





Abstract
Epidemiological studies indicate that the use of artificial sweeteners doubles the risk for Crohn’s disease (CD). Herein, we experimentally quantified the impact of 6-week supplementation with a commercial sweetener (Splenda; ingredients sucralose maltodextrin, 1:99, w/w) on both the severity of CD-like ileitis and the intestinal microbiome alterations using SAMP1/YitFc (SAMP) mice.
Metagenomic shotgun DNA sequencing was first used to characterize the microbiome of ileitis-prone SAMP mice. Then, 16S rRNA microbiome sequencing, quantitative polymerase chain reaction, fluorescent in situ hybridization (FISH), bacterial culture, stereomicroscopy, histology, and myeloperoxidase (MPO) activity analyses were then implemented to compare the microbiome and ileitis phenotype in SAMP with that of control ileitis-free AKR/J mice after Splenda supplementation.
Metagenomics indicated that SAMP mice have a gut microbial phenotype rich in Bacteroidetes, and experiments showed that Helicobacteraceae did not have an exacerbating effect on ileitis. Splenda did not increase the severity of (stereomicroscopic/histological) ileitis; however, biochemically, ileal MPO activity was increased in SAMP treated with Splenda compared with nonsupplemented mice (P < 0.022) and healthy AKR mice. Splenda promoted dysbiosis with expansion of Proteobacteria in all mice, and E. coli overgrowth with increased bacterial infiltration into the ileal lamina propria of SAMP mice. FISH showed increase malX gene–carrying bacterial clusters in the ilea of supplemented SAMP (but not AKR) mice.
Splenda promoted gut Proteobacteria, dysbiosis, and biochemical MPO reactivity in a spontaneous model of (Bacteroidetes-rich) ileal CD. Our results indicate that although Splenda may promote parallel microbiome alterations in CD-prone and healthy hosts, this did not result in elevated MPO levels in healthy mice, only CD-prone mice. The consumption of sucralose/maltodextrin-containing foods might exacerbate MPO intestinal reactivity only in individuals with a pro-inflammatory predisposition, such as CD.
INTRODUCTION
Recent self-assessment dietary surveys indicate that ~10% of patients suffering from inflammatory bowel disease (IBD) believe that “sugary foods” worsen the severity of their symptoms and trigger flare-ups.1–3 Epidemiological studies have correspondingly shown a strong association between the use of artificial sweeteners (AS) and an increased risk for IBD (odds ratio > 2.12; 95% confidence interval [CI], 1.1–4.2),4–6 but the mechanisms of diet-induced IBD are unknown. Although nutrition has long been considered critical to improving malnutrition and iron deficiency in severe IBD,7, 8 aside from Delphi expert opinions and systematic reviews, there are currently no evidence-based dietary guidelines to help IBD patients manage their own diet.4 The importance of novel dietary studies to promote gut health in IBD has been independently highlighted2 by both patients and clinicians who listed “diet” among the top 10 research IBD priorities in the 2017 James Lind Alliance report.9 Conducting and interpreting dietary studies in humans is challenging, however, due to genetic, microbiome, and dietary variability. As an alternative, animal models have been used to examine the effects of dietary ingredients, individually, on numerous diseases. Dietary habits deemed unhealthy (eg, “Western” diets, rich in fats and sugars) are believed to alter the gut microbiome and either directly or indirectly trigger IBD. Despite substantial progress in our knowledge of acute models of intestinal inflammation, the precise effect of “multi-ingredient” AS on exacerbation of IBD remains unclear.
