). Osteopenia was reported in eight patients, and bone mineral density measurement showed severe osteoporosis in three. Eight patients had fractures, either at birth or in infancy. PI:1 and PIII:1 were started on bisphosphonate treatment within the first 2 years of life; it was discontinued in PI:1 after a clear decrease in the number of fractures. Sporadic findings included radio-ulnar synostosis (PI:1) and craniosynostosis (PII:1).
Molecular studies
The complexity of the phenotype prompted whole-exome sequencing or panel sequencing in Families I and IX, and II and VIII, respectively. In all other families, immediate direct sequencing of B3GALT6 was performed. Supplementary Material, Table S1 provides an overview of the genetic tests performed. Overall, we identified eight compound heterozygous mutations and one homozygous mutation, comprising 11 missense variants, 2 frameshift variants, a deletion of 19 amino acids, and a start codon alteration (Table 2 and Fig. 2A and B). In silico predictions for all missense mutations are summarized in Table 2. Most mutations are located in the luminal domain of β3GalT6, and affect either the stem region, the predicted catalytic galactosyltransferase domain, or the very C-terminal part of the protein (Fig. 2B). In addition, we found a Leu-to-Pro substitution affecting the transmembrane region of the enzyme, and a start codon mutation. In those families for which parental DNA was available (I, V, VII, IX), mutations segregated as expected. Five variants were present in the queried databases; all were reported just once and in a heterozygous state (Table 2). Two of these were previously reported in compound heterozygosity with another variant in patients with a severe EDS-like disorder (24,27).
Overview of pathogenic B3GALT6 alleles
| Family | cDNA change | Protein change | PolyPhen-2 | SIFT | MutationTaster | Align GVGD | Amino acid conservation | MAF variant databases | EDS variant database ID | Reported by |
|---|---|---|---|---|---|---|---|---|---|---|
| I | c.795A>C | p.(Glu265Asp) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/12347 (ESP) | AN_005838 | Sellars et al. (27) |
| c.929A>G | p.(Tyr310Cys) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/30902 (gnomAD) | – | ||
| II | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005839 | – |
| c.925T>A | p.(Ser309Thr) | Possibly damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/104593 (ExAC) | Nakajima et al. (24) | ||
| III | c.477del | p.(Phe160Serfs*118) | NA | NA | NA | NA | NA | NP | AN_005840 | – |
| c.782G>A | p.(Arg261His) | Probably damaging | Deleterious | Disease causing | C25 | Highly conserved | NP | – | ||
| IV | c.197_253del | p.(Ala66_Arg84del) | NA | NA | NA | NA | NA | NP | AN_005841-3 | – |
| c.953C>T | p.(Pro318Leu) | Probably damaging | Deleterious | Disease causing | C35 | Highly conserved | NP | – | ||
| V | c.3G>A | p.(?) | NA | NA | NA | NA | Highly conserved | NP | AN_005844 | – |
| c.631C>T | p.(Pro211Ser) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/113633 (ExAC) | – | ||
| VI | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005845 | – |
| c.430G>A | p.(Asp144Asn) | Possibly damaging | Tolerated | Disease causing | C0 | Moderately conserved | NP | – | ||
| VII | c.545A>G (ho) | p.(Tyr182Cys) (ho) | probably damaging | Deleterious | Disease causing | C65 | Highly conserved | NP | AN_005846 | – |
| VIII | c.77T>C | p.(Leu26Pro) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | AN_005847 | – |
| c.808G>A | p.(Gly270Ser) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1–16836 (ExAC) | – | ||
| IX | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005848-9 | – |
| c.181G>C | p.(Val61Leu) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | – |
| Family | cDNA change | Protein change | PolyPhen-2 | SIFT | MutationTaster | Align GVGD | Amino acid conservation | MAF variant databases | EDS variant database ID | Reported by |
|---|---|---|---|---|---|---|---|---|---|---|
| I | c.795A>C | p.(Glu265Asp) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/12347 (ESP) | AN_005838 | Sellars et al. (27) |
| c.929A>G | p.(Tyr310Cys) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/30902 (gnomAD) | – | ||
| II | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005839 | – |
| c.925T>A | p.(Ser309Thr) | Possibly damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/104593 (ExAC) | Nakajima et al. (24) | ||
| III | c.477del | p.(Phe160Serfs*118) | NA | NA | NA | NA | NA | NP | AN_005840 | – |
| c.782G>A | p.(Arg261His) | Probably damaging | Deleterious | Disease causing | C25 | Highly conserved | NP | – | ||
| IV | c.197_253del | p.(Ala66_Arg84del) | NA | NA | NA | NA | NA | NP | AN_005841-3 | – |
| c.953C>T | p.(Pro318Leu) | Probably damaging | Deleterious | Disease causing | C35 | Highly conserved | NP | – | ||
| V | c.3G>A | p.(?) | NA | NA | NA | NA | Highly conserved | NP | AN_005844 | – |
| c.631C>T | p.(Pro211Ser) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/113633 (ExAC) | – | ||
| VI | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005845 | – |
| c.430G>A | p.(Asp144Asn) | Possibly damaging | Tolerated | Disease causing | C0 | Moderately conserved | NP | – | ||
| VII | c.545A>G (ho) | p.(Tyr182Cys) (ho) | probably damaging | Deleterious | Disease causing | C65 | Highly conserved | NP | AN_005846 | – |
| VIII | c.77T>C | p.(Leu26Pro) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | AN_005847 | – |
| c.808G>A | p.(Gly270Ser) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1–16836 (ExAC) | – | ||
| IX | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005848-9 | – |
| c.181G>C | p.(Val61Leu) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | – |
Nucleotide numbering of variants reflects cDNA numbering, with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence of B3GALT6 (RefSeq NM_080605.3). Amino acid residues are numbered from the first methionine residue of the protein reference sequence (RefSeq NP_542172.2). Variant nomenclature follows the HGVS guidelines (http://www.hgvs.org/mutnomen; date last accessed June 13, 2018). All variants were checked with Mutalyzer software (version 2.0.18; https://mutalyzer.nl; date last accessed June 13, 2018) and submitted to the EDS Variant Database (https://eds.gene.le.ac.uk; date last accessed June 13, 2018). A unique identifier was obtained for each proband following submission to the EDS Variant Database. The Ensembl (release 84, http://www.ensembl.org; date last accessed June 13, 2018), Genome Aggregation Databases (gnomAD, http://gnomad.broadinstitute.org; date last accessed June 13, 2018) and the EDS Variant database (https://eds.gene.le.ac.uk/home.php? select_db=B3GALT6; date last accessed June 13, 2018) were queried for the prevalence of the variations (45). Align GVGD classifiers indicate the interference with function and range from less likely (C0) to most likely (C65).
