





Abstract
To identify an effective misoprostol-only regimen for the termination of second trimester pregnancy, we compared sublingual and vaginal administration of multiple doses of misoprostol in a randomized, placebo-controlled equivalence trial.
Six hundred and eighty-one healthy pregnant women requesting medical abortion at 13–20 weeks' gestation were randomly assigned within 11 gynaecological centres in seven countries into two treatment groups: 400 µg of misoprostol administered either sublingually or vaginally every 3 h up to five doses, followed by sublingual administration of 400 µg misoprostol every 3 h up to five doses if abortion had not occurred at 24 h after the start of treatment. We chose 10% as the margin of equivalence. The primary end-point was the efficacy of the treatments to terminate pregnancy in 24 h. Successful abortion within 48 h was also considered as an outcome along with the induction-to-abortion-interval, side effects and women's perceptions on these treatments.
At 24 h, the success (complete or incomplete abortion) rate was 85.9% in the vaginal administration group and 79.8% in the sublingual group (difference: 6.1%, 95% CI: 0.5 to 11.8). Thus, equivalence could not be concluded overall; the difference, however, was driven by the nulliparous women, among whom vaginal administration was clearly superior to sublingual administration (87.3% versus 68.5%), whereas no significant difference was observed between vaginal and sublingual treatments among parous women (84.7% versus 88.5%). The rates of side effects were similar in both groups except for fever, which was more common in the vaginal group. About 70% of women in both groups preferred sublingual administration.
Equivalence between vaginal and sublingual administration could not be demonstrated overall. Vaginal administration showed a higher effectiveness than sublingual administration in terminating second trimester pregnancies, but this result was mainly driven by nulliparous women. Fever was more prevalent with vaginal administration. Registered with International Standard Randomized Controlled Trial number ISRCTN72965671.
Introduction
A combination of mifepristone followed by a prostaglandin analogue is the preferred non-surgical method for inducing abortion in the second trimester (WHO, 2003; RCOG, 2004). When mifepristone is not available, as is the situation in many countries, abortion can also be induced safely with prostaglandin analogues, such as misoprostol, alone (WHO, 1997). A large variety of different regimens have been described in the literature, and no commonly approved guidelines on misoprostol use were available. Thus, there was a need for a large multicentre trial to identify an effective misoprostol-alone regimen for the termination of pregnancy in the second trimester.
When choosing the dose of misoprostol, it seemed that doses higher than 400 µg did not significantly improve the efficacy but caused more side effects (Dickinson and Evans, 2002), and lower doses such as 200 µg were clearly less effective. When misoprostol is used alone without mifepristone pretreatment, several doses are usually needed to induce abortion. A previous study had demonstrated that 3 h intervals were significantly more effective (P < 0.02) than 6 h intervals between vaginal doses of misoprostol, and the median time to abortion was also significantly shorter in the 3 h group (Wong et al., 2000).
On the basis of the existing evidence, we chose 400 µg as the dose of misoprostol, and administered it every 3 h up to five doses either sublingually or vaginally. If abortion had not occurred in 24 h after the start of the treatment, a second course of 400 µg misoprostol was administered sublingually every 3 h up to five doses. The aim of this randomized, placebo-controlled trial was to study: (i) the effectiveness to induce complete or partial abortion; (ii) the induction-to-abortion interval; (iii) the frequency of side effects and (iv) women's perceptions of the treatment, for the termination of pregnancy when the gestational length is between 13 and 20 weeks (91–140 days).
Materials and Methods
Study population
This trial was carried out in 11 departments of obstetrics and gynaecology of teaching hospitals in Yerevan, Armenia; Tbilisi, Georgia; Szeged, Hungary; Mumbai, New Delhi and Trivandrum, India; Ljubljana, Slovenia; Johannesburg, South Africa; and Hanoi (two hospitals) and Ho Chi Minh City, Vietnam. Institutional review boards at all participating hospitals and the WHO Secretariat Committee on Research on Human Subjects gave ethics approval. This trial is registered as an International Standard Randomized Controlled Trial, number ISRCTN72965671 and this report follows CONSORT guidelines for reporting equivalence trials (Piaggio et al., 2006).
Women requesting pregnancy termination at 13–20 weeks' gestation were provided information about the study, screened for eligibility by clinical personnel if willing to participate and included if they were healthy, older than the age of legal consent, had a single intrauterine pregnancy of 13–20 weeks (91–140 days) duration as verified by ultrasound and had haemoglobin 100 g/l or higher. We excluded women who had: any indication of serious past or present illness; an allergy to misoprostol; a habit of heavy smoking (>20 cigarettes/day); a scar in the uterus or cervix or any gynaecological anomaly detected with ultrasound; mitral stenosis, glaucoma or sickle cell anaemia; diastolic blood pressure >90 mmHg; uncontrolled bronchial asthma; systolic blood pressure <90 mmHg; history or evidence of thromboembolism or liver disease; presence of an intrauterine device; or haemolytic disorders.
All participants provided written informed consent before enrolment. Medical, gynaecological and obstetric histories were recorded and bacteriological tests and Rhesus-typing was done according to the routine of the centre.
