




Abstract
A hexanucleotide repeat expansion mutation in the C9orf72 gene represents a prevalent genetic cause of several neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia. Non-canonical translation of this repeat gives rise to several distinct dipeptide protein species that could play pathological roles in disease. Here, we show in the model system Caenorhabditis elegans that expression of the arginine-containing dipeptides, but not alanine-containing dipeptides, produces toxic phenotypes in multiple cellular contexts, including motor neurons. Expression of either (PR)50 or (GR)50 during development caused a highly penetrant developmental arrest, while post-developmental expression caused age-onset paralysis. Both (PR)50- and (GR)50-green fluorescent protein tagged dipeptides were present in the nucleus and nuclear localization was necessary and sufficient for their toxicity. Using an inducible expression system, we discovered that age-onset phenotypes caused by (PR)50 required both continual (PR)50 expression and an aged cellular environment. The toxicity of (PR)50 was modified by genetic mutations that uncouple physiological aging from chronological aging. However, these same mutations failed to modify the toxicity of (GR)50, suggesting that (PR)50 and (GR)50 exert their toxicity through partially distinct mechanism(s). Changing the rate of physiological aging also mitigates toxicity in other C. elegans models of ALS, suggesting that the (PR)50 dipeptide might engage similar toxicity mechanisms as other ALS disease-causing proteins.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating and currently incurable neurodegenerative disorders. Most cases of both ALS and FTD with known genetic etiology are caused by the expansion of an intronic GGGGCC (G4C2) hexanucleotide repeat in the chromosome 9 open reading frame 72 (C9orf72) gene (1,2). Several mechanisms for why this repeat leads to disease have been proposed, including loss of C9orf72 function, nuclear G4C2 RNA foci formation and abnormal translation of dipeptide repeat proteins (DPRs) (3). Recent work shows that C9orf72-associated DPRs accumulate in the spinal fluid of C9 ALS/FTD patients and can cause cell death in in vitro and in vivo settings (4–8). While this suggests an important role for DPRs in C9 ALS/FTD pathogenesis, analysis of post-mortem brain samples shows that DPRs are present at very low levels in brain regions that undergo the most severe neurodegeneration (i.e. spinal cord motor neurons), thus calling into question the relevance of DPR proteins in disease pathogenesis (9,10). While this discrepancy could be due to a variety of factors (poor antibody detection of toxic species, loss of DPR-expressing neurons in end-stage samples), understanding the relationship between the individual DPRs and neurodegeneration in a variety of settings will be necessary to parse out the role of the DPRs in the development of C9orf72-associated diseases.
Translation of the intronic G4C2 repeat occurs through a process termed repeat-associated non-AUG-dependent (RAN) translation (11). RAN translation of the G4C2 repeats can give rise to five distinct DPRs—proline–alanine (PA), glycine–alanine (GA), proline–arginine (PR), glycine–proline (GP) and glycine–arginine (GR). While PA, GP and GA are not thought to cause significant toxicity, PR and to a lesser extent GR have been shown to be toxic in yeast, Drosophila, and mammalian cell models (6–8). Recent studies implicate several pathways in the toxicity of the PR RAN dipeptide (8,12,13). Given that both ALS and FTD exhibit age-related onset and that age-related phenotypes are not easily modeled in cellular systems, organismal models are needed to examine the role of cell non-autonomous aging regulatory pathways on the onset and severity of DPR toxicity. In this study, we establish Caenorhabditis elegans as a new model for investigating DPR toxicity mechanisms in vivo. We expressed codon-optimized transgenes for four DPRs (GA, PA, GR, PR) and characterized their toxicity and expression patterns. We found that (i) arginine-containing DPRs exhibited potent toxicity in neuronal and non-neuronal contexts; (ii) nuclear localization was necessary and sufficient for this toxicity; (iii) PR and GR toxicity was age-dependent and PR, but not GR, toxicity was influenced by mutations that uncouple physiological aging from chronological aging. Given that the toxicities of other ALS disease proteins are similarly modified by these mutations, our findings suggest that PR and other forms of ALS may share common disease mechanisms.
Results
Arginine-rich dipeptides are toxic in C.elegans motor neurons and muscle