ho, homozygous; NP, not present; NA, not applicable; ESP, NHLBI Exome Sequencing Project; ExAC, Exome Aggregation Consortium.
Overview of pathogenic B3GALT6 alleles
| Family | cDNA change | Protein change | PolyPhen-2 | SIFT | MutationTaster | Align GVGD | Amino acid conservation | MAF variant databases | EDS variant database ID | Reported by |
|---|---|---|---|---|---|---|---|---|---|---|
| I | c.795A>C | p.(Glu265Asp) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/12347 (ESP) | AN_005838 | Sellars et al. (27) |
| c.929A>G | p.(Tyr310Cys) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/30902 (gnomAD) | – | ||
| II | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005839 | – |
| c.925T>A | p.(Ser309Thr) | Possibly damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/104593 (ExAC) | Nakajima et al. (24) | ||
| III | c.477del | p.(Phe160Serfs*118) | NA | NA | NA | NA | NA | NP | AN_005840 | – |
| c.782G>A | p.(Arg261His) | Probably damaging | Deleterious | Disease causing | C25 | Highly conserved | NP | – | ||
| IV | c.197_253del | p.(Ala66_Arg84del) | NA | NA | NA | NA | NA | NP | AN_005841-3 | – |
| c.953C>T | p.(Pro318Leu) | Probably damaging | Deleterious | Disease causing | C35 | Highly conserved | NP | – | ||
| V | c.3G>A | p.(?) | NA | NA | NA | NA | Highly conserved | NP | AN_005844 | – |
| c.631C>T | p.(Pro211Ser) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/113633 (ExAC) | – | ||
| VI | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005845 | – |
| c.430G>A | p.(Asp144Asn) | Possibly damaging | Tolerated | Disease causing | C0 | Moderately conserved | NP | – | ||
| VII | c.545A>G (ho) | p.(Tyr182Cys) (ho) | probably damaging | Deleterious | Disease causing | C65 | Highly conserved | NP | AN_005846 | – |
| VIII | c.77T>C | p.(Leu26Pro) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | AN_005847 | – |
| c.808G>A | p.(Gly270Ser) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1–16836 (ExAC) | – | ||
| IX | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005848-9 | – |
| c.181G>C | p.(Val61Leu) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | – |
| Family | cDNA change | Protein change | PolyPhen-2 | SIFT | MutationTaster | Align GVGD | Amino acid conservation | MAF variant databases | EDS variant database ID | Reported by |
|---|---|---|---|---|---|---|---|---|---|---|
| I | c.795A>C | p.(Glu265Asp) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/12347 (ESP) | AN_005838 | Sellars et al. (27) |
| c.929A>G | p.(Tyr310Cys) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/30902 (gnomAD) | – | ||
| II | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005839 | – |
| c.925T>A | p.(Ser309Thr) | Possibly damaging | Tolerated | Disease causing | C0 | Highly conserved | 1/104593 (ExAC) | Nakajima et al. (24) | ||
| III | c.477del | p.(Phe160Serfs*118) | NA | NA | NA | NA | NA | NP | AN_005840 | – |
| c.782G>A | p.(Arg261His) | Probably damaging | Deleterious | Disease causing | C25 | Highly conserved | NP | – | ||
| IV | c.197_253del | p.(Ala66_Arg84del) | NA | NA | NA | NA | NA | NP | AN_005841-3 | – |
| c.953C>T | p.(Pro318Leu) | Probably damaging | Deleterious | Disease causing | C35 | Highly conserved | NP | – | ||
| V | c.3G>A | p.(?) | NA | NA | NA | NA | Highly conserved | NP | AN_005844 | – |
| c.631C>T | p.(Pro211Ser) | Probably damaging | Deleterious | Disease causing | C65 | Highly conserved | 1/113633 (ExAC) | – | ||
| VI | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005845 | – |
| c.430G>A | p.(Asp144Asn) | Possibly damaging | Tolerated | Disease causing | C0 | Moderately conserved | NP | – | ||
| VII | c.545A>G (ho) | p.(Tyr182Cys) (ho) | probably damaging | Deleterious | Disease causing | C65 | Highly conserved | NP | AN_005846 | – |
| VIII | c.77T>C | p.(Leu26Pro) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | AN_005847 | – |
| c.808G>A | p.(Gly270Ser) | Probably damaging | Tolerated | Disease causing | C0 | Highly conserved | 1–16836 (ExAC) | – | ||
| IX | c.513_520del | p.(Glu174Alafs*266) | NA | NA | NA | NA | NA | NP | AN_005848-9 | – |
| c.181G>C | p.(Val61Leu) | Benign | Deleterious | Disease causing | C0 | Moderately conserved | NP | – |