The ongoing and increased prevalence of obesity, which coincides with the rise of IBD diagnoses, indicates that there is a possible link between the risk factors of both diseases. Due to the growing obesity epidemic and the pressure to avoid sugar to reduce caloric intake, the food industry and consumers are using AS as substitutes for “table sugar.” The main ingredient of sugar is often sucrose, a disaccharide of glucose and fructose. Instead of sucrose, AS (eg, Splenda) use “sucralose” as a noncaloric sweetening ingredient, typically at 1% concentration, mixed with a filling ingredient (99%) that provides texture and volume.10 The most commonly used filler is maltodextrin, a nutritive polysaccharide regarded as inert and affirmed as “generally regarded as safe” (GRAS) by the US Food and Drug Administration (code 21CFR184.1444).11 However, studies indicate that neither sucralose nor maltodextrin is biologically inert.12–17 The potential adverse effects of AS across various diseases are controversial as they vary with the diseases and human populations studied (eg, obesity, diabetes),15 with some reports indicating mild or no negative effects.12, 17
Because the dietary modulation of IBD is a realistic goal that may involve modifications in patient behavior, especially in Crohn’s Disease (CD), which is one of the IBDs that has the strongest correlation with dietary habits, it is important to determine the effect of multi-ingredient commercial AS (eg, sucralose and maltodextrin in Splenda) on IBD. The information derived from studying any commercial mixture of dietary ingredients may assist clinicians and patients in making informed and corrective dietary decisions that could potentially alleviate IBD symptoms and prevent flare-ups. Using mice, in this study, we first determined the features and variability of the gut microbiome in a spontaneous mouse model of CD-like ileitis (strain SAMP1/YitFc [SAMP]) compared with its parental ileitis-free control mouse strain AKR/J (AKR), and then we quantified the inflammatory effects of 6-week chronic supplementation of a commercial AS (Splenda) on IBD using the SAMP ileitis mouse model. The objectives of this study were (1) to characterize the fecal metagenome (ie, the collective composition profiling of the bacterial genomes in the gut) of experimental SAMP mice and the ability of animal cohousing in transmitting SAMP ileitis to healthy AKR mice; and then (2) to quantify the effects of Splenda supplementation on CD ileitis using SAMP and AKR mice and a fecal homogenization protocol18 where all mice prior to experimentation were exposed to (via oral gavage) the pool of fecal microbes present in all mice. As a CD-relevant morphological feature, a recent three-dimensional (3D) stereomicroscopic (SM) assessment and pattern profiling (3D-SMAPgut) phenotyping protocol revealed that SAMP mice naturally develop segmental enteritis with cobblestone lesion formation in a fashion resembling CD (100% penetrance).18 Herein, we discovered that ileitis-prone SAMP mice have a microbiome enriched with Bacteroidetes species (as described in CD) and that the chronic ileitis phenotype is not transmissible to AKR or affected by the presence of Helicobacter in the gut as a commensal. In this microbial context, we then determined that 6 weeks of Splenda supplementation in the drinking water of adult inflamed mice did not worsen the morphological severity of ileitis in SAMP mice, based on histological or stereoenterotype 3D-SMAPgut score indexes. However, Splenda did cause gut microbiota dysbiosis in both SAMP and AKR mice, with a predominant increase of Proteobacteria, and a significant increase of pro-inflammatory myeloperoxidase (MPO) activity and penetrating bacteria primarily in the gut tissues of SAMP mice (ileitis). Interestingly, Splenda did not induce such an increase in MPO activity or bacterial tissue abundance in healthy mice (AKR), indicating that Splenda might have pro-inflammatory implications only if consumers have susceptibility to CD, potentially aggravating the severity of symptoms and flare-ups, which would be in agreement with observations reported by IBD patients.1–3
METHODS
Mice and Husbandry