Unconventional translation of the C9orf72-associated G4C2 repeats can produce several types of dipeptide repeat proteins (DPRs) (14). The nematode C. elegans contains a C9orf72 homolog, alfa-1, which is involved in aging and stress responses, as well as stress-induced neurodegeneration (15). However, alfa-1 does not contain G4C2 repeats and therefore does not model either RNA toxicity or RAN translation-induced DPR toxicity. To develop a C. elegans model for DPR expression, we generated constructs that express 50 repeats of 4 DPRs (GA, PA, GR and PR). Each of these constructs utilizes codon-optimized sequences that encode for one dipeptide but eliminates the G4C2 repetitive sequence. Thus, our worm system is a ‘pure’ dipeptide model and does not contain repetitive GGGGCC sequences. To facilitate biochemical and cell biological detection of the DPRs, we inserted an in frame N-terminal FLAG epitope and a C-terminal green fluorescent protein (GFP) (Fig. 1A). Since ALS is a motor neuron disease, we used the motor neuron-specific unc-47 promoter to control expression and generated transgenic animals expressing each individual DPR.
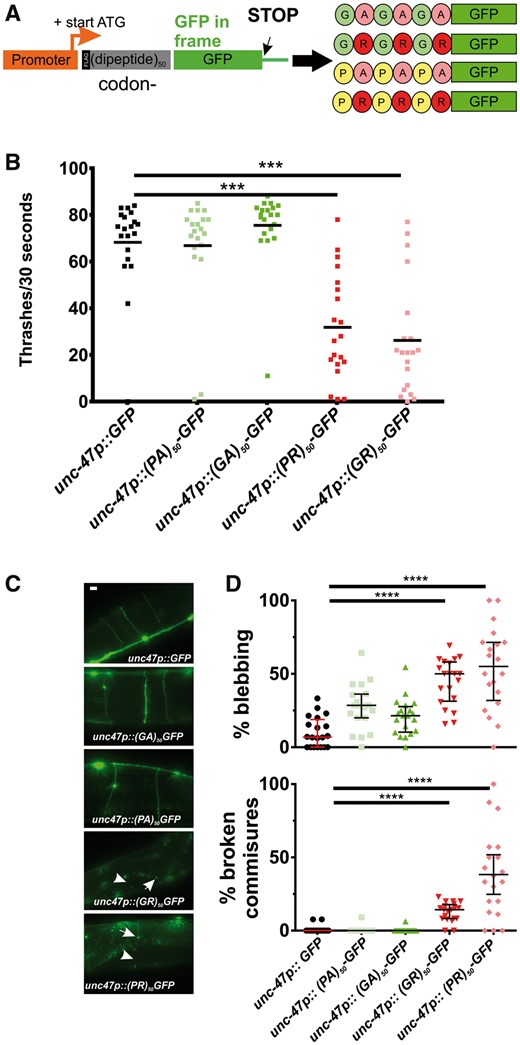
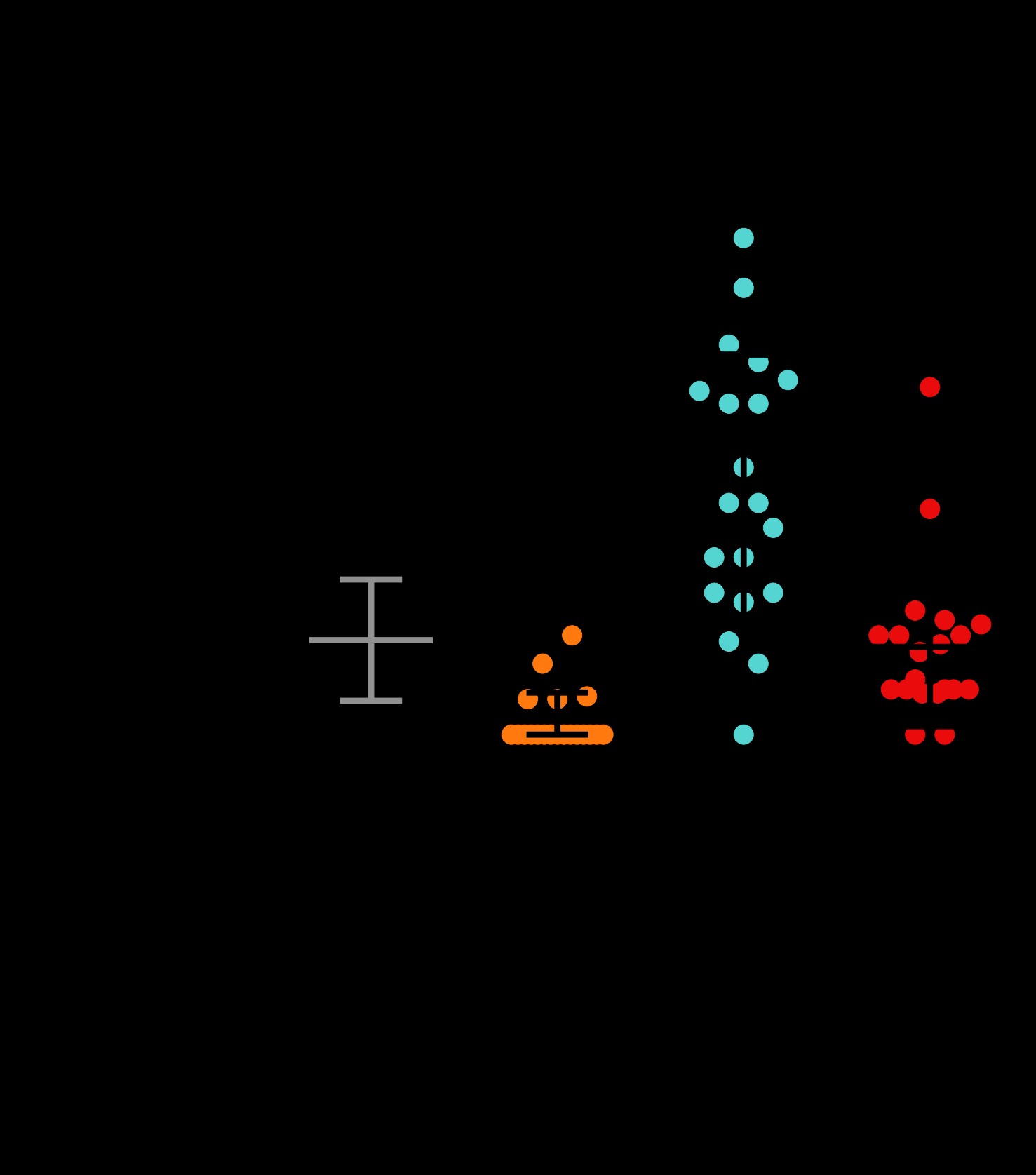
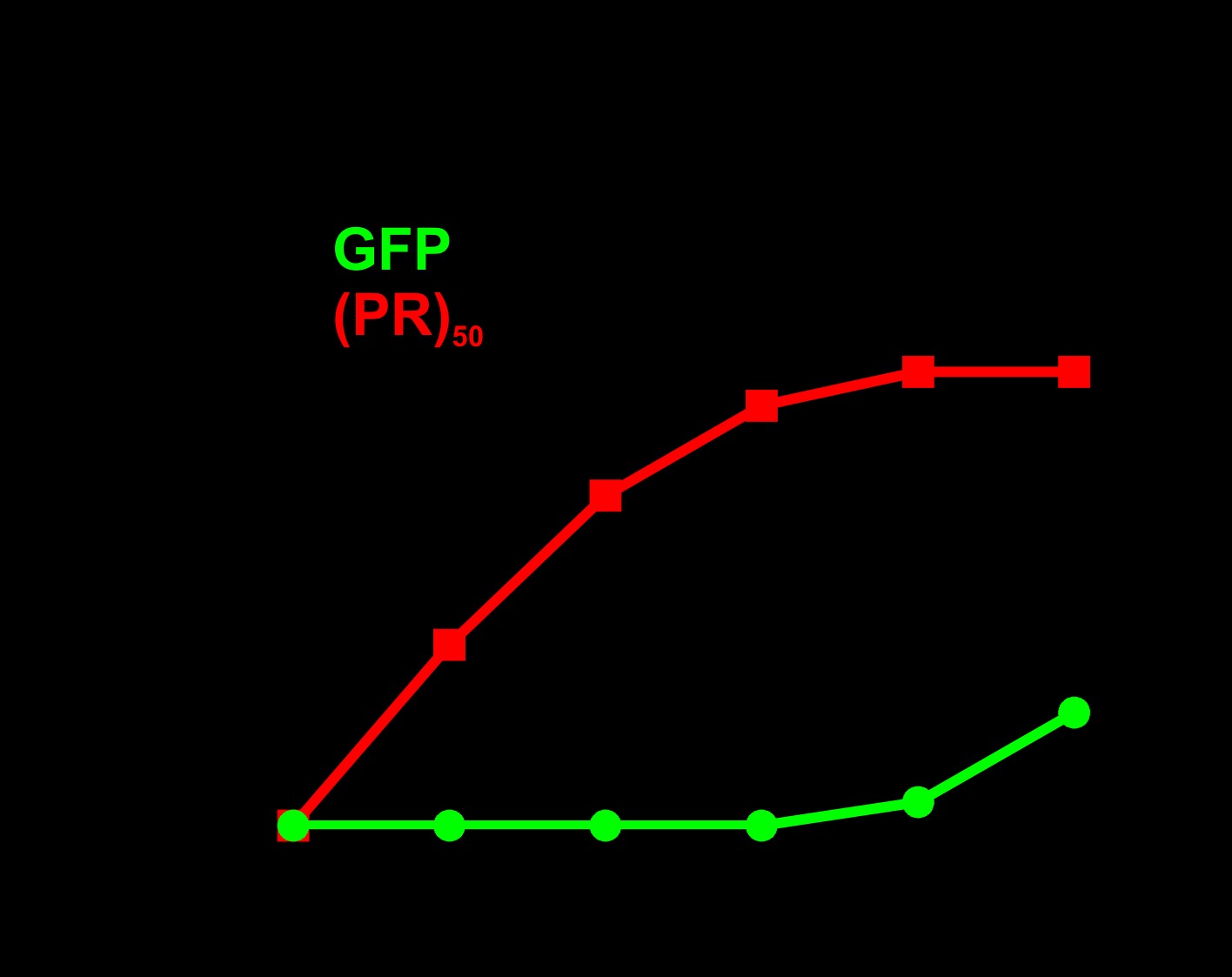
Arginine-containing DPRs are toxic in motor neurons. (A) Molecular strategy for expression of codon-varied dipeptide-repeat proteins in C. elegans. (B) Liquid thrashing quantification of transgenic animals expressing the indicated DPR under the motor neuron-specific unc-47 promoter. N = 20 animals per genotype. Each symbol represents quantification for one animal, horizontal line indicates population median. ***P < 0.001 versus GFP control (one-way non-parametric ANOVA with Dunn’s post hoc test). (C) Representative images of unc-47+ motor neurons in animals expressing the indicated DPR. Arrow points to an example of membrane blebbing. Arrowhead points to examples of commissure breaks. Scale bar = 10 μm. (D) Quantification of membrane blebbing and commissure breaks in each of the indicated strains. For each animal, we counted the total number of commissures with blebbing or breaks and divided by the total number of detectable commissures. N = 20 animals per genotype. Each symbol represents quantification for one animal expressed as a percentage. The horizontal line indicates population median with interquartile range. ****P < 0.0001 versus GFP control (one-way non-parametric ANOVA with Dunn’s post hoc test). N = 20 animals per genotype.
Consistent with previous studies in yeast, Drosophila, and mammalian cells (5–8), we found that the arginine-rich DPRs were highly toxic in C. elegans. Animals expressing arginine-rich DPRs were viable, consistent with previous studies showing that unc-47+ motor neurons are not required for viability in C. elegans (16). However, motor neuron expressed (PR)50 and (GR)50 each led to significant reductions in motility (Fig. 1B). Motor neurons expressing (PR)50 exhibited morphological signs of degeneration commonly observed in other C. elegans models of neurodegenerative diseases (17–19), including commissure degeneration and membrane blebbing that were not observed in control animals or in animals expressing either (PA)50 or (GA)50 (Fig. 1C and D). These data show that arginine-containing dipeptides cause motility defects and neurodegenerative morphological changes when expressed in C. elegans motor neurons.
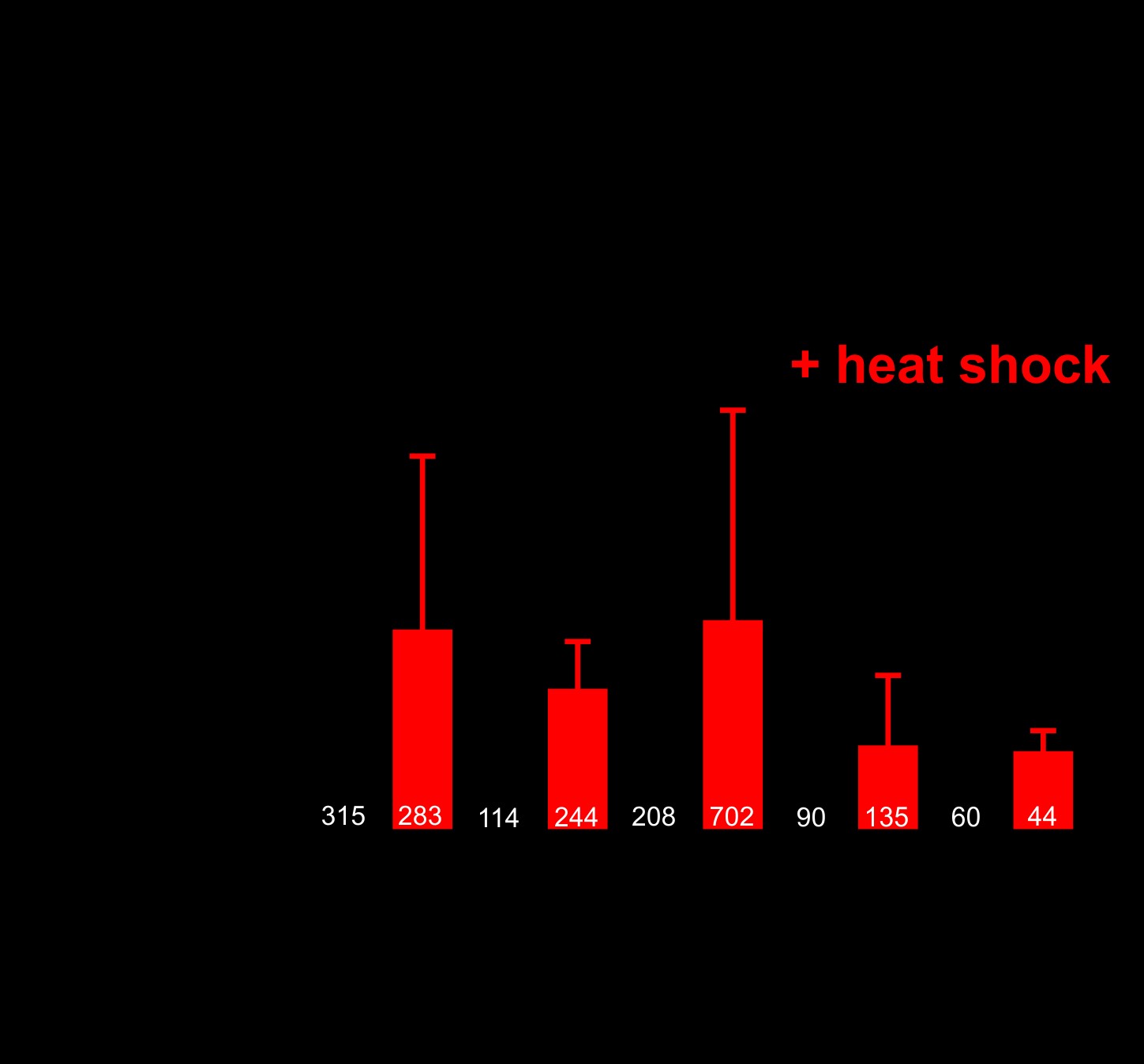
Caenorhabditis elegans motor neurons are extremely small (2–3 μm in diameter), making cell biological analysis of DPR localization in these cells difficult. Additionally, C. elegans neurons are highly resistant to RNA-interference-mediated gene knockdown, which is commonly used to explore mechanisms of protein toxicity in C. elegans disease models (20,21). To overcome these limitations, we expressed the DPRs in the larger muscle cells, which regulate motility and are a common site of expression for other C. elegans disease models (22–24). As we observed in motor neurons, muscle expression of arginine-rich dipeptides was highly toxic. Expression of either (PR)50 or (GR)50 in muscle led to completely penetrant embryonic and larval lethality that could be suppressed by feeding animals gfp(RNAi)-expressing bacteria (Fig. 2A;Supplementary Material, Table S1). gfp(RNAi)-sensitive embryonic or larval lethality was not observed in animals expressing either (PA)50 or (GA)50. (PR)50 or (GR)50 animals that were removed from gfp(RNAi) after embryonic development survived and went on to develop into adults. However, these DPR expressing adults exhibited age-onset paralysis (Fig. 2B). (PR)50 animals showed a significantly reduced lifespan that was not observed in (GR)50 expressing animals (Supplementary Material, Fig. S1). The enhanced paralysis observed in (PR)50 and (GR)50 animals was not due to higher mRNA expression, since qPCR showed that all DPRs were expressed at similar levels (Supplementary Material, Fig. S2 and Table S2). It was also not due to higher protein levels since in vivo imaging and flow cytometry quantification showed that (PR)50-GFP and (GR)50-GFP fluorescence levels were significantly below those of all other DPRs (Supplementary Material, Fig. S3). The toxicity phenotype required protein production and was not due to any potential RNA