Nucleotide numbering of variants reflects cDNA numbering, with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence of B3GALT6 (RefSeq NM_080605.3). Amino acid residues are numbered from the first methionine residue of the protein reference sequence (RefSeq NP_542172.2). Variant nomenclature follows the HGVS guidelines (http://www.hgvs.org/mutnomen; date last accessed June 13, 2018). All variants were checked with Mutalyzer software (version 2.0.18; https://mutalyzer.nl; date last accessed June 13, 2018) and submitted to the EDS Variant Database (https://eds.gene.le.ac.uk; date last accessed June 13, 2018). A unique identifier was obtained for each proband following submission to the EDS Variant Database. The Ensembl (release 84, http://www.ensembl.org; date last accessed June 13, 2018), Genome Aggregation Databases (gnomAD, http://gnomad.broadinstitute.org; date last accessed June 13, 2018) and the EDS Variant database (https://eds.gene.le.ac.uk/home.php? select_db=B3GALT6; date last accessed June 13, 2018) were queried for the prevalence of the variations (45). Align GVGD classifiers indicate the interference with function and range from less likely (C0) to most likely (C65).
ho, homozygous; NP, not present; NA, not applicable; ESP, NHLBI Exome Sequencing Project; ExAC, Exome Aggregation Consortium.

Overview of identified and reported B3GALT6 mutations. (A) Schematic representation of the domain structure of β3GalT6. The pathogenic variants reported here are depicted in the upper bar as black-filled icons. The lower diagram gives an overview of all previously reported pathogenic variants in EDS, spondyloepimetaphyseal dysplasia with joint hypermobility type 1 (SEMD), and Al-Gazali syndrome (AGS) (black-hollow icons). Stars, triangles and squares depict frameshift, missense and indel mutations, respectively. TM, transmembrane region. (B) Schematic representation of the predicted effect of all B3GALT6 mutations reported here.
B3GALT6 mutations reduce the amount and activity of β3GalT6
To gain further insights into the functional consequences of these mutations we performed additional experiments on fibroblast cultures from PI:1 [p.(Glu265Asp), p.(Tyr310Cys)], PIII:1 [p.(Phe160Serfs*118), p.(Arg261His)], PVII:1 [homozygous p.(Tyr182Cys)], and PIX:1 [p.(Val61Leu), p.(Glu174Alafs*266)].
Reverse-transcription quantitative PCR (RT-qPCR) analysis showed no apparent difference in B3GALT6 expression between patient and control samples (Fig. 3A). At protein level, β3GalT6 co-localized with GOLPH4 to the Golgi complex in both patient and control fibroblasts (Fig. 3B). Semi-quantitative immunofluorescence microscopy did, however, show a significant reduction in the amount of β3GalT6 protein in all patient fibroblasts. This reduction was more pronounced in fibroblasts from patients with one frameshift and one missense variant (PIII:1 and PIX:1) compared with those with two missense variants (Fig. 3B).
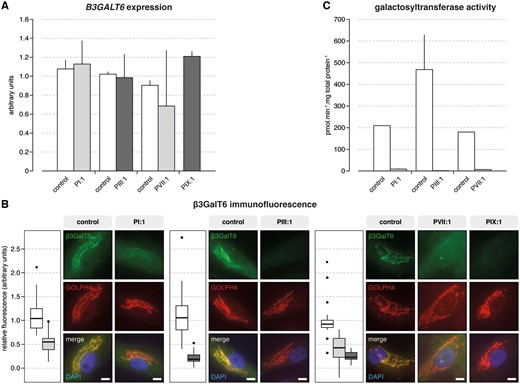
B3GALT6 mutations affect B3GALT6 expression, β3GalT6 protein level and galactosyltransferase activity. (A) RT-qPCR analysis on cDNA derived from cultured fibroblasts from PI:1, PIII:1, PVII:1, PIX:1, and age- and sex-matched controls showed no statistically significant difference in B3GALT6 expression. Assays were run in duplicate. Data are expressed as the mean±SEM. (B) Semi-quantitative immunofluorescence showed a significant reduction in the amount of β3GalT6 protein in patients compared with control fibroblast cell cultures. This reduction was function of the type of mutation and was most profound in the case of compound heterozygous frameshift and missense variants (compared with two missense variants). Nuclei were counterstained with DAPI. Scale bars represent 10 µm. (D) Galactosyltransferase activity of β3GalT6 toward the Gal-Xyl(2P)-OMN acceptor substrate was strongly reduced in patient’s fibroblasts. Notably, in all patient-derived fibroblasts, the activity was <10 pmol.min−1.mg total protein−1, whereas the activity reached 250 to 400 pmol.min−1.mg total protein−1 in control fibroblasts. These data are the result of two independent experiments with assays performed in duplicate. Data are expressed as mean±SEM.