This study was conducted with protocols approved by the Institutional Animal Care and Use Committee of Case Western Reserve University (IACUC-CWRU), using SAMP, AKR, and C57BL/6J (B6) mice, which are maintained in specific pathogen–free (SPF) colonies at the Animal Resource Center (ARC-CWRU, Cleveland, OH, USA). Mice were housed in shoe box cages with pine shavings for bedding, offered reverse osmosis drinking water (eg, free of salt, bacteria, viruses), fed ad libitum irradiated standard laboratory chow (Prolab RMH 3000; porcine animal–derived fat preserved with BHA; 6.8% content by acid hydrolysis) and kept on 12-hour light/dark cycles.18 Age- and sex-matched mice (5–7/group) were used in all experiments. SAMP mice originated from selective sibling breeding of AKR mice, with an outcrossing to B6, as previously described.18–20 SAMP mice are prone to developing progressive ileitis with “typical cobblestone lesions” resembling the 3D features of CD.18–20 Preliminary whole-genome sequencing analysis indicates that SAMP ileitis is a polygenic disease, as is CD in humans.21 As needed, AKR and B6 mice were used as parental genetic ileitis-free controls. Splenda experiments were conducted with mice produced in the main SAMP breeding colony (herein designated as facility A). A parallel alternate colony (facility B) was used for verification purposes of metagenomic findings in facility A. To maintain phenotypic and genetic homogeneity of SAMP in both facilities, periodic exchange of breeders occurred between the 2 facilities every 12 months. “Breeder mice” typically set at a 1:2 male:female ratio produced offspring “experimental mice,” which are weaned at 3–4 weeks of age and maintained next to the “breeder mice” cages. Helicobacter-negative SPF SAMP mice, rederived as at the National Cancer Institute (Center for Cancer Research; Frederick, MD, USA), were maintained and tested on a high-level of isolation unit within the ARC-CWRU, using the husbandry standards described above.
Fecal Metagenomic Shotgun DNA Sequencing
Due to the unresolved issues associated with the metagenomic shotgun DNA sequencing analysis of all kingdoms present in the gut microbiota, which could be relevant as a cause or consequence of IBD,22, 23 herein we decided to focus primarily on bacteria and screen only for viruses. To prevent sequencing and postsequencing analytical variability, validated bioinformatic methods were used on sequencing raw and high-quality filtered data (ie, Illumina and Metaphlan).24 We focused on bacteria because wholesome “kingdom-agnostic” metagenomics requires controlling for large differences in cellular integrity across all members of the gut flora,22, 23 which was not widely validated or available when the study was initiated.
For DNA extraction, flash-frozen fecal samples were homogenized in 1x PBS (1:5, grams:vol) with ceramic beads and mechanical disruption using Fast Prep FP120 homogenizer (setting 5 for 30 seconds at 4oC twice, with 1 freezing cycle in between). Homogenates, after a 20-second centrifugation at 10,000 × g at 4oC, were shipped overnight to the laboratory of Dr. Skip Virgin at Washington University, St. Louis, MO. DNA extraction and purification from the homogenates were performed using the commercial Qiagen DNA extraction mini-kit following the manufacturer instructions. The integrity (and quantity) of the DNA was monitored by spectrophotometry and gel electrophoresis. dsDNA was quantified with an Invitrogen Qubit Fluorometer (8.62 ± 5 µg/mL; Quant-iT). Shotgun metagenome analysis was conducted following library preparation using Illumina reagents and Miseq sequencing validated protocols. Standard Fastaq files were then used to quantify the abundance of bacterial reads using MetaPlhan alignment and analytical pipelines developed by Dr. Huttenhower at the Harvard School of Public Health.24 Briefly, raw FASTQ files were transferred to a local server where Metaphlan pipelines were run on Galaxy, where the R1 and R2 fasta files were concatenated, then assembled and blasted using bowtie2 very-sensitive-local search. Metaphlan output files were then merged using the merge_metaphlan_tables.py script, and were normalized and analyzed using Metaphlan and R visualization tools.