Study design
A computer-generated randomization sequence was produced by WHO staff in Geneva to assign participants within each centre to sublingual or vaginal treatment group by randomly permuted blocks with a fixed block size of six. The number of women per centre varied according to the demand for second trimester abortion: Ljubljana planned to recruit 22 women, Johannesburg planned to recruit 140 and other centres recruited between these numbers. Allocation was concealed by using sealed, opaque, sequentially numbered envelopes, which were filled and labelled in accordance with the list of randomization for each centre by Magistra, Geneva, Switzerland.
For each woman, there were two blisters of tablets, 10 tablets of 200 µg of misoprostol (Cytotec, Pfizer) and 10 placebo tablets (manufactured by Labatec, Geneva, Switzerland; similar shape and colour as misoprostol tablets). The blisters were labelled indicating which tablets were to be taken sublingually and which tablets vaginally. Additional misoprostol tablets were provided to the centres to be used sublingually for those women who did not abort within 24 h.
Previous experience indicated that ∼80% of women receiving 400 µg of misoprostol vaginally every 3 h for termination of pregnancy in the second trimester will abort within 24 h (Tang et al., 2004). To establish the equivalent efficacy of the two regimens, we require the 95% confidence interval for the difference in abortion rates to be within the margin of equivalence of 10% with a probability of 80%. It was estimated that if the abortion rates in the two regimens are both equal to 80%, 340 women will be required for each group, i.e. a total of ∼680 women for the whole study.
The primary outcome measure was successful abortion (including complete and incomplete abortion) within 24 h. In addition, successful abortion within 48 h was also considered as an outcome, along with the induction-to-abortion interval from the start of treatment to expulsion of fetus. The rates of side effects and women's perceptions of the method were compared between treatment groups as were possible complications up to the follow-up visit ∼2 weeks after abortion.
Procedures
Each treatment dose consisted of two sublingual and two vaginal tablets (two tablets of 200 µg misoprostol and two placebo tablets), which was repeated at 3 h intervals up to five doses until abortion took place. The treatment was withheld if the patient had strong uterine contractions. Side effects, uterine contractions, blood pressure and pulse were recorded 1 and 3 h after every dose, along with any medication given. A vaginal examination was performed before each dose. Also, the time of expulsion was recorded. After expulsion of the fetus, one additional dose of the tablets was administered.
The patient was reassessed if abortion had not occurred after 24 h. If there were no signs and symptoms suggestive of imminent abortion, a second course of treatment, two tablets sublingually (2 × 200 µg) at 3 h intervals for a maximum of five doses, was given. If abortion still failed to occur, the pregnancy was terminated as judged best by the investigator. The outcome of treatment was assessed at 24 and 48 h from the start of treatment.
After abortion, the products of gestation were examined to see whether the abortion was complete. If necessary, or if it was a local routine practice, exploration and evacuation of the uterus was performed. The patient was discharged 24 h after abortion if there were no complications. Post-abortion contraceptive counselling was also given. The assessment of the outcome was done before the woman left the hospital. At this point, also the women's perceptions about the treatment were assessed using a questionnaire. Women had a follow-up visit 2 (range 1–3 weeks) weeks after abortion.
Statistical analysis
Data were analysed with the SAS software (version 9.1.3) centrally at WHO. An independent Data Monitoring Committee (DMC) reviewed the data from interim analysis performed after 429 women were included in the study. Descriptive statistics were calculated for all baseline characteristics for all subjects recruited, by treatment group, to assess comparability of the groups.
We considered abortion, whether complete or incomplete, as successful treatment outcome while treatment failures included missed abortion, continuing pregnancy and undetermined outcomes.
We included in the analysis all randomized women according to the intention-to-treat (ITT) principle. When protocol violations consist of switched treatment allocation, a per-protocol analysis excluding subjects who switched treatments is the most desirable in equivalence trials as a protection from the ITT's increase in the probability of type I error. In this trial, there was no switch of treatment allocation. In this situation, the benefit of conducting a non-ITT analysis is not obvious and we preferred ITT analysis in order to preserve the advantages of randomization.
However, for side effects, one woman in the sublingual group had missed observations and thus, she was excluded from the side effects analysis. We conducted two sensitivity analyses: one excluding one centre that was accounting for a treatment by centre interaction; another one excluding two centres that started the second course of treatment after 15 h instead of 24 h (per protocol analysis excluding protocol violations). The equivalence of the treatment outcome was assessed by computing the difference in the proportions of abortion failures between treatment groups along with 95% confidence intervals. Additionally, relative risks were computed to compare treatment failures between the groups. Interactions between treatment and centre and treatment and parity status were assessed with a logistic regression model. In cases where the standard logistic regression model failed to produce estimates due to convergence problems, an exact logistic regression method was used to approximate the significance of the interaction terms. Time to fetal expulsion was computed and then analysed using standard survival analysis techniques. For this analysis, subjects with treatment failure were considered censored with censoring time equal to the time from onset of treatment to surgical termination of the pregnancy. Median times to expulsion were derived from Kaplan–Meier estimates of the survival function, and treatment groups were compared using the log-rank test. Interactions between treatment and centre and treatment and parity status were assessed using the Cox regression model. Comparisons of side effects and women's perceptions on the regimens were carried out using Fisher exact tests.
We also conducted a stratified analysis by parity because there was a highly significant interaction of treatment by parity, for which the reporting might have clinical relevance.
Role of the funding source
The donors and sponsors of the study had no role in the study design, data collection, data analysis, data interpretation, writing of the report or the decision to submit the paper for publication. The corresponding author had full access to all final data in the study and had the final responsibility for the decision to submit for publication.