To assess the in vitro galactosyltransferase activity of β3GalT6 in patients’ fibroblast extracts we used the Galβ1–4Xyl(2-O-phosphate)-O-methoxy-naphthyle [Gal-Xyl-(2P)-OMN] substrate. B3GALT6 mutations caused a dramatic reduction (PI:1 and PVII:1) or even a complete loss (PIII:1) of galactosyltransferase activity towards this acceptor substrate (Fig. 3C).
β3GalT6-deficiency disturbs GAG synthesis
To assess the effect of B3GALT6 mutations at the cellular level, we first analyzed the capacity of fibroblasts to prime GAG synthesis on the exogenous xyloside 4-methylumbelliferyl-β-d-xylopyranoside (β-d-xyloside 4-MUX). Figure 4A and Supplementary Material, Figure S1 show a concentration-dependent increase in GAG synthesis in control fibroblasts, whereas in all patient samples GAG synthesis was strongly reduced. Notably, the barely undetectable in vitro galactosyltransferase activity did not translate to a complete loss of GAG synthesis in cellulo. At the highest concentration of 4-MUX (10 µM), GAG synthesis in patient fibroblasts was reduced by ∼70–80%.
![B3GALT6 mutations affect GAG synthesis. (A) GAG synthesis was strongly reduced in patient fibroblasts compared with controls. GAG synthesis rate was evaluated by [35SO4]-incorporation in cultured cells as a function of xyloside concentration. These data are the result of three independent experiments. Statistical analysis was performed using Student’s t-test. Data are expressed as mean±SEM. **P < 0.01 and ***P < 0.001. (B) Glycanation of decorin core protein was decreased in patients’ fibroblast cultures. Control samples showed a broad band of glycanated decorin around ∼75 kDa (ranging from 60 to 95 kDa). In patient samples, this glycanated decorin band is decreased in intensity, and a second band (∼50 kDa) is observed, corresponding to the decorin core protein devoid of its CS/DS chain. In agreement with the reported reduction in GAG chain length with age, the molecular weight was noticeably higher in fetal fibroblasts (PIX:1 and a sex-matched control sample), and ranged from 75 to 100 kDa (44). In PIX:1 and its control, a third, low-molecular band (∼32 kDa), possibly a degradation product, is seen. (C) Semi-quantitative immunofluorescence microscopy showed a reduction in the amount of HS chains at the membrane of fibroblasts from patients compared with controls. Nuclei were counterstained with TO-PRO-3. Scale bars represent 10 µm. Data are expressed as mean±SEM. ***P < 0.001 (Student’s t-test). (D) In vitro wound closure was delayed in β3GalT6-deficient fibroblasts compared to controls. Wound closure was reduced by 23 and 38% in PVII:1 after a 20 and 40 h time period, respectively. No significant difference was observed in PIII:1 at 20 h, but closure was reduced by ∼20% at 40 h. The delay in wound closure was the most dramatic for PI:1 (reduction of ∼60 and 57% at 20 and 40 h, respectively). These data are the result of three independent experiments. Statistical analysis was performed using Student’s t-test. Data are expressed as mean±SEM. *P < 0.05 and ***P < 0.001. (E) TEM of the dermis showed dispersed collagen fibrils in the reticular dermis, compared with the regular and tight assembly in controls. Collagen fibril diameter varied, with small and large-sized fibrils and occasional fibrils with very irregular contours. In patient samples, granulofilamentous deposits were observed between the collagen fibrils. In addition, elastic fibers were highly fragmented into small and ‘crowded’ portions. Scale bars represent 200 nm.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/27/20/10.1093_hmg_ddy234/2/m_ddy234f4.jpeg?Expires=1716397144&Signature=eN2g~jIQRKQUhwOYSG0hFwh-6E-V3oYbVvwhKUA7ZlgliPGbD4pqPJEvAzcvOOaLrDFGwrR2DyO9nBdr681uZCqnIKO58-40~eZBh7jo6vT-2Ck855psM8xvZhJpSXAegH~p~sLyMAp6zBGArh3cQz4V9WQcrWQHXlKuOLqS7NYgLZCbi8XDofHBNjztVOQ2LkzxR1i3YHbaJw9iI5iuZ2o-o9~IPlyi4O5DkV0uWs~N6qG6-NkJNFv9xdoqiEkHvOVV6OmLoFHft6eRE9fnwRNbOgc3e6uKUYIL7u9jqi04nT2arW9mzWEeNG7jzO~~~SbWpsWLWKk~hRMmf3GmMw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
B3GALT6 mutations affect GAG synthesis. (A) GAG synthesis was strongly reduced in patient fibroblasts compared with controls. GAG synthesis rate was evaluated by [35SO4]-incorporation in cultured cells as a function of xyloside concentration. These data are the result of three independent experiments. Statistical analysis was performed using Student’s t-test. Data are expressed as mean±SEM. **P < 0.01 and ***P < 0.001. (B) Glycanation of decorin core protein was decreased in patients’ fibroblast cultures. Control samples showed a broad band of glycanated decorin around ∼75 kDa (ranging from 60 to 95 kDa). In patient samples, this glycanated decorin band is decreased in intensity, and a second band (∼50 kDa) is observed, corresponding to the decorin core protein devoid of its CS/DS chain. In agreement with the reported reduction in GAG chain length with age, the molecular weight was noticeably higher in fetal fibroblasts (PIX:1 and a sex-matched control sample), and ranged from 75 to 100 kDa (44). In PIX:1 and its control, a third, low-molecular band (∼32 kDa), possibly a degradation product, is seen. (C) Semi-quantitative immunofluorescence microscopy showed a reduction in the amount of HS chains at the membrane of fibroblasts from patients compared with controls. Nuclei were counterstained with TO-PRO-3. Scale bars represent 10 µm. Data are expressed as mean±SEM. ***P < 0.001 (Student’s t-test). (D) In vitro wound closure was delayed in β3GalT6-deficient fibroblasts compared to controls. Wound closure was reduced by 23 and 38% in PVII:1 after a 20 and 40 h time period, respectively. No significant difference was observed in PIII:1 at 20 h, but closure was reduced by ∼20% at 40 h. The delay in wound closure was the most dramatic for PI:1 (reduction of ∼60 and 57% at 20 and 40 h, respectively). These data are the result of three independent experiments. Statistical analysis was performed using Student’s t-test. Data are expressed as mean±SEM. *P < 0.05 and ***P < 0.001. (E) TEM of the dermis showed dispersed collagen fibrils in the reticular dermis, compared with the regular and tight assembly in controls. Collagen fibril diameter varied, with small and large-sized fibrils and occasional fibrils with very irregular contours. In patient samples, granulofilamentous deposits were observed between the collagen fibrils. In addition, elastic fibers were highly fragmented into small and ‘crowded’ portions. Scale bars represent 200 nm.