effects, since (PR)50 transgenic animals lacking a start ATG exhibited no detectable toxicity (Supplementary Material, Fig. S4). (PR)50 toxicity was dependent on the number of repeats, as animals expressing 5, 15 or 25 repeats did not exhibit embryonic lethality [number of viable transgenic progeny (>L4) after 72 h—(PR)5–GFP—139/145; (PR)15–GFP—68/75; (PR)25–GFP—114/117; P > 0.1, Fisher’s exact test]. Adults expressing (PR)5–GFP exhibited robust expression that appeared concentrated within almost all nuclei (Fig. 2C). Adults expressing both (PR)15–GFP and (PR)25–GFP expressed much lower levels of GFP when compared with (PR)5–GFP, although the protein that was present also appeared to be concentrated within the nucleus and nucleolus (Fig. 2C and D). Adults expressing (PR)25 and (PR)50, but not those expressing (PR)5 or (PR)15, exhibited accelerated age-onset paralysis (Fig. 2E), despite the fact that (PR)25 was present at dramatically lower levels than (PR)5 (Fig. 2D). Together, these studies show that arginine containing dipeptides exhibit developmental and post-developmental, age-dependent toxicity in C. elegans and in the case of the PR dipeptide, this toxicity is repeat-length dependent.
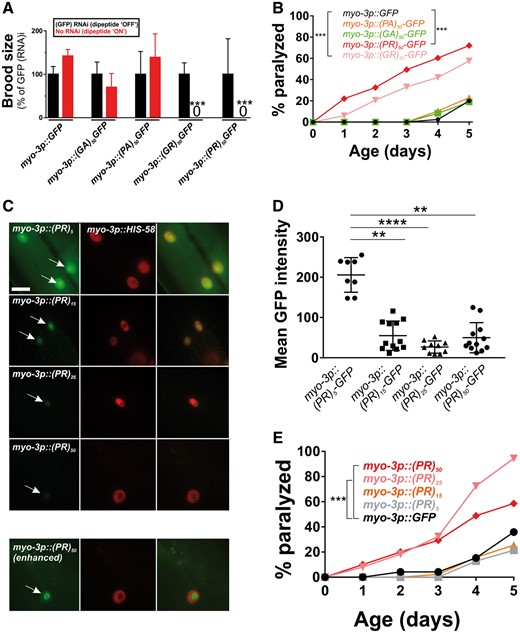
Muscle expressed arginine-containing dipeptides are toxic in C. elegans. (A) Brood size for worms expressing each integrated dipeptide protein in the presence or absence of gfp(RNAi) at 25 °C. Data for each strain are normalized to the mean brood size of animals grown on gfp(RNAi). ***P < 0.001 (one-way ANOVA with post hoc Tukey’s test). Raw brood size data for 25 °C are included in Supplementary Material, Table S1. (B) Paralysis assay for adult animals raised in the absence of gfp(RNAi). N ≥ 43–49 animals per genotype. ***P < 0.001 (Log-rank test with Bonferroni adjusted P-value). (C) Fluorescent microscopy of Day 1 adult hermaphrodites expressing (PR)5-GFP, (PR)15-GFP, (PR)25-GFP or (PR)50-GFP (green) in the muscle. Muscle DNA (marked by a myo-3p::his-58-mCherry reporter) is in red. Arrows point to sites of nuclear GFP puncta. Scale bar = 10 μm. All images were acquired using identical exposure settings. The row labeled ‘myo-3p::(PR)50 (enhanced)’ was adjusted for brightness and contrast in the GFP channel differently from the other images so that the (PR)50-GFP signal is observable. (D) Quantification of the GFP levels from the indicated PR repeat animals. Each point shows the measured value for a single nucleus, horizontal line indicates population median with interquartile range. **P < 0.01, ****P < 0.0001 versus GFP control (one-way non-parametric ANOVA with Dunn’s post hoc test). (E) Paralysis assay of animals expressing the indicated number of (PR) repeats under the control of the myo-3 promoter. ‘Day 0’ animals were isolated as L4 stage animals. N = 48–50 animals per genotype. ***P < 0.001 versus GFP control (Log-rank test with Bonferroni adjusted P-value).
Toxic (GR)50 and (PR)50 dipeptides are localized to the nucleus and are not aggregated
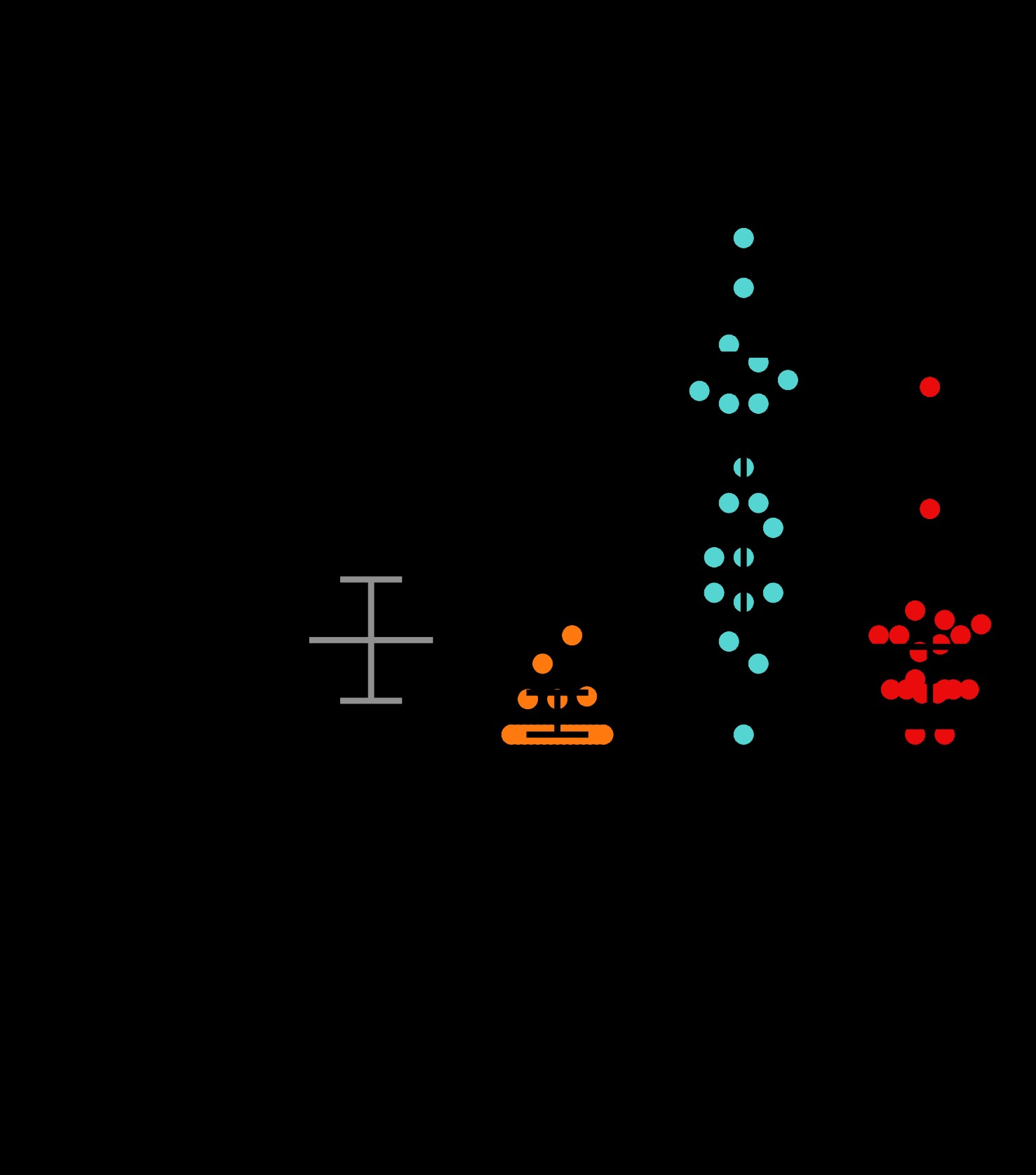
To gain insight into the cell biological properties of (PR)50 and (GR)50 proteins, we examined their subcellular localization patterns in live animals expressing each DPR in the muscle. Animals expressing both GFP and non-toxic (PA)50–GFP exhibited diffuse nuclear and non-nuclear GFP expression that was always excluded from the histone-free nucleolar region (Fig. 3A and B). Muscle expressed (GA)50–GFP formed intense non-nuclear puncta. In some cases, we noted (GA)50–GFP signal in the nucleus but this nuclear localization was always diffuse and never in puncta (Fig. 3A and B). (GA)50 non-nuclear exhibited little fluorescence recovery after photobleaching (FRAP; Fig. 4A and B), suggesting these puncta are highly immobilized protein aggregates. Muscle expressed (GR)50-GFP and (PR)50-GFP both exhibited substantially lower GFP levels than either of the other DPRs, despite their similar mRNA expression levels (Supplementary Material, Fig. S3), and appeared to be predominantly localized to the histone-free nucleolar region of the nucleus (Fig. 3A and B). Non-nuclear localization was always observed in (GR)50 animals but was never observed in (PR)50 animals (Fig. 3A and B). Unlike what we observed for (GA)50, FRAP analysis revealed that the nucleolar accumulations of (GR)50 and (PR)50 exhibited substantial fluorescence recovery after photobleaching that was significantly different from that of (GA)50 (Fig. 4). The significant mobility of both (PR)50 and (GR)50 nuclear foci is inconsistent with the known biophysical properties of bona fide aggregates (25). Since both (PR)50 and (GR)50 appeared to be localized to the nucleolus, we compared their FRAP dynamics with those of FIB-1, which is homologous to the mammalian nucleolar protein fibrillarin (26). FIB-1 exhibited recovery kinetics that were similar to those of (GR)50 and (PR)50, suggesting that the dynamics of these two DPRs behave similarly to a bona fide nucleolar protein (Fig. 4C).