Results
Between July 2002 and March 2004, a total of 681 women were enrolled in the study. Of the 921 women screened, 123 were not eligible. Main reasons for non-eligibility were gestational age outside the 13–20 weeks range (38.2%), not willing to return to the follow-up visits (29.3%), haemoglobin level below 100 g/l (16.3%) and not in good health (15.5%). Additionally, 117 women were eligible but chose not to participate. A flowchart for participants is shown in Fig. 1.
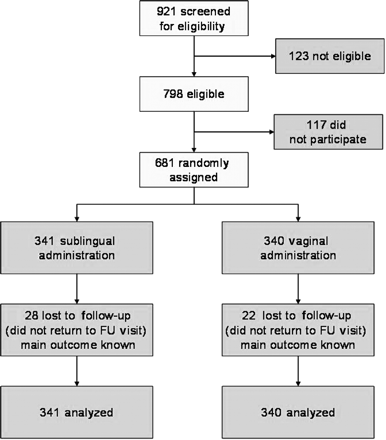
Trial profile.
The baseline characteristics were similar in both treatment groups (Table I). Average length of gestation (assessed by ultrasound) was 16.5 weeks. Approximately 45% of the women did not have previous deliveries (nulliparous) and 16% had had a previous induced abortion.
Demographic characteristics
| Sublingual group (n = 341) | Vaginal group (n = 340) | |
|---|---|---|
| Age (year) | 26.2 [6.7] | 26.2 [6.6] |
| Weight (kg) | 54.6 [12.2] | 54.1 [13.6] |
| Height (cm) | 157.5 [7.9] | 157.2 [7.6] |
| Gestational age (weeks) | 16.5 [2.0] | 16.6 [2.1] |
| Haemoglobin level (g/l) | 114 [11] | 115 [12] |
| Previous induced abortion | 54 (15.8) | 57 (16.8) |
| Previous delivery | 192 (56.3) | 190 (55.9) |
| Sublingual group (n = 341) | Vaginal group (n = 340) | |
|---|---|---|
| Age (year) | 26.2 [6.7] | 26.2 [6.6] |
| Weight (kg) | 54.6 [12.2] | 54.1 [13.6] |
| Height (cm) | 157.5 [7.9] | 157.2 [7.6] |
| Gestational age (weeks) | 16.5 [2.0] | 16.6 [2.1] |
| Haemoglobin level (g/l) | 114 [11] | 115 [12] |
| Previous induced abortion | 54 (15.8) | 57 (16.8) |
| Previous delivery | 192 (56.3) | 190 (55.9) |
Values are expressed as mean [SD] or n (%).
Demographic characteristics
| Sublingual group (n = 341) | Vaginal group (n = 340) | |
|---|---|---|
| Age (year) | 26.2 [6.7] | 26.2 [6.6] |
| Weight (kg) | 54.6 [12.2] | 54.1 [13.6] |
| Height (cm) | 157.5 [7.9] | 157.2 [7.6] |
| Gestational age (weeks) | 16.5 [2.0] | 16.6 [2.1] |
| Haemoglobin level (g/l) | 114 [11] | 115 [12] |
| Previous induced abortion | 54 (15.8) | 57 (16.8) |
| Previous delivery | 192 (56.3) | 190 (55.9) |
| Sublingual group (n = 341) | Vaginal group (n = 340) | |
|---|---|---|
| Age (year) | 26.2 [6.7] | 26.2 [6.6] |
| Weight (kg) | 54.6 [12.2] | 54.1 [13.6] |
| Height (cm) | 157.5 [7.9] | 157.2 [7.6] |
| Gestational age (weeks) | 16.5 [2.0] | 16.6 [2.1] |
| Haemoglobin level (g/l) | 114 [11] | 115 [12] |
| Previous induced abortion | 54 (15.8) | 57 (16.8) |
| Previous delivery | 192 (56.3) | 190 (55.9) |
Values are expressed as mean [SD] or n (%).
All women included in the study received at least one dose of misoprostol. A total of 117 (17.2%) women required a second course of treatment, 69 (20.2%) in the sublingual group and 48 (14.1%) in the vaginal group (P = 0.042). In all, 52 women did not return for the follow-up visit; however, the outcome of treatment was known for them. For two women, the outcome of treatment was undetermined: one of them discontinued from the study but aborted with two additional misoprostol doses; and the other one was treated by a doctor who was not aware of the study and evacuated the uterus surgically without a clear indication. These women were included among failures.
Overall, treatment success was 82.8% at 24 h and 94.3% at 48 h. Table II shows success rates by treatment group. At 24 h, the vaginal administration group had a higher success rate than the sublingual administration group (85.9% versus 79.8%, difference: 6.1%, 95% CI: 0.5 to 11.8). Equivalence of the two arms cannot be concluded since the confidence interval crosses the 10% pre-established margin (Fig. 2). Both treatments can be claimed to be equivalent at 48 h within the 10% margin established [96.2% (vaginal) versus 92.4% (sublingual), difference: 3.8%, 95% CI: 0.3 to 7.3]. Treatment by centre interaction was statistically significant (P = 0.027), mainly due to different results in one centre (New Delhi). A sensitivity analysis excluding that centre produced similar results (difference: 8.8%, 95% CI: 2.7 to 14.9).