β3GalT6 deficiency reduces CS/DS and HS synthesis
To determine whether the reported B3GALT6 mutations affect both HS and CS/DS synthesis, we first examined the glycanation of the CS/DS small leucine rich proteoglycan (SLRP) decorin by immunoblotting (Fig. 4B). As expected, a single, broad band of ∼75 kDa, corresponding to the GAG-substituted decorin, was detected in control fibroblasts. The glycanation of decorin was impaired in PIII:1 and PIX:1 but was more severely affected in fibroblasts from PI:1 and PVII:1 as shown by the almost complete loss of decorin GAG substitution. All patient samples showed the presence of a second, faster migrating band (∼50 kDa), corresponding to the decorin core protein devoid of its CS/DS chain, that was not found or almost absent in controls.
Next, we examined the synthesis of HS chains on the cell surface of fibroblasts by indirect immunofluorescence microscopy (Fig. 4C). A strong cell membrane-associated, mostly continuous, staining of the HS chains was observed in all three control cell lines. Analysis of the patients’ fibroblasts revealed a characteristic ‘patchy’ and punctuate distribution of HS at the cell membrane. Semi-quantitative analysis showed a similar reduction in staining intensity of ∼50% in all three analyzed patients.
β3GalT6 deficiency disturbs cell migration and collagen fibrillogenesis
To provide further insights into the phenotypic consequences of deficient GAG synthesis, we employed an in vitro wound healing assay. After 40 h, wound closure was significantly delayed in PI:1, PIII:1 and PVII:1 (reduction between by 20 and 57%). This indicates that altered GAG synthesis has important effects on cell behavior (Fig. 4D).
Finally, transmission electron microscopy (TEM) was performed to examine the effect of β3GalT6 deficiency on connective tissue architecture (Fig. 4E). This showed collagen abnormalities including increased interfibrillar space with granulofilamentous deposits, and more variable fibril diameters (Fig. 4E). In addition, elastic fibers were highly fragmented into small and ‘crowded’ portions.
Discussion
In 2013, biallelic B3GALT6 mutations were reported to cause two different disorders. Nakajima and colleagues (24) identified B3GALT6 mutations in ten Japanese families with either SEMDJL1 or the so-called ‘progeroid’ EDS. Coincidentally, our group identified B3GALT6 mutations in three families with a severe and pleiotropic EDS-like disorder characterized by SEMD, kyphoscoliosis, joint hypermobility and contractures, fractures, skin fragility and intellectual disability (23). Subsequent studies expanded the total number of patients with B3GALT6 mutations to 40 (31 families), and thereby established it as the most frequent genetic defect affecting the synthesis of the GAG linker region (16,23,24,27–32).
The current study presents clinical, molecular and biochemical data on 12 additional patients (9 families) with biallelic B3GALT6 mutations. The most salient clinical features are: (i) post-natal growth restriction, kyphoscoliosis, joint contractures, joint hypermobility, congenital hip dysplasia and hypotonia with delayed motor development; (ii) severe osteoporosis with fractures after minor trauma; (iii) SEMD with metaphyseal widening and bowing of long bones, hypoplasia of the iliac bones, acetabular/femoral head dysplasia and vertebral body changes; (iv) a hyperextensible sometimes lax skin with a soft and doughy texture; and (v) a recognizable facial gestalt with frontal bossing, midfacial hypoplasia, low-set and posteriorly rotated ears, prominent eyes and blue sclerae, a long philtrum and a short nose with anteverted nares. In addition, this report describes several less frequent but severe and potentially life-threatening features. An aneurysm/dilatation of the ascending part of the TAA was observed in three patients. Previously, TAA had not been reported in the context of B3GALT6 mutations. Another unreported feature was instability of the cervical spine. This was reported in three patients and caused hydrocephalus in two. Chronic respiratory insufficiency was reported in two patients. Finally, one patient suffered from a cerebrovascular accident, and one was born with an intracranial hemorrhage. The latter is the second major bleeding to be reported in B3GALT6-related disease, after we previously reported a patient with a cerebral hemorrhage after minor head trauma (23).