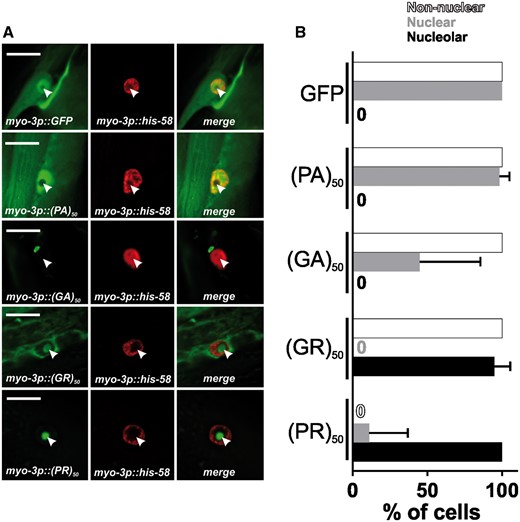
(PR)50 and (GR)50 are localized to the nucleolus in C. elegans muscle cells (A) Imaging of live, anesthetized Day 1 adult C. elegans hermaphrodites expressing the indicated dipeptide. Green shows dipeptide-GFP localization expressed in muscle, red shows histone-tagRFP within the muscle nucleus. Arrowhead points to the histone-free nucleolar region. Scale bar=10 μm. (B) Quantification of DPR–GFP signal that is observed in nucleoplasm, nucleolus, and non-nuclear cellular compartments. N = 55–78 nuclei from five to six animals. Data shown are means ± S.D.
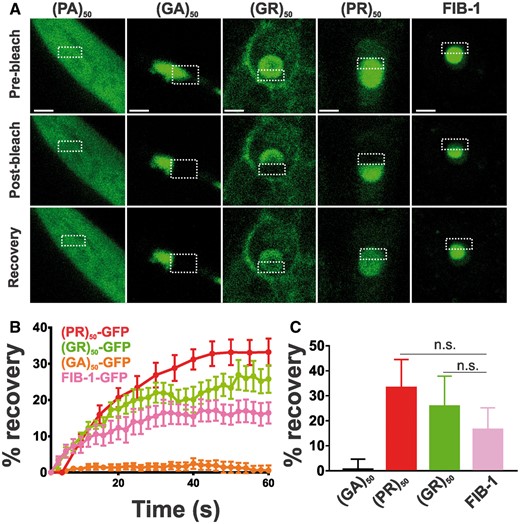
Arginine-containing DPRs are not aggregates. (A) Representative images from FRAP analysis of subcellular localized DPR proteins expressed in muscle. Dashed outline indicates site of photobleaching and post-bleaching quantification. Recovery images are 150 s post-bleach. Scale bar = 2 μm. (B) Quantification of FRAP imaging. Data shown are mean ± S.E.M. from 9 to 10 datasets per genotype. (C) Average equilibrium fluorescence recovery after 60 s. Data shown are mean ± S.D. from 9 to 10 datasets per genotype. n.s. versus FIB-1-GFP (one-way ANOVA with Dunn’s multiple comparison test).
Nuclear localization is required for (PR)50 and (GR)50 toxicity
Given that toxic (PR)50 and (GR)50 in muscle were primarily localized to the nucleus, we asked if nuclear localization was necessary for their toxicity. To test this, we tethered each of the arginine-containing DPRs to a signal sequence-transmembrane domain (SS-TM) tag that restricts the DPR to cellular membranes with the DPR oriented on the cytosolic side of the membrane. In this context, the membrane-localized (PR)50 and (GR)50 were excluded from the nucleus (Fig. 5A). In contrast to soluble (PR)50 and (GR)50, membrane-localized (PR)50 and (GR)50 were viable in the absence of gfp(RNAi) and showed no age-dependent paralysis (Fig. 5B). We further tested whether nuclear localization was sufficient for toxicity by fusing the (PR)50 DPR to the coding sequence for the his-58 gene, which encodes a DNA-binding histone only found in the nucleus. GFP-his-58 expressing animals were viable and motile and exhibited strong nuclear GFP expression (Fig. 5C and D). However, GFP-his-58-(PR)50 expressing animals were not viable unless they were cultured under gfp(RNAi) conditions [number of viable transgenic progeny (>L4)/total progeny in the absence of GFP after 72 h—GFP-his-58–348/406; GFP-his-58-(PR)50–9/56; P < 0.0001, Fisher’s exact test]. Upon removal from gfp(RNAi), his-58-(PR)50 animals exhibited age-dependent paralysis (Fig. 5D) and this paralysis occurred with more rapid onset than in (PR)50 animals (Fig. 5E), suggesting that nuclear localization further enhances (PR)50 toxicity. The enhanced his-58-(PR)50 toxicity was not due to higher expression levels of the transgene since GFP fluorescence levels of his-58-(PR)50 was not higher than (PR)50 (Supplementary Material, Fig. S5). Overall, these data show that nuclear localization is necessary and sufficient for (PR)50 toxicity in C. elegans.
![Nuclear localization of muscle expressed arginine-containing DPRs is necessary and sufficient for toxicity. (A) Representative images of the indicated soluble or membrane anchored DPR (green), the HIS-58-mCherry nuclear marker (red), and a merged image. Arrows point to nuclear regions defined by the HIS-58 mCherry signal. Coincident green and red signals in the ‘TM-(PR)50 image is intestinal autofluorescence. Scale bar = 10 μm. (B) Paralysis assay comparing animals expressing soluble or transmembrane localized (GR)50 (top) or (PR)50 (bottom). N = 48–50 animals per genotype, ***P < 0.001, **P < 0.01, *P < 0.05 (Log-rank test with Bonferroni adjusted P-value). (C) Representative images of HIS-58 anchored (PR)50 (green; top) or unanchored (PR)50 (green; bottom), HIS-58-mCherry nuclear marker (red) and a merged image. Scale bar = 5 μm. (D) Paralysis assay comparing animals expressing nuclear localized GFP or (PR)50.n ≥ 30 animals per genotype, **P < 0.01 (Log-rank test with Bonferroni adjusted P-value). (E) Paralysis assay comparing soluble (PR)50 with his-58-(PR)50. n≥ 50 animals [(PR)50] or 80 animals [his-58-(PR)50], ***P < 0.001 (Log-rank test with Bonferroni adjusted P-value).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/26/24/10.1093_hmg_ddx372/4/m_ddx372f5.jpeg?Expires=1716398582&Signature=L9y-XstkbyI3qh~RNweC0NMOMEZm1I9j0tOfpYaFCmcQP88uHh2Eg9elCPwoXeHJ9fEHK8EBxLQ7dzKYO0uoltn8zMAjXeVOWf-TotKrN681geuPUF6xPQzTQrJlgAmc8MdHzYlQLmOqACIuLMap0nDL94-P96fCk8Sto9fHEQy4PgSsL1A-yi42-ss5ZrMtWUsyjidGBPOkeXwwy6b7K5ST2VdbupQntWQT0Ao5Jq61ZiCIWPq3DMrfUpHT7ksFdN3UmHVnm78dG4C-Je1OA0q7xSEFdZZDj3H~RzKW9iCu~fcuj8PgYEcjWowswl8Cw0B5ubW6tWThjAiupTcWcg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Nuclear localization of muscle expressed arginine-containing DPRs is necessary and sufficient for toxicity. (A) Representative images of the indicated soluble or membrane anchored DPR (green), the HIS-58-mCherry nuclear marker (red), and a merged image. Arrows point to nuclear regions defined by the HIS-58 mCherry signal. Coincident green and red signals in the ‘TM-(PR)50 image is intestinal autofluorescence. Scale bar = 10 μm. (B) Paralysis assay comparing animals expressing soluble or transmembrane localized (GR)50 (top) or (PR)50 (bottom). N = 48–50 animals per genotype, ***P < 0.001, **P < 0.01, *P < 0.05 (Log-rank test with Bonferroni adjusted P-value). (C) Representative images of HIS-58 anchored (PR)50 (green; top) or unanchored (PR)50 (green; bottom), HIS-58-mCherry nuclear marker (red) and a merged image. Scale bar = 5 μm. (D) Paralysis assay comparing animals expressing nuclear localized GFP or (PR)50.n ≥ 30 animals per genotype, **P < 0.01 (Log-rank test with Bonferroni adjusted P-value). (E) Paralysis assay comparing soluble (PR)50 with his-58-(PR)50. n≥ 50 animals [(PR)50] or 80 animals [his-58-(PR)50], ***P < 0.001 (Log-rank test with Bonferroni adjusted P-value).