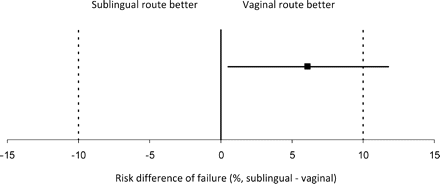
Failure to achieve abortion at 24 h.
Treatment outcomes
| Sublingual group n/N (%) | Vaginal group n/N (%) | Relative risk of failure to abort (95% CI) | Risk difference (95% CI) | |
|---|---|---|---|---|
| Abortion rate in 24 h | ||||
| Overall1 | 272/341 (79.8) | 292/340 (85.9) | 1.43 (1.02, 2.01) | 6.1 (0.5, 11.8) |
| Overall excluding New Delhi2 | 237/306 (77.5) | 263/305 (86.2) | 1.64 (1.15, 2.32) | 8.8 (2.7, 14.9) |
| Overall excluding Johannesburg and Hanoi OGH | 190/230 (82.6) | 202/230 (87.8) | 1.43 (0.91, 2.23) | 5.2 (−1.4, 11.7) |
| By parity3 | ||||
| Nulliparous | 102/149 (68.5) | 131/150 (87.3) | 2.49 (1.54, 4.03) | 18.9 (9.7, 28.0) |
| Parous | 170/192 (88.5) | 161/190 (84.7) | 0.75 (0.45, 1.26) | −3.8 (−10.6, 3.0) |
| Abortion rate at 48 h | ||||
| Overall4 | 315/341 (92.4) | 327/340 (96.2) | 1.99 (1.04, 3.81) | 3.8 (0.3, 7.3) |
| By parity5 | ||||
| Nulliparous | 133/149 (89.3) | 149/150 (99.3) | 16.1 (2.16, 119.9) | 10.1 (4.9, 15.2) |
| Parous | 182/192 (94.8) | 178/190 (93.7) | 0.82 (0.37, 1.86) | −1.1 (−5.8, 3.6) |
| Time to fetal expulsion (h) | Median (range) | Median (range) | P-value | |
| Overall6 | 12.0 (4.1–61.8) | 12.3 (3.2–48.0) | 0.227 | |
| By parity7 | ||||
| Nulliparous | 14.4 (5.3–61.8) | 13.0 (3.2–48.0) | 0.002 | |
| Parous | 11.0 (4.1–45.0) | 11.8 (4.5–43.8) | 0.190 | |
| Sublingual group n/N (%) | Vaginal group n/N (%) | Relative risk of failure to abort (95% CI) | Risk difference (95% CI) | |
|---|---|---|---|---|
| Abortion rate in 24 h | ||||
| Overall1 | 272/341 (79.8) | 292/340 (85.9) | 1.43 (1.02, 2.01) | 6.1 (0.5, 11.8) |
| Overall excluding New Delhi2 | 237/306 (77.5) | 263/305 (86.2) | 1.64 (1.15, 2.32) | 8.8 (2.7, 14.9) |
| Overall excluding Johannesburg and Hanoi OGH | 190/230 (82.6) | 202/230 (87.8) | 1.43 (0.91, 2.23) | 5.2 (−1.4, 11.7) |
| By parity3 | ||||
| Nulliparous | 102/149 (68.5) | 131/150 (87.3) | 2.49 (1.54, 4.03) | 18.9 (9.7, 28.0) |
| Parous | 170/192 (88.5) | 161/190 (84.7) | 0.75 (0.45, 1.26) | −3.8 (−10.6, 3.0) |
| Abortion rate at 48 h | ||||
| Overall4 | 315/341 (92.4) | 327/340 (96.2) | 1.99 (1.04, 3.81) | 3.8 (0.3, 7.3) |
| By parity5 | ||||
| Nulliparous | 133/149 (89.3) | 149/150 (99.3) | 16.1 (2.16, 119.9) | 10.1 (4.9, 15.2) |
| Parous | 182/192 (94.8) | 178/190 (93.7) | 0.82 (0.37, 1.86) | −1.1 (−5.8, 3.6) |
| Time to fetal expulsion (h) | Median (range) | Median (range) | P-value | |
| Overall6 | 12.0 (4.1–61.8) | 12.3 (3.2–48.0) | 0.227 | |
| By parity7 | ||||
| Nulliparous | 14.4 (5.3–61.8) | 13.0 (3.2–48.0) | 0.002 | |
| Parous | 11.0 (4.1–45.0) | 11.8 (4.5–43.8) | 0.190 | |
1Interaction treatment by centre: P ≈ 0.027; 2Interaction treatment by centre: P ≈ 0.454; 3Interaction treatment by parity: P < 0.001; 4Interaction treatment by centre: P ≈ 0.106; 5Interaction treatment by parity: P = 0.006; 6Interaction treatment by centre: P = 0.009; 7Interaction treatment by parity: P = 0.001.