This pleiotropic phenotype evokes a wide differential diagnosis that includes not only other types of EDS, but also osteogenesis imperfecta, cutis laxa, chondrodysplasias, and familial TAA aneurysm and dissection as well. It also argues for the use of next generation sequencing techniques in the diagnostic workup of complex heritable connective tissue disorders: panel sequencing, whole-exome and whole-genome sequencing appear to be more efficient and less expensive than conventional Sanger sequencing for the diagnosis of such complex and multisystemic phenotypes (16,33).
Biallelic B3GALT6 mutations have been reported to cause two distinct disorders. The 2017 International Classification of the EDS associates these mutations with spondylodysplastic EDS (spEDS). This is a rare AR subtype of EDS caused by biallelic mutations in B4GALT7 (named spEDS-B4GALT7), B3GALT6 (named spEDS-B3GALT6) or SLC39A13 (named spEDS-SLC39A13) (25,26). In contrast, the 2015 revision of the nosology and classification of genetic skeletal disorders associates B3GALT6 mutations with SEMD with joint hypermobility (SEMDJL1 or SEMDJL Beighton type), a clinically distinct skeletal dysplasia (34). The classification of individual patients, however, seems arbitrary and mostly based on the dominant feature(s) at presentation. Upon review of patients previously reported to have SEMDJL1, we noted that some had features of EDS such as a soft and hyperextensible skin (Table 1). Although these EDS features were often less severe and not consistently present, this does suggest that biallelic B3GALT6 mutations cause a single clinical entity with variable signs of EDS and SEMDJL1, and the ‘spondylodysplastic EDS’ moniker might therefor be more apt to describe this pleiotropic but recognizable disorder.
Hitherto, 26 pathogenic B3GALT6 variants have been reported. Most of these affect the N-terminal end or the luminal domain of β3GalT6. We now further expand the molecular spectrum by reporting eight compound heterozygous mutations and one homozygous mutation. Notably, we describe one mutation affecting the transmembrane region of the enzyme, an important domain for Golgi trafficking and retention (35).
B3GALT6 encodes the Golgi-resident galactosyltransferase β3GalT6, which is essential for the synthesis of the GAG linker region. In the four patient fibroblasts studied, PI:1 [p.(Glu265Asp), p.(Tyr310Cys)], PIII:1 [p.(Phe160Serfs*118), p.(Arg261His)], PVII:1 [homozygous p.(Tyr182Cys)] and PIX:1 [p.(Val61Leu), p.(Glu174Alafs*266)], B3GALT6 was expressed at normal levels. At protein level, β3GalT6 localized to the Golgi compartment in both patient and control fibroblasts. Semi-quantitative immunofluorescence microscopy analysis did, however, show a significant reduction in the amount of protein in all patient cell lines and especially in those with a frameshift variant (PIII:1 and PIX:1). This is in agreement with our previous work on three other patients that showed a reduced protein level without mislocalization (23). Together, these findings suggest that the mutant β3GalT6 enzymes are expressed, but are either inefficiently translated or prematurely degraded. Note that two other reports have showed mislocalization of the recombinant mutant proteins in HeLa cells (24,32).
Two studies have assayed the in vitro galactosyltransferase activity of mutant β3GalT6 using different acceptor substrates (Gal-O-naphthyl and Gal-Xyl-O-methoxynaphthyl); in both instances, activity in either fibroblasts or in recombinant cells was severely reduced for most of the mutant enzymes (23,24). A recent report showing that β3GalT6 activity is greatly increased after xylose phosphorylation by the kinase Fam20B, prompted us to use the synthesized phosphorylated disaccharide analog Gal-Xyl(2P)-OMN as acceptor substrate (36). We observed a severely reduced or even absent enzyme activity in all assayed patients’ fibroblasts. Although some differences in residual enzymatic activity could be observed, they were deemed too small to explain the differences in severity of the disorder.
GAG chains exhibit structural diversity that results from a complex and tightly regulated biosynthetic pathway. β3GalT6 catalyzes the second step in the synthesis of the GAG linker region that is essential prior to polymerization of either HS or CS/DS chains. Notably, the barely undetectable in vitro galactosyltransferase activity did not translate to a complete loss of GAG synthesis in cellulo. Using the exogenous 4-MUX substrate we showed that the ability of patient fibroblasts to prime GAG synthesis was strongly decreased, but not absent. To further assess how this affects the different types of GAGs attached onto the core protein, we examined the glycanation of the CS/DS-SLRP decorin as well as the HS expression in patients’ fibroblasts. Immunoblotting showed striking differences in decorin glycanation: substitution of decorin was moderately impaired in PIII:1 and PVII:1 but was almost completely absent in PI:1 and PIX:1. On the other hand, expression of HS on the cell membrane was reduced by half in all patients’ fibroblasts. The differential effects on GAG synthesis did, however, not translate to differences in disease severity. Previous studies have already shown that different mutations can have different effects on HS and CS/DS GAG synthesis, which is remarkable since defective β3GalT6 activity and thus linker region synthesis could be assumed to affect synthesis of both GAG types to a similar degree (23,24). Several, not mutually exclusive, explanations can account for these seemingly paradoxical results. First, some mutations may have a greater deleterious effect on the in vitro than on the in cellulo β3GalT6 activity. Second, the enzyme EXTL3, which adds a GalNAc residue onto the linker region to initiate CS synthesis, might be able to take up an uncomplete linker structure such as Gal-Xyl-decorin or GlcA-Gal-Xyl-decorin. Finally, it is plausible that compensatory mechanisms such as an increase in the expression of CS synthesizing enzymes or linker modifications favoring CS synthesis take place to counteract the loss of GAG chains. These explanations are, however, speculative and require further investigation.