Post-mitotic age-dependent toxicity of (PR)50 requires continuous dipeptide expression
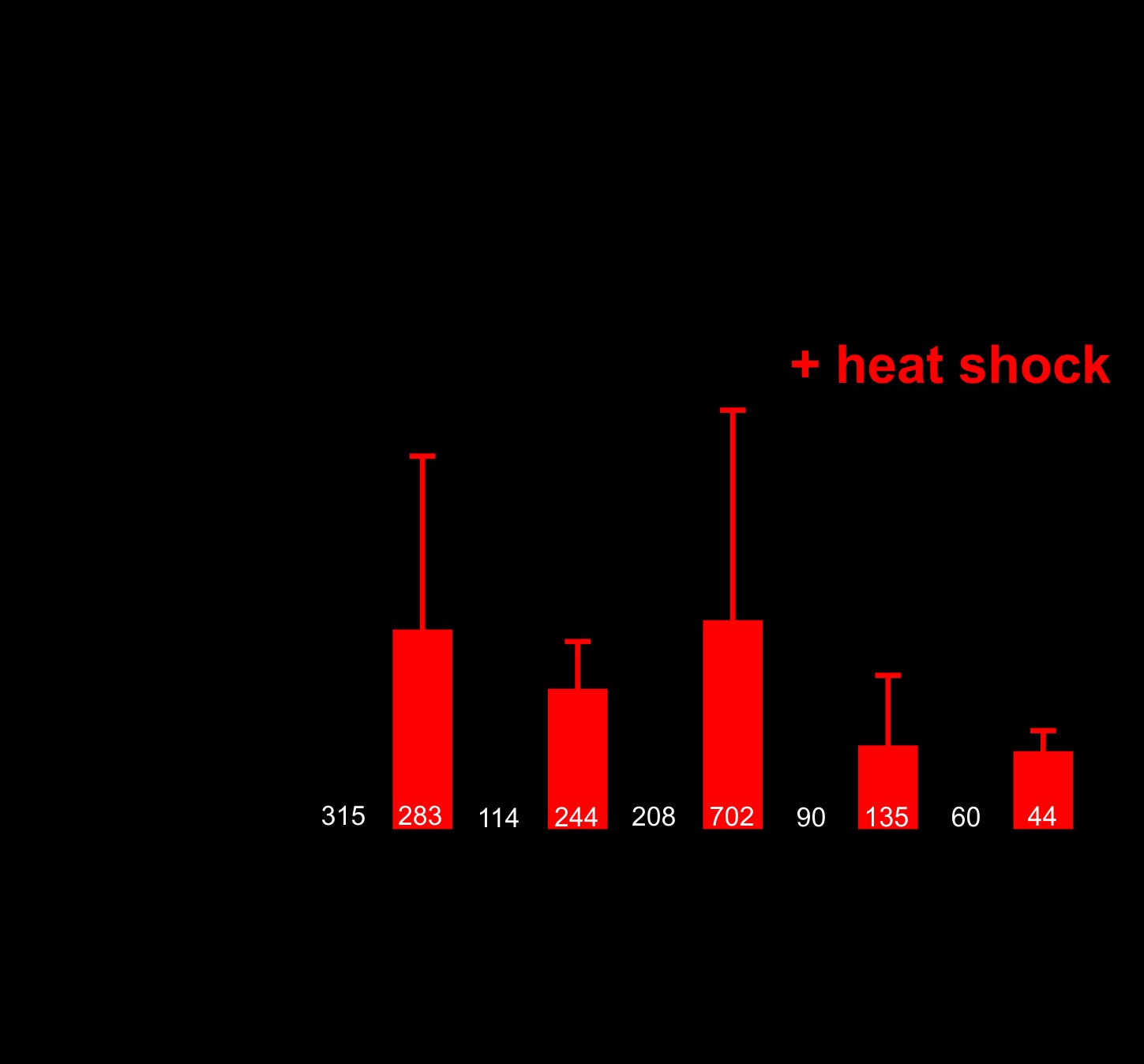
Nuclear (PR)50 exhibits age-dependent toxicity in C. elegans. This could be because (PR)50 requires time to accumulate to sufficiently high levels necessary for toxicity. Alternatively, it could be because the cellular environment in aged animals is more permissive for toxicity. To test these models, we generated animals expressing DPR–GFP under the control of a heat shock inducible promoter (hsp-16.2). Following a brief (30 min) heat shock, we observed robust GFP expression of all DPRs in young adult animals. In most cases, the subcellular expression patterns for each DPR matched what we had observed previously, although the proteins were expressed more broadly across tissues due to the global activity of the heat shock promoter (Fig. 6A). Induction of (PR)50 during embryonic development caused significant toxicity that was not observed in the absence of induction (Fig. 6B). Animals that had been induced to express (PR)50 on Day 1 of adulthood continued to exhibit normal motility and survival across their lifespan (Fig. 6C). However, continual daily induction of (PR)50 protein eventually led to increased paralysis and death that was not observed in the control animals or in other DPR expressing animals, including those expressing (GR)50. A single induction of (PR)50 expression later in life (Day 4 of adulthood) also failed to elicit any detectable phenotypes. The toxicity of (PR)50 was not due to higher induction levels than other DPRs (Supplementary Material, Fig. S6). Therefore, the age-onset phenotypes of nuclear (PR)50 in post-mitotic adult C. elegans require both continuous expression throughout life and an aged cellular environment.
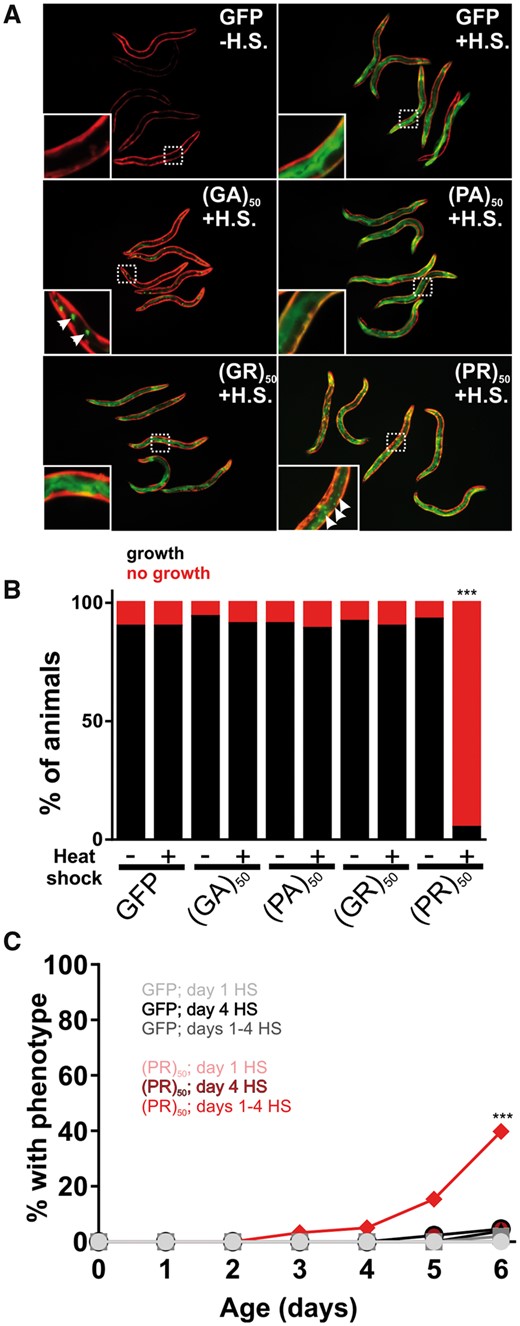
Age-onset toxicity requires continuous DPR expression. (A) Images of worms expressing the indicated DPR under the control of a heat shock inducible promoter. Red fluorescence shows the myo-3p::mCherry transgenic marker. Green fluorescence shows the hsp-16.2p::DPR–GFP signal. All DPRs are shown 2–4 h after heat shock. Insets show an enlarged view of the region identified by the dashed boxes. Arrows in the (GA)50 inset point to putative protein aggregates, suggesting that (GA)50 rapidly aggregates after synthesis. Arrows in the (PR)50 inset point to sites of nuclear localization in the intestine. No such nuclear localization was observed in the (GR)50 line after heat shock. (B) Developmental toxicity of transgenic embryos expressing the indicated hsp-16.2p::DPR–GFP. Embryos were heat shocked at 35 °C for 30 min and then returned to 20 °C. The number that reached ≥L4 after 48 h were scored as ‘growth’ while the remaining animals were scored as ‘no growth’. N ≥ 81 transgenic animals per condition. ***P < 0.001 (Fisher’s exact test versus GFP + H.S.). (C) Adult toxicity of animals expressing either hsp-16.2p::GFP or hsp-16.2p:(PR)50-GFP. Animals were subjected to either a heat shock (30 min, 35 °C) on Day 1, Day 4 or on Days 1, 2, 3 and 4. Every 24 h, animals were scored as either mobile, censored or affected (paralyzed + dead = ‘% with phenotype’). Heat shock induction of (GA)50, (PA)50 and (GR)50 produced phenotypes identical to that of GFP. N ≥ 60 animals per genotype. ***P < 0.001 versus GFP; Days 1–4 H.S (Log-rank test with Bonferroni adjusted P-value).