Treatment outcomes
| Sublingual group n/N (%) | Vaginal group n/N (%) | Relative risk of failure to abort (95% CI) | Risk difference (95% CI) | |
|---|---|---|---|---|
| Abortion rate in 24 h | ||||
| Overall1 | 272/341 (79.8) | 292/340 (85.9) | 1.43 (1.02, 2.01) | 6.1 (0.5, 11.8) |
| Overall excluding New Delhi2 | 237/306 (77.5) | 263/305 (86.2) | 1.64 (1.15, 2.32) | 8.8 (2.7, 14.9) |
| Overall excluding Johannesburg and Hanoi OGH | 190/230 (82.6) | 202/230 (87.8) | 1.43 (0.91, 2.23) | 5.2 (−1.4, 11.7) |
| By parity3 | ||||
| Nulliparous | 102/149 (68.5) | 131/150 (87.3) | 2.49 (1.54, 4.03) | 18.9 (9.7, 28.0) |
| Parous | 170/192 (88.5) | 161/190 (84.7) | 0.75 (0.45, 1.26) | −3.8 (−10.6, 3.0) |
| Abortion rate at 48 h | ||||
| Overall4 | 315/341 (92.4) | 327/340 (96.2) | 1.99 (1.04, 3.81) | 3.8 (0.3, 7.3) |
| By parity5 | ||||
| Nulliparous | 133/149 (89.3) | 149/150 (99.3) | 16.1 (2.16, 119.9) | 10.1 (4.9, 15.2) |
| Parous | 182/192 (94.8) | 178/190 (93.7) | 0.82 (0.37, 1.86) | −1.1 (−5.8, 3.6) |
| Time to fetal expulsion (h) | Median (range) | Median (range) | P-value | |
| Overall6 | 12.0 (4.1–61.8) | 12.3 (3.2–48.0) | 0.227 | |
| By parity7 | ||||
| Nulliparous | 14.4 (5.3–61.8) | 13.0 (3.2–48.0) | 0.002 | |
| Parous | 11.0 (4.1–45.0) | 11.8 (4.5–43.8) | 0.190 | |
| Sublingual group n/N (%) | Vaginal group n/N (%) | Relative risk of failure to abort (95% CI) | Risk difference (95% CI) | |
|---|---|---|---|---|
| Abortion rate in 24 h | ||||
| Overall1 | 272/341 (79.8) | 292/340 (85.9) | 1.43 (1.02, 2.01) | 6.1 (0.5, 11.8) |
| Overall excluding New Delhi2 | 237/306 (77.5) | 263/305 (86.2) | 1.64 (1.15, 2.32) | 8.8 (2.7, 14.9) |
| Overall excluding Johannesburg and Hanoi OGH | 190/230 (82.6) | 202/230 (87.8) | 1.43 (0.91, 2.23) | 5.2 (−1.4, 11.7) |
| By parity3 | ||||
| Nulliparous | 102/149 (68.5) | 131/150 (87.3) | 2.49 (1.54, 4.03) | 18.9 (9.7, 28.0) |
| Parous | 170/192 (88.5) | 161/190 (84.7) | 0.75 (0.45, 1.26) | −3.8 (−10.6, 3.0) |
| Abortion rate at 48 h | ||||
| Overall4 | 315/341 (92.4) | 327/340 (96.2) | 1.99 (1.04, 3.81) | 3.8 (0.3, 7.3) |
| By parity5 | ||||
| Nulliparous | 133/149 (89.3) | 149/150 (99.3) | 16.1 (2.16, 119.9) | 10.1 (4.9, 15.2) |
| Parous | 182/192 (94.8) | 178/190 (93.7) | 0.82 (0.37, 1.86) | −1.1 (−5.8, 3.6) |
| Time to fetal expulsion (h) | Median (range) | Median (range) | P-value | |
| Overall6 | 12.0 (4.1–61.8) | 12.3 (3.2–48.0) | 0.227 | |
| By parity7 | ||||
| Nulliparous | 14.4 (5.3–61.8) | 13.0 (3.2–48.0) | 0.002 | |
| Parous | 11.0 (4.1–45.0) | 11.8 (4.5–43.8) | 0.190 | |
1Interaction treatment by centre: P ≈ 0.027; 2Interaction treatment by centre: P ≈ 0.454; 3Interaction treatment by parity: P < 0.001; 4Interaction treatment by centre: P ≈ 0.106; 5Interaction treatment by parity: P = 0.006; 6Interaction treatment by centre: P = 0.009; 7Interaction treatment by parity: P = 0.001.
Three centres (Johannesburg, Szeged and Yerevan) performed routine curettage after fetal expulsion. Excluding these centres, complete abortion rates were 70.9% (163/302) in the vaginal and 71.9% (166/302) in the sublingual groups. Despite two courses of misoprostol treatment, 2.1% (7/340) of the pregnancies continued in the vaginal group and 5.6% (19/341) in the sublingual group.
When success rates at 24 h were analysed according to parity, vaginal administration was clearly superior to sublingual administration in nulliparous women (87.3% versus 68.5%) but the difference between treatments was smaller and reversed in parous women [84.7% (vaginal) versus 88.5% (sublingual)] (P = 0.006 for the interaction). A similar conclusion can be drawn for the outcome at 48 h (Table II).
Median time to fetal expulsion was ∼ 12 h, with no difference between the treatments (P = 0.227). Nulliparous women tended to have longer intervals to fetal expulsion than parous women. The vaginal route of administration appeared to be faster than the sublingual route in nulliparous women but not in parous women (P = 0.001 for the interaction).
Two centres (Johannesburg and Hanoi OGH) started the second course of misoprostol 15 h after the onset of treatment (opposed to 24 h as requested by protocol). When we removed those two centres from the analysis, the results did not change.