Decorin is a key regulator of collagen fibril and extracellular matrix assembly (37). TEM demonstrated an abnormal dermal collagen fibril architecture characterized by loosely packed collagen fibrils of variable size and shape in patients. We previously attributed these abnormalities to the effect of the abnormal GAGs on decorin and possibly other SLRPs (23).
In summary, we have redefined the phenotype associated with B3GALT6 mutations, and demonstrated that they cause a disorder with both signs of EDS and a moderate to severe SEMD, thereby adhering to the spondylodysplastic EDS moniker. We also expand the clinical spectrum by describing several severe complications. Finally, the combination of these data with an in-depth characterization of B3GALT6 expression, β3GalT6 protein level and activity, and GAG synthesis significantly improves our understanding of this rare and severe condition.
Materials and Methods
Family ascertainment
This study includes data on 12 patients (nine families). Informed consent was obtained from the patients and/or parents participating in this study. The study was approved by the Ethics Committee of the Ghent University Hospital (Ghent, Belgium). Genomic DNA was isolated from blood samples according to the standard procedures. Skin biopsies were obtained for biochemical (PI:1, PIII:1, PVII:1, PIX:1) and ultrastructural studies (PIV:1–3, PV:1). Dermal fibroblasts were cultured in Dulbecco’s modified Eagle’s medium supplemented with fetal bovine serum, non-essential amino acids, and antibiotics (all ThermoFisher Scientific, Waltham, MA, USA).
Molecular analyses
Using different platforms, whole exome sequencing and next generation sequencing panel sequencing were performed in Families I and IX, and II and VIII, respectively (details on request). For direct sequencing, coding and flanking 5′ and 3′ untranslated regions of B3GALT6 were PCR amplified starting from genomic DNA. Amplicons were sequenced using the BigDye Terminator Cycle Sequencing kit (Life Technologies, Carlsbad, CA, USA) and run on an ABI 3730XL DNA Analyzer (Life Technologies).
Expression analysis
Fetal control fibroblasts were not available for this experiment. Total RNA from dermal fibroblast cultures was extracted in triplicate using the RNeasy kit, and submitted to DNase digestion according to the manufacturer’s instructions (both Qiagen, Hilden, Germany). cDNA was synthesized with the iScript cDNA Synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA), and used for RT-qPCR using the RealTime ready DNA Probes Master mix supplemented with ResoLight Dye (Roche, Basel, Switzerland). For each sample, assays were run in duplicate on a Roche LightCycler 480 System. Data were normalized to the expression of HPRT1, RPL13A and YWHAZ reference genes, and analyzed using qbase+ (version 3.0, Biogazelle, Zwijnaarde, Belgium).
In vitro β3GalT6 activity assay
To assay β3GalT6 galactosyltransferase activity in cell homogenates Gal-Xyl(2P)-OMN was synthesized as a specific acceptor substrate mimicking the linker region primer disaccharide (36). Fifty micrograms of total protein from cell homogenates were incubated with 2 mM Gal-Xyl(2P)-OMN acceptor substrate and 2 mM UDP-Gal donor substrate in 50 mM Bis–Tris buffer (pH 7.2) containing 5 mM MnCl2 for 2 h at 37°C. The reaction products were separated by high-performance liquid chromatography using reverse phase XBridge BEH C18 column (4.6×150 mm, 5 µm; Waters, Milford, MA, USA) on an Alliance e2695 Separations Module coupled to a 486 Tunable Absorbance Detector (both Waters). Reaction product formation was detected at a wavelength of 320 nm. The mobile phase was composed of 18% (v/v) acetonitrile and 0.02% (v/v) trifluoroacetic acid in water and run at a flow rate of 1 ml/min. The amount of reaction product was quantified to calibration curves drawn with increasing concentrations of Gal-Gal-Xyl(2P)-OMN standard and analyzed under similar chromatographic conditions.
Immunocytochemistry
Immunocytochemistry was performed using a modified version of the previously described method (23). Fibroblasts were seeded at 3 × 105 cells/well and grown overnight under standard conditions. Cells were subsequently fixed with 4% (w/v) paraformaldehyde (Sigma-Aldrich, St Louis, MO, USA), and permeabilized with 0.2% (v/v) Triton X-100 in 1× phosphate buffered saline (PBS; Sigma-Aldrich) for staining of intracellular proteins. After blocking with bovine serum albumin [BSA, 5% or 10% (w/v) in PBS; Sigma-Aldrich] samples were incubated with primary antibodies against β3GalT6 (1:50; H00126792-B01P, Abnova, Taipei City, Taiwan) and GOLPH4 (1:400 dilution; ab28049, Abcam, Cambridge, UK) or HS (1:50 dilution; H1890, 10E4 epitope, United States Biological, Salem, MA, USA). Bound primary antibody was detected with Alexa Fluor 488 (1:750 dilution; A-11001), Alexa Fluor 594 (1:750 dilution; A-21207) or Alexa Fluor 555 (1:200 dilution; A-31570) conjugated secondary antibodies (all ThermoFisher Scientific). Antibodies were diluted in a 1% (anti-HS) or 2% (anti-β3GalT6, anti-GOLPH4) BSA/PBS (w/v) solution. Nuclei were counterstained with 4′-6-diamidino-2-phenylinodole hydroxychloride (DAPI; Life Technologies) or TO-PRO-3 Iodide (642/661, Life Technologies) after permeabilization with 0.1% (v/v) Triton X-100 in a 0.2% (w/v) BSA/PBS solution.