Mutations that alter the rate of physiological aging delay (PR)50 age-dependent toxicity
The toxicity of (PR)50 in an aged cellular environment could be due to the stochastic accumulation of toxic proteins levels or conformations over long periods of time. Alternatively, the process of aging could lead to a decline in cellular processes that oppose (PR)50 toxicity. To differentiate between these two hypotheses, we tested the role of the aging process in the toxicity of (PR)50. If (PR)50 toxicity is due to accumulation of toxic protein levels/conformation over time, lifespan-extending conditions should have no effect on the rate of toxicity. However, if the aging process actively regulates (PR)50 toxicity, alterations in the rate of aging should enhance (lifespan shortening) or suppress (lifespan extending) the toxicity of (PR)50. The insulin signaling pathway is the most well characterized pathway that regulates lifespan and youthfulness from worms to humans (27). Mutations in this pathway are thought to extend lifespan by promoting a youthful cellular physiological state and therefore separate chronological and physiological states of aging. In C. elegans, the daf-2 gene encodes the homolog of the insulin/IGF receptor. Loss of function mutations in daf-2 exert their effects via signaling-dependent activation of the FOXO transcription factor daf-16, as well as the heat shock transcription factor hsf-1 (28). Mutations in daf-16 and hsf-1 suppress phenotypes associated with daf-2 mutations and accelerate the rate of aging (27).
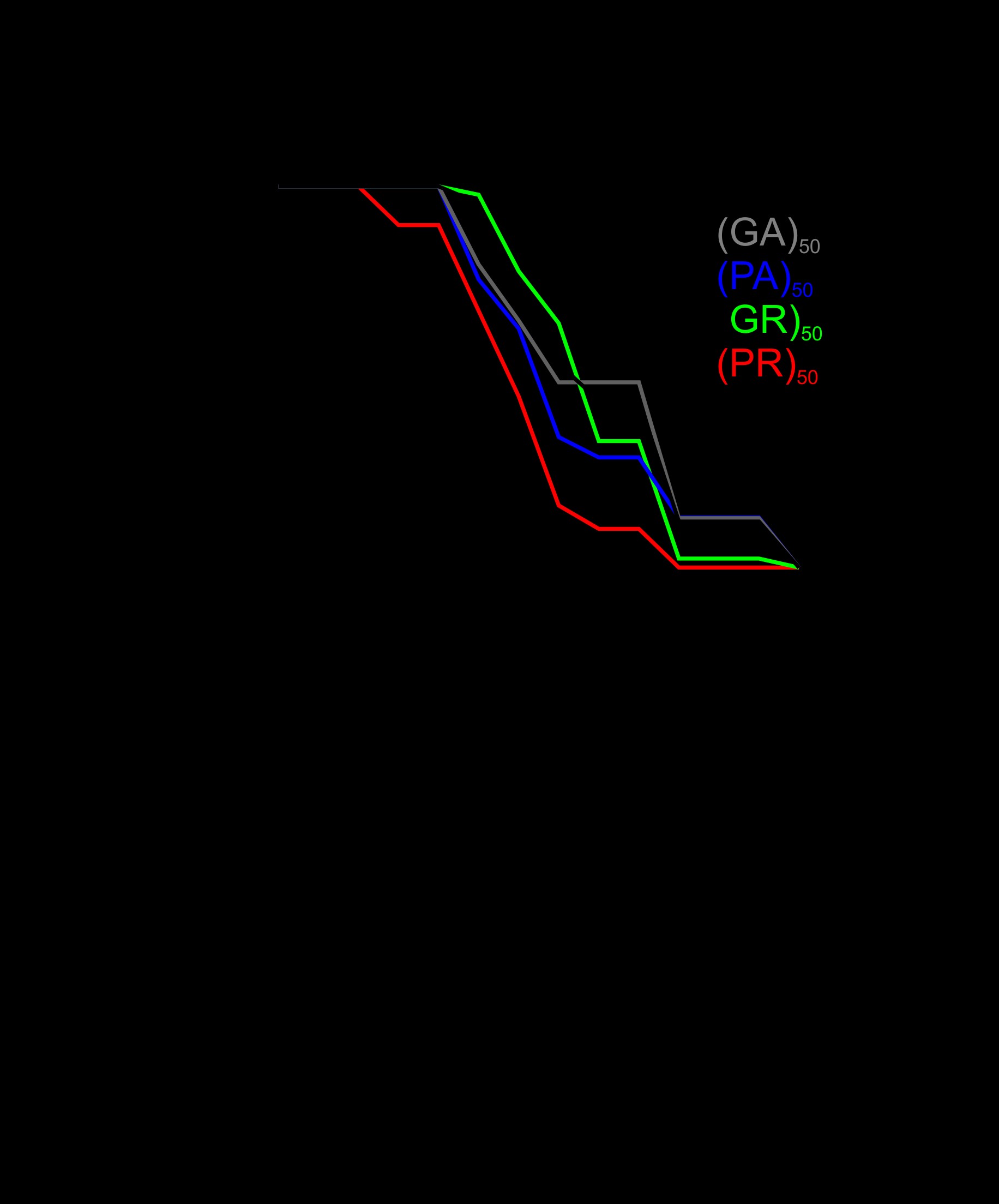
To determine whether the toxicity of (PR)50 is stochastic or enabled by declining physiological processes, we crossed the (PR)50 transgene into the daf-2(e1370); daf-16(mu86) and segregated all possible (PR)50 genotypes. If (PR)50 toxicity is stochastic, then reduced insulin signaling should not alter the age-related phenotypes. However, if (PR)50 toxicity is due to declining cellular physiology, then alterations in the rate of aging should slow the initiation and progression of toxicity. We found that in the daf-2 mutant background, age-dependent declines in (PR)50 motility were significantly reduced (Fig. 7A). These daf-2-dependent phenotypes were suppressed in a daf-2; daf-16 double mutant and were accelerated in the daf-16 mutant background (Fig. 7B). Likewise, a mutation in the heat shock transcription factor hsf-1, which accelerates the rate of aging (29), enhanced the toxicity of (PR)50 (Fig. 7C). Interestingly, the protective effects of the daf-2 mutant were not observed in (GR)50 expressing animals (Fig. 7A). The protective effects of insulin signaling appear to act downstream from DPR accumulation and nuclear localization, as DPR abundance and subcellular distribution did not appear to be altered in any of these genetic backgrounds (Fig. 7D–F). We also found that in motor neurons expressing (PR)50, daf-2 reduced the levels of neurodegeneration-associated membrane blebbing (Supplementary Material, Fig. S7). These data demonstrate that alterations in the rate of aging impact (PR)50 toxicity in both C. elegans muscle cells and motor neurons and suggest that (PR)50 toxicity is not stochastic but is dependent on the cellular aging process. Additionally, they suggest that (PR)50 and (GR)50 are toxic through partially distinct mechanisms since enhanced DAF-16/FOXO activation protects (PR)50 animals but has no protective effects on (GR)50 animals.
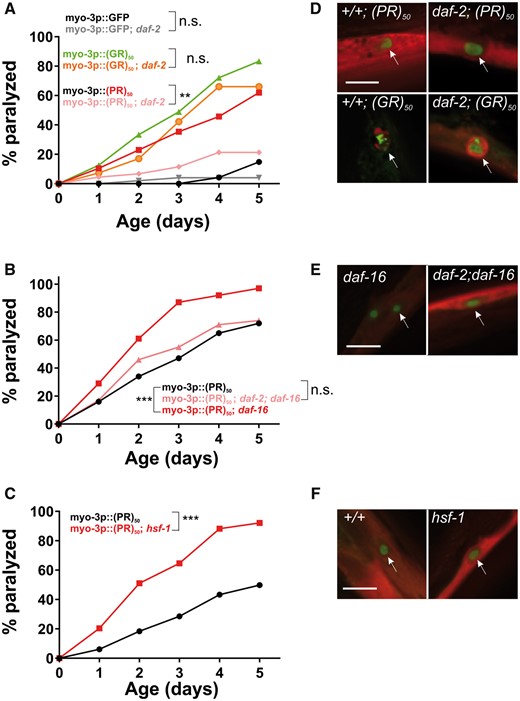
Altering the rate of aging affects (PR)50 but not (GR)50 toxicity. (A) Paralysis assay of animals expressing GFP, (GR)50 or (PR)50 under the control of the myo-3 promoter in the wild-type or daf-2(e1370) background. ‘Day 0’ animals were isolated as L4 stage animals. N = 48–50 animals per genotype. n.s., ‘not significant’; **P < 0.01 versus GFP control (Log-rank test with Bonferroni adjusted P-value). (B) Paralysis assay of animals expressing (PR)50 under the control of the myo-3 promoter in the wild-type, daf-16(mu86), or daf-2(e1370); daf-16(mu86) background. N = 48–50 animals per genotype. n.s., ‘not significant’; ***P < 0.001 versus (PR)50 (Log-rank test with Bonferroni adjusted P-value). (C) Paralysis assay of animals expressing (PR)50 under the control of the myo-3 promoter in the wild-type or hsf-1(sy441) mutant background. N = 48–50 animals per genotype. ***P < 0.001 versus wild-type (Log-rank test with Bonferroni adjusted P-value). (D) Fluorescent microscopy images of day 1 adult worms expressing either (GR)50 or (PR)50 (green) and soluble muscle mCherry (red) in the wild-type or daf-2(e1370) mutant background. Arrow points to site of nuclear DPR accumulation. Scale bar=10 μm. (E) Fluorescent microscopy images of Day 1 adult worms expressing (PR)50 (green) and soluble muscle mCherry (red) in the daf-16(mu86) or daf-2(e1370); daf-16(mu86) mutant background. Arrow points to site of nuclear DPR accumulation. Scale bar=10 μm . (F) Fluorescent microscopy images of day 1 adult worms expressing (PR)50 (green) and soluble muscle mCherry (red) in the wild-type or hsf-1(sy441) mutant background. Arrow points to site of nuclear DPR accumulation. Scale bar=10 μm.