Expected side effects are presented in Table III. The most common side effects were chills/shivering (38%), fever (35%) and diarrhoea (24%). No statistically significant differences between treatment groups were observed for any side effect except for fever which was more common in the vaginal group, and which just failed to be significant at 5% when adjusting for multiple inferences.
Side effects any time during the first course of treatment
| Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value* | |
|---|---|---|---|
| Nausea | 51/341 (15.0) | 56/340 (16.5) | 0.600 |
| Vomiting | 43/341 (12.6) | 40/340 (11.8) | 0.815 |
| Diarrhoea | 83/341 (24.3) | 80/340 (23.5) | 0.858 |
| Dizziness | 28/341 (8.2) | 33/340 (9.7) | 0.506 |
| Headache | 53/341 (15.5) | 60/340 (17.7) | 0.473 |
| Chills/Shivering | 130/341 (38.1) | 128/340 (37.7) | 0.937 |
| Fever (>38°C) | 102/341 (29.9) | 135/340 (39.7) | 0.008 |
| Any side effect | 218/341 (63.9) | 226/340 (66.5) | 0.520 |
| Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value* | |
|---|---|---|---|
| Nausea | 51/341 (15.0) | 56/340 (16.5) | 0.600 |
| Vomiting | 43/341 (12.6) | 40/340 (11.8) | 0.815 |
| Diarrhoea | 83/341 (24.3) | 80/340 (23.5) | 0.858 |
| Dizziness | 28/341 (8.2) | 33/340 (9.7) | 0.506 |
| Headache | 53/341 (15.5) | 60/340 (17.7) | 0.473 |
| Chills/Shivering | 130/341 (38.1) | 128/340 (37.7) | 0.937 |
| Fever (>38°C) | 102/341 (29.9) | 135/340 (39.7) | 0.008 |
| Any side effect | 218/341 (63.9) | 226/340 (66.5) | 0.520 |
*Significant at 5% if p-value <0.006 using Bonferroni criterion.
Side effects any time during the first course of treatment
| Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value* | |
|---|---|---|---|
| Nausea | 51/341 (15.0) | 56/340 (16.5) | 0.600 |
| Vomiting | 43/341 (12.6) | 40/340 (11.8) | 0.815 |
| Diarrhoea | 83/341 (24.3) | 80/340 (23.5) | 0.858 |
| Dizziness | 28/341 (8.2) | 33/340 (9.7) | 0.506 |
| Headache | 53/341 (15.5) | 60/340 (17.7) | 0.473 |
| Chills/Shivering | 130/341 (38.1) | 128/340 (37.7) | 0.937 |
| Fever (>38°C) | 102/341 (29.9) | 135/340 (39.7) | 0.008 |
| Any side effect | 218/341 (63.9) | 226/340 (66.5) | 0.520 |
| Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value* | |
|---|---|---|---|
| Nausea | 51/341 (15.0) | 56/340 (16.5) | 0.600 |
| Vomiting | 43/341 (12.6) | 40/340 (11.8) | 0.815 |
| Diarrhoea | 83/341 (24.3) | 80/340 (23.5) | 0.858 |
| Dizziness | 28/341 (8.2) | 33/340 (9.7) | 0.506 |
| Headache | 53/341 (15.5) | 60/340 (17.7) | 0.473 |
| Chills/Shivering | 130/341 (38.1) | 128/340 (37.7) | 0.937 |
| Fever (>38°C) | 102/341 (29.9) | 135/340 (39.7) | 0.008 |
| Any side effect | 218/341 (63.9) | 226/340 (66.5) | 0.520 |
*Significant at 5% if p-value <0.006 using Bonferroni criterion.
Approximately 32% of the women received some medication (mostly analgesics) during the treatment period [36% (vaginal) versus 29% (sublingual), P = 0.09]. There were 10 women who received a blood transfusion and three women required hospitalization after discharge, two of them for surgical evacuation of the uterus and one for reasons unrelated to the study. During the study, 12 adverse events (none serious) were reported and they were equally distributed between the two groups. Excessive bleeding was the characteristic of all of them except one where uterine hyper-contractility was reported.
There was no difference between treatment groups in the duration of post-abortion bleeding (∼ 7 days) or in the haemoglobin levels at the follow-up visit. The duration of bleeding was >14 days in 5.8% (18/309) of the women in the sublingual group and 7.0% (22/315) in the vaginal group (P = 0.625).
Most women in both groups preferred sublingual administration of tablets (Table IV), mainly because they felt that sublingual route was more convenient (321/439; 73.1%) or due to the discomfort of vaginal examination (159/439; 36.2%). However, about one-quarter of the women in both groups preferred vaginal administration mainly as they regarded this route to be more convenient (64/167; 38.3%). Although more than 40% of the women said that abortion was less painful than they had expected, pain was also ranked as the worst side effect of the treatment by 41% (262/630) of the women in both groups, whereas 26% (163/630) regarded the long duration of the abortion process as the worst feature of the method.