Images of β3GalT6 and GOLPH4 staining were captured with an Axio Observer.Z1 fluorescence microscope (Zeiss, Oberkochen, Germany). Quantification of β3GalT6 immunofluorescence was performed as previously described (38–41). Using Fiji (version 2.0.0-rc-49/1.51a), an outline was drawn around the Golgi complex and integrated density, area and mean fluorescence of β3GalT6 and GOLPH4 stainings were measured, along with five adjacent background readings. Using these reading, the total corrected fluorescence [integrated density−(area of selected cell×mean fluorescence of background readings)] was calculated. The mean β3GalT6 total corrected fluorescence was then normalized against the mean GOLPH4 total corrected fluorescence. This process was repeated on at least 20 different Golgi complexes from 2 separate experiments.
Images of HS staining were captured with a Leica TCS SP5× confocal microscope (Leica, Wetzlar, Germany). Following conversion of the colored images into grayscale, quantification of the immunofluorescence signal in patients’ and control cells was carried out using ImageJ (version 1.52d 11) on at least eight different areas of each picture taken from two separate experiments (38–40,42).
In cellulo GAG synthesis analysis
GAG synthesis in dermal fibroblast cultures was analyzed using a modified version of the previously described method (23). Briefly, fibroblasts were seeded onto 12-well plates at 1.5 × 105 cells well−1. After 48 h, cells were incubated in Fisher’s medium (Life Technologies) containing 2% (v/v) FBS, Na2[35SO4] (10 µCi ml−1; PerkinElmer, Waltham, MA, USA) and increasing concentrations of xyloside 4-methylumbelliferyl-β-d-xylopyranoside (4-MUX: 0–10 µM; Sigma-Aldrich) or vehicle only (dimethylsulfoxide, DMSO). Next, neosynthesized [35SO4]-labeled GAG chains secreted in the medium were precipitated by adding four volumes of ice-cold 95% ethanol for 2 h and resuspended in water. The suspension was applied to glass microfiber filters (GF/C diameter 25 mm; GE Healthcare Life Sciences) to separate radiolabeled GAG chains from non-incorporated Na2[35SO4]. Radiolabeling was quantified by scintillation counting.
Immunoblot analysis
Serum-free conditioned medium from dermal fibroblast cultures was collected at Day 14 (PI:1, PIII:1, PIV:1, PV:1) or Day 21 (PIX:1) and concentrated with Centriprep Centrifugal Filter Devices with Ultracel 30K membranes (Millipore, Billerica, MA, USA). Concentrated medium was subjected to SDS-PAGE and immunoblotting with an anti-human decorin primary antibody (1:250 dilution; Clone 115402, R&D Systems, Minneapolis, MN, USA), as described previously (23). For loading control, total protein amount of the concentrated medium was visualized with Imperial Protein Stain (ThermoFisher Scientific) after SDS-PAGE.
Transmission electron microscopy
TEM was performed as described previously (43). Briefly, small fragments of skin were first fixed in 2.5% glutaraldehyde (v/v) and 3% (w/v) formaldehyde in 0.1 M sodium cacodylate buffer (pH 7.2), and later post-fixed in 1% (w/v) OsO4 with K3Fe(CN)6 in 0.1 M sodium cacodylate buffer. Samples were put through a graded ethanol series. They were stained with 2% uranyl acetate at the 50% ethanol step and embedded in Spurr’s resin. Ultrathin sections of a gold interference color were cut, and post-stained with uranyl acetate and lead stain. Sections collected on formvar-coated copper slot grids were imaged with a JEM 1400plus transmission electron microscope (JEOL, Tokyo, Japan).
Cell migration experiments
Fibroblasts were seeded at 35 × 103 cells well−1 in two-well silicone inserts with a 500 µm cell-free gap (Biovalley, Marne-La-Vallée, France) and grown under standard conditions for 16 h. Upon removal of the insert, cell migration in the denuded area was evaluated by phase-contrast microscopy, and photographed with a Zeiss AxioCam ERc 5s microscope camera immediately, and 20 and 40 h later, as previously described (23). Evaluation of the number of migrated cells in the wound area was performed using Adobe Photoshop CS6 (Adobe, San Jose, CA, USA) on a minimum of eight different fields for each cell line.
Acknowledgements
SFG is grateful to Valérie Gisclard and Elodie Chasseraud from UNSED for their support and stimulating discussions. The Gal-Xyl(2P)-OMN substrate was kindly provided by Chrystel Lopin-Bon from the Institute of Organic and Analytical Chemistry (ICOA, Orléans, France).
Conflict of Interest statement. None declared.
Funding
This work was supported by Research Foundation Flanders [12Q5917N to D.S., 1842318N to F.M.], Ghent University [08/01M01108 to A.D.P.] and Union Nationale des Syndromes d’Ehlers-Danlos [to S.F.G.].
References
Author notes
The authors wish it to be known that, in their opinion, Tim Van Damme and Xiaomeng Pang should be regarded as joint First Authors.
The authors wish it to be known that, in their opinion, Sylvie Fournel-Gigleux and Fransiska Malfait should be regarded as joint Last and Corresponding Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}