Discussion
In this study, we developed the first C. elegans model for C9orf72 dipeptide toxicity. We discovered that (1) (PR)50 and (GR)50 dipeptides are toxic in neuronal and non-neuronal contexts, (2) both toxic dipeptides exhibit a shared nucleolar localization, (3) nuclear localization of both (PR)50 and (GR)50 dipeptides is required for toxicity, (4) post-mitotic toxicity of (PR)50 is age-dependent, (5) age-dependent toxicity of (PR)50 is modified by genetic pathways that accelerate or decelerate the rate of physiological aging in both muscle and motor neurons while (GR)50 toxicity is not modified by these pathways. While some of these findings (i.e. Points 1 and 2) are consistent with recent studies from yeast, Drosophila, and mammalian cells, others leverage the unique strengths of C. elegans to examine questions that either have not or cannot be addressed in other systems (i.e. Points 3–5). While many of our findings are consistent with studies using other C9 animal models, there are also significant differences. For example, a recent report describes toxicity associated with (GA) dipeptides in zebrafish (30). However, we find no evidence of (GA) toxicity in C. elegans when expressed in either motor neurons or muscle or more globally after heat shock, even though (GA) aggregates are observed in all of these conditions. The lack of toxicity of GA in worms compared with fish could be due to differences in repeat number (50 in worms versus 80 in fish) or differences in expression levels. In another example, we found prominent nucleolar (GR)50 localization, whereas a recent post-mortem study of C9orf72 carrier brains revealed perinuclear GR aggregates (31). Such discrepancies could reflect differences in dipeptide repeat length (50 repeats in our model versus unknown repeats in patient cells) or in live animal versus post-mortem sample fixation and preparation. Overall, our findings suggest that this new C. elegans model, in conjunction with high throughput genetic and pharmacological screening methods, will be a useful and complementary system for exploring the mechanisms underlying C9orf72-associated DPR toxicity.
Results from our work suggest that the localization of (PR) and (GR) dipeptides to the nucleus is essential for their toxicity. This is consistent with several recent studies that suggest both PR and GR DPRs exert their toxicity by interfering with nuclear functions and nuclear pores (8,12,13,32,33). For example, in yeast, overexpression of nuclear import pathways, ribosome biogenesis pathways and nuclear RNA-binding proteins strongly modify (PR)50 toxicity (8). Likewise, knockdown of Drosophila genes implicated in nuclear transport also modify PR toxicity (34). In most systems, including our new C. elegans model, both (PR)50 and (GR)50 DPRs exhibit prominent nucleolar localization. However, notable non-nuclear localization is also observed, especially in the case of the (GR)50 dipeptide. Our FRAP data suggest that while (GA)50 dipeptides are clearly protein aggregates, nuclear (PR)50 and (GR)50 are not protein aggregates, since, unlike (GA)50, they exhibit significant recovery upon photobleaching. This localization pattern, as well as the pattern of genetic modifiers identified from yeast and Drosophila screens, has led to the hypothesis that (PR)50 and (GR)50 toxicity may occur through direct interactions within the nucleus. Alternatively, (PR)50 and (GR)50 could cause toxicity via non-nuclear interactions that then impinge on nuclear functions. Findings from our C. elegans model strongly support the former hypothesis, since we found that preventing nuclear localization completely abrogated the toxicity of the arginine-containing DPRs. We also found that forcing (PR)50 into the nucleus maintained, and perhaps enhanced, toxicity. Our findings do not preclude the additional possibility that freely soluble cytoplasmic DPRs could also mediate toxicity, since our method of preventing nuclear accumulation restricted DPRs to cytosolic membranes, which would also remove freely soluble cytoplasmic DPRs. Moreover, our method for restricting DPRs to the nucleus could result in unusual toxicity mediated by charge interactions between positively charged DPRs and negatively charged DNA. Unfortunately, attempts at using traditional NLS/NES sequence to drive DPRs into or out of the nucleus were unsuccessful for unknown reasons. Nevertheless, our findings are consistent with recent work showing that these DPRs specifically interact with low-complexity nuclear and nucleolar proteins that shape the formation of non-membrane bound organelles within the nucleus (12).
Worms have been utilized to model other genetic forms of ALS, including those associated with mutant forms of proteins such as superoxide dismutase 1 (SOD1) (17,23,35–38), TAR DNA-binding protein 43 (TDP-43) (39–43) and fused in sarcoma (FUS) (39). In each of these cases, the worm model expresses a mutant protein that forms protein aggregates. The toxicity associated with the SOD1 and TDP-43 models is responsive to the activation state of the daf-2/insulin signaling pathway (41,44). One of the major mechanisms by which the daf-2/insulin signaling pathway is thought to oppose these proteotoxic disease models is via the upregulation of genes that maintain protein folding (i.e. chaperones) or enhance the clearance of misfolded/aggregated proteins (i.e. proteases) (21). The overall effect of these targets is to delay the proteotoxic consequences of aging, thus making mutants in the daf-2/insulin pathway tools for separating physiological aging from chronological aging. Like SOD1 and TDP-43, we found that (PR)50 can also be suppressed by activating insulin signaling. However, unlike SOD1 and TDP-43, (PR)50 did not form detectable protein aggregates. Given that mutations in genes comprising the daf-2/insulin signaling pathway can modify all three disease models, this suggests that (PR)50 may share common toxicity mechanisms with SOD1 and TDP-43, for example, via disruptions in protein homeostasis. Interestingly, insulin signaling did not appear to alter DPR abundance or subcellular localization in our model, suggesting that this pathway may act downstream from DPR nuclear functions to provide toxicity protection. Future genetic modifier screens should reveal the identity of such mechanisms, as well as whether DPRs and other ALS disease genes share common disease mechanisms. The powerful phenotypes elicited by (PR)50 in our C. elegans model, as well as the tractability of genome-wide RNAi approaches, will facilitate such screens.
Despite the fact that both (PR)50 and (GR)50 are required in the nucleus to exert their toxicity, we found important differences between their mechanisms of toxicity. For example, the toxicity associated with (PR)50 was sensitive to manipulations in the activation state of the daf-2/insulin signaling pathway. In the daf-2 mutant background, (PR)50 toxicity was delayed, whereas in the daf-16 and hsf-1 mutant background, (PR)50 toxicity was enhanced. Curiously, (PR)50 toxicity in the daf-2; daf-16 background was restored although not to the levels seen with daf-16 alone. This could be due to variability in assay conditions. Alternatively, it could suggest that daf-2 engages other factors, such as skn-1 (45) and/or hsf-1 (28,46), in addition to daf-16 to mediate protection against (PR)50 toxicity. However, (GR)50 toxicity was unaffected by the daf-2/insulin pathway activation state. Additionally, acute induction of (PR)50 using a heat shock-inducible promoter led to toxicity, while acute induction of (GR)50 did not produce toxicity. These observations suggest that (PR)50 and (GR)50 exert different types of toxicity and that only (PR)50 toxicity is susceptible to the consequences of daf-2/insulin pathway activation. The ability of insulin/IGF signaling to modify toxicity of one dipeptide but not another may partially explain the failure of insulin/IGF therapies in ALS clinical trials among patients with C9orf72 mutations (47–49). Perhaps additional therapies targeting insulin/IGF-independent toxicity like that associated with (GR)50 could be used in combinatorial therapies to protect against the multiple forms of toxicity associated with C9orf72 mutations. Comparative genetic modifier screens, as well as comparative RNAseq studies, should be useful in determining how cells differentially respond to these two DPRs.
RAN translation products are an emerging component of several nucleotide repeat expansion disorders, including C9orf72-associated FTD/ALS and CAG-associated Huntington’s disease (50). Caenorhabditis elegans is a particularly attractive model for identifying the mechanisms underlying the toxicity of these various RAN proteins. Given the strong phenotypes described here for the C9orf72-associated (PR)50 and (GR)50 RAN products, genetic, and pharmacological screens should prove to be a valuable approach for identifying the genes and pathways that are required for these and perhaps other RAN protein-mediated toxicity. In addition to delineating mechanisms of DPR toxicity, C. elegans may also prove useful in defining the requirements for RAN translation. Worms expressing pure GGGGCC repeats were recently described (51,52). These studies suggest that G4C2 repeat expression produces RNA foci, dipeptide protein expression and toxicity in the form of a shortened lifespan and paralysis, although whether such toxicity was due to the expression of dipeptides was not determined. If such RAN products were tagged with GFP and these RAN–GFP products were easily detectable, high-throughput genetic approaches could be used to decipher the genetic, cellular and age-dependent requirements for RAN translation in vivo.