Women's expectations and preferences
| Expectations | Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value |
|---|---|---|---|
| Pain | |||
| Less than expected | 135/315 (42.9) | 131/315 (41.6) | 0.809 |
| More than expected | 69/315 (21.9) | 82/315 (26.0) | 0.263 |
| Bleeding | |||
| Less than expected | 65/315 (20.6) | 61/315 (19.4) | 0.765 |
| More than expected | 33/315 (10.5) | 32/315 (10.2) | 1.000 |
| Side effects | |||
| Fewer than expected | 91/315 (28.9) | 93/315 (29.5) | 0.930 |
| More than expected | 18/315 (5.7) | 25/315 (7.9) | 0.343 |
| Preference | |||
| Sublingual | 214/306 (69.9) | 225/304 (74.0) | 0.280 |
| Vaginal | 91/306 (29.7) | 76/304 (25.0) | |
| No preference | 1 (0.3) | 3 (1.0) |
| Expectations | Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value |
|---|---|---|---|
| Pain | |||
| Less than expected | 135/315 (42.9) | 131/315 (41.6) | 0.809 |
| More than expected | 69/315 (21.9) | 82/315 (26.0) | 0.263 |
| Bleeding | |||
| Less than expected | 65/315 (20.6) | 61/315 (19.4) | 0.765 |
| More than expected | 33/315 (10.5) | 32/315 (10.2) | 1.000 |
| Side effects | |||
| Fewer than expected | 91/315 (28.9) | 93/315 (29.5) | 0.930 |
| More than expected | 18/315 (5.7) | 25/315 (7.9) | 0.343 |
| Preference | |||
| Sublingual | 214/306 (69.9) | 225/304 (74.0) | 0.280 |
| Vaginal | 91/306 (29.7) | 76/304 (25.0) | |
| No preference | 1 (0.3) | 3 (1.0) |
Women's expectations and preferences
| Expectations | Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value |
|---|---|---|---|
| Pain | |||
| Less than expected | 135/315 (42.9) | 131/315 (41.6) | 0.809 |
| More than expected | 69/315 (21.9) | 82/315 (26.0) | 0.263 |
| Bleeding | |||
| Less than expected | 65/315 (20.6) | 61/315 (19.4) | 0.765 |
| More than expected | 33/315 (10.5) | 32/315 (10.2) | 1.000 |
| Side effects | |||
| Fewer than expected | 91/315 (28.9) | 93/315 (29.5) | 0.930 |
| More than expected | 18/315 (5.7) | 25/315 (7.9) | 0.343 |
| Preference | |||
| Sublingual | 214/306 (69.9) | 225/304 (74.0) | 0.280 |
| Vaginal | 91/306 (29.7) | 76/304 (25.0) | |
| No preference | 1 (0.3) | 3 (1.0) |
| Expectations | Sublingual group, n/N (%) | Vaginal group, n/N (%) | P-value |
|---|---|---|---|
| Pain | |||
| Less than expected | 135/315 (42.9) | 131/315 (41.6) | 0.809 |
| More than expected | 69/315 (21.9) | 82/315 (26.0) | 0.263 |
| Bleeding | |||
| Less than expected | 65/315 (20.6) | 61/315 (19.4) | 0.765 |
| More than expected | 33/315 (10.5) | 32/315 (10.2) | 1.000 |
| Side effects | |||
| Fewer than expected | 91/315 (28.9) | 93/315 (29.5) | 0.930 |
| More than expected | 18/315 (5.7) | 25/315 (7.9) | 0.343 |
| Preference | |||
| Sublingual | 214/306 (69.9) | 225/304 (74.0) | 0.280 |
| Vaginal | 91/306 (29.7) | 76/304 (25.0) | |
| No preference | 1 (0.3) | 3 (1.0) |
Discussion
Our findings show that when repeated doses of 400 µg misoprostol are used to induce abortion in the second trimester, vaginal and sublingual administration are not equivalent within a 10% margin: vaginal administration appears to result in a lower failure rate at 24 h. This outcome was mainly driven by the better efficacy of vaginal administration among nulliparous women. The finding is very similar to that from a smaller study among Chinese women (Tang et al., 2004).
The primary outcome was successful abortion, complete or incomplete at 24 h. In many countries, exploration and evacuation of the uterus is performed routinely after fetal expulsion in second trimester abortion, and this was a routine practice also in three centres in this study. Thus, we did not aim to make a distinction between complete and incomplete abortion. Post-abortion aspiration/curettage was performed in 309 cases (45.4%); 103 of them were due to recognized or suspected incomplete abortion at the eight centres that did not use routine curettage, which is ∼22% (103/461) of the cases at those centres. The use of curettage to ensure that the uterus is empty reflects local practices and inexperience with the method rather than the actual need for post-abortion surgical evacuation.
Median time to fetal expulsion was ∼12 h after both vaginal and sublingual administration of misoprostol. This is similar to other studies with 3 h administration (Tang et al., 2004), whereas it takes longer to abort when the administration interval is longer (Wong et al., 2000). When mifepristone pretreatment precedes misoprostol, the median abortion- induction interval, calculated from the start of misoprostol administration, is ∼6 h which allows most abortions to be managed as day cases (Ashok et al., 2004). In this study, ∼50% of the women aborted before the fifth dose, i.e. within 12 h. In all, 117 (17.2%) women were administered the second course of treatment, i.e. 400 µg misoprostol sublingually at 3 h intervals up to five doses. For practical reasons, we had decided to administer the second course of tablets sublingually.
Both vaginal and sublingual administration of misoprostol lead to softening and dilation of uterine cervix (Fiala et al., 2007). However, regular uterine contractions are sustained for a longer period of time (>4 h) after vaginal administration than after the sublingual route (∼2–3 h) (Aronsson et al., 2004), which may lead to stronger contractility during the treatment period in the vaginal administration group. Further, a direct transport of prostaglandins seems to occur from vagina to the uterus (Krzymowski et al. 1989), which can also contribute to the better efficacy after vaginal administration.