In conclusion, we developed the first C. elegans model for C9orf72-associated DPR toxicity. Our findings reveal prominent roles for protein localization and aging in the toxicity of arginine-containing dipeptides and suggest significant differences in the response of the DPRs to protective genetic pathways. Using these phenotypes, our future studies will utilize the genetic and pharmacological accessibility of worms to better define the mechanisms of DPR toxicity. Given the conserved nature of worm genetics, we anticipate that our findings may guide new ideas for treatments or biomarkers in C9orf72 associated diseases.
Materials and Methods
Caenorhabditiselegans strains and culture
Strains were cultured on standard NGM media with E.coli OP50 bacteria. Strains expressing (PR)50 and (GR)50 DPRs were cultured on gfp(RNAi) bacteria at 20 °C until the experiment, when they were shifted to OP50. The following strains were used; daf-2(e1370), daf-16(mu86), hsf-1(sy441). Standard genetic approaches were utilized to cross mutants into the DPR backgrounds. The homozygous genotype of every strain was confirmed by DNA sequencing of the mutant lesion. Wild-type animals were re-isolated in every cross and utilized as the DPR only control in resulting experiments. DPR toxicity studies were carried out using animals grown at 25 °C, unless otherwise noted.
Molecular biology and transgenics
Codon-varied dipeptide sequences were isolated from previously described plasmids (6) (nucleotide sequences can be found in Supplementary Material, Table S3). (PR)5, (PR)15 and (PR)25 codon-varied plasmids were synthesized (GeneArt, ThermoFisher Scientific, Waltham, MA, USA). Dipeptide sequences were isolated as a HindIII/BamHI fragment and subcloned into the C. elegans expression vector pPD95.79 to generate dipeptide with a C-terminal GFP tag. Promoters were PCR amplified, incorporating both a 3XFLAG epitope immediately downstream from the start ATG and HindIII sites flanking the fragment. Promoters were subcloned into the DPR-pPD95.79 vectors as HindIII fragments. To make N-terminal GFP–(PR)50 and GFP–(GR)50 fusions, GFP was PCR amplified and cloned in frame with the start codon of the myo-3 promoter. The dipeptide sequences were subcloned as a HindIII/BamHI fragment into the pPD49.26 expression vector. The myo-3-GFP fragment was then subcloned into the DPR-pPD49.26 as a HindIII fragment. The membrane-bound DPRs were generated by a PCR fusion method. We first amplified the signal sequence and transmembrane domain from the pat-3 gene as supplied in vector pPD122.39. The membrane domain was then fused to the myo-3 promoter in-frame with the start ATG and then subcloned as a HindIII fragment into the DPR-pPD95.79 vectors. The resulting clones produce a membrane-localized DPR with the DPR facing the cytoplasm. To make the his-58–DPR fusions, the his-58 genomic sequence was PCR amplified and Gibson cloned (New England Biolabs, Ipswich, MA, USA) into the myo-3p::GFP sequence downstream of and in-frame with the GFP sequence. The resulting myo-3p::GFP-his-58 fragment was subcloned as a HindIII fragment into the DPR-pPD49.26 vectors. Primer sequences and plasmids utilized for this study are available upon request. A list of nucleotide sequences for promoters used in this study can be found in Supplementary Material, Table S4.
Transgenic worms were generated by injecting the DPR construct (20 ng/µl) and the myo-3p::mCherry pCFJ104 or myo-3p::dsRed2 marker plasmid (100 ng/µl) into the gonad of wild-type animals. Transgenes were integrated using a standard gamma-ray (Cs137) mutagenesis, followed by selection of animals exhibiting 100% transmission of the mCherry marker. Integrated strains were outcrossed six times to wild-type animals. In the case of (PR)50 and (GR)50, injected animals were maintained on gfp(RNAi) plates until the experimental assay was performed. All procedures involving recombinant or synthetic nucleic acid molecules and materials were approved by the University of Pittsburgh Institutional Biosafety Committee.
Microscopy
Worms were anesthetized (10 mM levamisole) and mounted on agar pads for fluorescence microscopy. Images were collected on either a Leica MZ16FA stereo dissecting scope or a Leica DMI4000 inverted microscope and a Leica DFC 340Fx digital camera (Leica Microsystems, Wetzlar, Germany). Z-stack images were deconvolved using Leica AF6000 software. Unless noted, images within an experiment were collected using the same exposure settings and processed with identical deconvolution parameters.
FRAP studies were carried out on levamisole anesthetized day 1 adult hermaphrodites using a Leica DMI8 confocal microscope at the University of Pittsburgh Center for Biologic Imaging. Images were captured at 20–30 Hz. Following imaging of baseline fluorescence, a region of interest corresponding to a portion of the nuclear or cytoplasmic foci was photobleached and fluorescence recovery within the photobleached area was monitored over at least 60 s. Data were normalized so that the image preceding the photobleach was set to 100% and the first image following the photobleach was set to zero percent. Imaging conditions over the time course of the experiment caused minimal loss of signal, suggesting an absence of photobleaching during the monitoring period. Photobleaching of the entire foci led to no recovery of signal over the imaging time period, suggesting that any resulting new signal was owing to diffusion of molecules within the foci rather than exchange of new molecules outside of the foci.
To quantify DPR subcellular localization of the DPRs, we co-expressed each DPR with a myo-3p::tagRFP-his-58 transgene to mark the nucleus. Z-stack images were collected for 1–21 nuclei across 5–6 individual animals, looking at at least 55 nuclei per DPR. We scored each nucleus for DPR expression in the histone-free region of the nucleus (Nucleolar), histone positive region of the nucleus (Nuclear) and outside of the histone marker (non-nuclear). The percentage of each category in each worm was determined and used to calculate and mean ± SD for each category.
Paralysis and thrashing assays
For all assays except for that shown in Figure 5D and E, gravid DPR expressing transgenic animals were moved from gfp(RNAi) to OP50 plates and allowed to lay eggs for 24 h. The resulting progeny were allowed to grow up on OP50, permitting DPR accumulation. Ten L4 animals were placed on each of three to five plates (N = 30–50 per assay). Each day, animals that failed to move at least half a body length in response to manual stimulation with a platinum wire but were still alive (pharyngeal pumping, movement of less than half a body length) were scored as paralyzed. Animals that died, desiccated on the plate edges or exhibited internal hatching of progeny were censored from the assay. Each day, mobile animals were transferred to a new plate and paralyzed, dead and censored animals were removed from the assay. For the assay in Figure 5D and E, his-58-(PR)50 progeny from animals moved from gfp(RNAi) to OP50 were not viable, presumably owing to the enhanced toxicity of the nuclear localized (PR)50 protein. Therefore, for these assays, animals were removed from gfp(RNAi) as L4 stage animals, placed on OP50 at 25 °C, and motility in the animals moved from gfp(RNA) was monitored as described above. Since animals are removed from the assay once they are scored as paralyzed, it is not appropriate to utilize statistical approaches that compare means and errors between time points (i.e. T-tests, ANOVA tests). Instead, we utilized the Log-rank statistical method, a cumulative statistical approach that compares changes in population sizes over time for a specified endpoint (53). Because this end-point assay is cumulative and not replicative, data points do not contain error bars. For each assay, 2–3 independent trials with 45–50 animals per assay were performed and the results from one representative trial are shown.
To measure thrashing, animals were maintained at 25 °C on OP50, and transgenic animals for each strain were picked as L4’s the day before the experiment. The following day, worms were placed on clean NGM plates and allowed to move freely for 10 min so that most of the bacteria came off the animal before the experiment. Worms were then placed individually into 3 cm petri dishes containing M9 buffer and allowed to adjust to the new environment for 5 min. The worms were then observed for 30 s and the number of thrashes (reversal of body bend that crosses the midline) was counted. We were unable to score thrashing in daf-2(e1370) animals since this mutation on its own causes significant defects in liquid thrashing.
Brood size assays
For brood size assays, at least 10 L4 stage animals were individually picked to OP50-containing NGM plates at 25°C and transferred to a new plate every 24 h until cessation of egg laying. Each plate was allowed to age for 48–72 h and the number of animals ≥ L4 stage of development was counted. Brood sizes were normalized within each strain to the mean brood size of animals grown on gfp(RNAi) bacteria.
Neurodegeneration assays
L4 animals of the indicated genotype were isolated at 25 °C and imaged 24 h later as ‘Day 1 adults’. All of the strains contained an unc-47p::GFP marker to reveal GABA motor neuron morphology. Animals were anesthetized in levamisole and Z-series images of GABAergic commissures were collected. Commissure breaks were identified as interruptions in the GFP signal surrounded by dorsal and ventral GFP in the commissures. Blebbing was scored only in the commissures and was identified by the presence of one or more GFP varicosities. For assays involving daf-2, we were unable to score either membrane blebbing or commissure breakage in (GR)50 expressing animals because the neuronal GFP marker used to score such events was undetectable, likely due to significant neurodegeneration that continued to occur in (GR)50; daf-2 animals.
Statistical analysis
Paralysis assays were analyzed using the Kaplan–Meier log-rank function (OASIS) (54,55). Comparisons of means were analyzed with either a two-tailed Student’s t-test (two groups) or ANOVA (three or more groups) using the Tukey’s or Dunn’s post-test analysis as indicated in GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA). P-values of <0.05 were considered significant.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by grants from the NIH [NS094921 (U.B.P. and T.L.), 1S10OD021540 (S.C.W.), R01NS081303, R21NS100055, R21NS098379, R21NS101661 (U.B.P.), and R01GM105655 and NS096319 (T.L.)], the Robert Packard Center for ALS at Johns Hopkins (U.B.P.), the Muscular Dystrophy Association (U.B.P.), and the Israeli Binational Science Foundation Grant [2011323 (T.L.)]. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Funding to pay the Open Access publication charges for this article was provided by NIH NS094921 and NS096319.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}