Chills, shivering and fever >38°C were the most common side effects of misoprostol treatment and there was no difference between the groups except in the occurrence of fever: from the third dose on, significantly more women had fever in the vaginal administration group, with the rates being highest after the third dose (28.2% in the vaginal group versus 18.5% in the sublingual group). Overall, 39.7% of the women had fever at any point during the vaginal treatment period compared with 29.9% of women in the sublingual group (P = 0.008). This finding is somewhat unexpected because in studies where misoprostol is administered after mifepristone pretreatment (Tang et al., 2003) or alone in repeated doses in the first trimester (von Hertzen et al., 2007) side effects, especially fever, chills and vomiting were more common after sublingual administration. Serum levels of misoprostol acid are higher after a single sublingual dose compared with a vaginal dose (Tang et al., 2007), which may explain higher side effect rates after a single sublingual dose. When the dose is repeated, however, the pharmacokinetics may be different, but no pharmacokinetic studies have as yet been published on repeat administration of misoprostol.
Women in our study received both sublingual and vaginal tablets, so that the tablets for one of the routes contained misoprostol and for the other they were placebo tablets. Thus, every woman had experience on both sublingual and vaginal administration. Both routes were acceptable to women, although in both groups a higher percentage of women preferred sublingual administration. Also in other studies comparing these two routes, most women have preferred sublingual administration of misoprostol (Tang et al., 2004; Hamoda et al., 2005).
We only included healthy women in this study and had therefore, rather strict exclusion criteria, all of which do not need to apply for routine practices. However, health-related reasons for exclusion were uncommon as most women who could not be enrolled had pregnancies outside the gestational age range of this study or they were not willing to return to the follow-up visit or they did not want to join the study despite being eligible.
We believe that this trial has internal validity as women were randomly assigned to the two treatment groups, random allocation was concealed and the sample size was calculated according to the pre-stated hypothesis. Women were enrolled for this study from several different populations, a feature which increases external validity.
Our study demonstrates that, when mifepristone is not available, repeated administration of 400 µg misoprostol either vaginally or sublingually is an effective and acceptable option. However, vaginal administration appears to produce better results among nulliparous women.
Authors' contribution
H.v.H., in collaboration with the Steering Committee, was responsible for the conception of the trial and selection of centres. H.v.H. and G.P. prepared the protocol. G.O., A.K., A.K., S.M., R.N., R.D., A.P-D., K.D., N.D.H., N.H.B. and H.T.D.T. contributed to the final protocol and implemented the trial in their respective countries. H.v.H., L.M. and G.P. supervised the trial. G.P. and D.W. were responsible for the statistical analysis, and N.T.M.H. and A.P. were responsible for the data management. H.v.H., D.W. and G.P. wrote the paper with inputs from all the other authors.
Funding
The UNDP/UNFPA/WHO/World Bank Special Programme of Research, Development and Research Training in Human Reproduction funded this study including the misoprostol and placebo tablets.
Acknowledgements
We thank the members of the Data Monitoring Committee for their contribution to interim analyses and their valuable comments on the report.
Conflict of interest: We declare that we have no conflict of interest.
References
Appendix
Coordination of the trial
Overall co-ordination of the trial: H.v.H.; Data co-ordination: G.P., N.T.M.H. and A.P.; Steering Committee: G. Bàrtfai, L. Cheng, K. Gemzell-Danielsson and P.C. Ho (chair); Data Monitoring Committee: M. Bygdeman, D. Elbourne (chair) and D. Grimes.
Collaborators
Research Center of Maternal and Child Health Protection, Yerevan, Armenia (G. Okoev, K. Arustamyan, S. Kirakosyan and G. Martirosyan); Zhordania Institute of Human Reproduction, Tbilisi, Georgia (A. Khomassuridze, G. Tsertsvadze, T. Tsereteli and M. Parkauli); University of Szeged, Hungary (L. Kovács, A. Kereszturi and Z. Pal); All India Institute of Medical Sciences, New Delhi, India (S. Mittal, S. Kumar and N. Gupta); SAT Hospital, Trivandrum, India (R. Nair, P. Sobhana, P. Varghese and A. Harshan); National Institute for Research in Reproductive Health, Indian Council of Medical Research and Sir JJ Group of Hospitals, Mumbai, India (R. Daver, R. Shah, L. Savardekar, A. Peddawad and K. Tilwani); University Department of Obstetrics and Gynaecology, Ljubljana, Slovenia (A. Pretnar-Darovec, B. Cvjeticanin and Mojka Pirc); Reproductive Health and HIV Research Unit, Soweto, South Africa (K.E. Dickson, C. von Mollendorf, L. Mavuya and B. Nkosi); Institute for the Protection of Mother and Newborn, Hanoi, Vietnam (N.DE. Hinh, P.V. Quy, N.T.L. Huong, N.T.H. Minh, D.L. Dung and P.D. Dung); Hanoi Obstetrics and Gynaecology Hospital, Hanoi, Vietnam (N.H. Bao, N.M. Tri, D.K. Huynh, L.Q. Khai and D.T. Nghia); Tu Du Hospital, Ho Chi Minh City, Vietnam (H.T.D. Tuyet, P.V. Thanh, N.T.N. Phuong and D.P. Mai).

{kind=link}
{kind=link}