SAMP Ileitis Transmissibility, Cohousing, and Fecal Microbiota Homogenization
To determine if the passive exposure of fecal pathogens in a confined space during cage cohousing could alter the expected morphological mucosal pattern (presence/absence of cobblestone ileitis) of both AKR and SAMP colonies, 3-week-old SAMP and age/sex-matched AKR mice were weaned and subjected to a 24-week period of cage cohousing. As our metagenome data indicated that there was a relevant microbiota variability within the colony over time, Splenda experiments were conducted 7 days after implementing a fecal flora (microbiota) homogenization protocol (IsPreFeH),18, 25 where all mice were exposed to a composite (pool) of feces from all intended experimental mice, by gavaging 400 µL of fecal suspensions to each mouse.18, 25
Artificial Sweetener Experiments in Mice
To determine the effects of Splenda supplementation on SAMP ileitis and gut microbiota, we conducted 3 separate experiments with incremental adjustments. In experiment 1, we added a “low dose” of Splenda (1.08 mg/mL) to the drinking water for 6 weeks and compared it to “plain water” using only SAMP mice (6/group). Body weight, microbiological culture–based data (feces and spleen), and ileal histological and SM scores were the main study outcomes. In experiment 2, we repeated the same protocol but increased the dose to the maximum recommended by the US Food and Drug Administration (FDA; 3.5 mg/mL) and included AKR mice (6/group). The fecal bacteriome changes were assessed using 16S rRNA microbiome, glucose tolerance tests, and a well-validated method of biochemical inflammation based on the enzymatic detection of MPO activity in both the ileum and colon.18, 25 Finally, systemic inflammation was evaluated by serum tumor necrosis factor–alpha (TNFα) levels (Ready-Set-Go ELISA kit eBioscience, San Diego, CA, USA), and glycemic responses were assessed by glucose tolerance tests. In experiment 3, we repeated protocol 2 but administered Splenda at a dose that was 10 times higher than the dose used in experiment 2 (ie, 35 mg/mL). The main outcomes were MPO activity and intestinal inflammation (histology and SM). To minimize cage-to-cage clustered variability, all animals were housed individually, and all water bottles were replaced every 48 hours to provide fresh supplementation and prevent bacterial over growth. An irradiated/autoclaved standard diet was offered to all mice during experimentation to prevent external microbial confounders. We have recently reported the effect of soiled bedding in the mouse microbiome as a source of “cyclical bedding-dependent bias” in microbiome research, which could be varied depending on a number of factors, including the number of mice, microbiota, and humidity.26 In the present study, we controlled for “cyclical bias” by (1) housing all mice individually, (2) maintaining all cages in HEPA-filtered pressurized standard dorms (low cage humidity), (3) replacing all cages simultaneously weekly, and (4) collecting all data (food consumption, fecal samples, euthanasia) and samples (live and postmortem) for all animals the same day. Stereomicroscopic 3D pattern profiling (to determine the presence of SM abnormalities on the mucosal surface) and histological assessment were conducted on Bouin’s fixed intestinal tissues. Histological evaluation of inflammation severity was determined by hematoxylin and eosin–stained 5-µm-thick sections using a semiquantitative scoring system. A board-certified pathologist determined all scores in a blinded fashion. The detailed 3D-SMAPgut protocol’s considerations, along with the scoring forms and criteria, have been validated and described elsewhere.18
Relative Multidilution Enumeration of Fecal Bacterial Communities Using a “Parallel Lanes Plating” Method
Traditionally, the enumeration of total bacteria from feces has been conducted using spread plate methods, which require up to ten 10-fold serial dilutions to inoculate 100 μL of each dilution in individual agar plates.27, 28 For feces, which contain up to 8–9 log10 colony-forming units (CFUs) of bacteria per gram, a full quantitating spectrum requires up to 10 agar plates. To reduce costs, scientists often plate dilutions 7, 8, and 9 to estimate the fecal CFU counts, leaving more concentrated dilutions (1 to 6) unexamined. Other methods used for enumeration of mono-strain bacterial suspensions in food science use 5-μL drops of each 10-fold serial dilution to be “spotted”/incubated on an agar plate. Despite their simplicity, spotted platting only allows the collection of binary data (presence/absence of growth in each spot) to obtain “approximate” CFU log10 range counts (most probable number/dilution), with no capabilities to compare the relative growth or inhibition potential of cocultured strains.27 As we were interested in estimating the relative concentrations of mouse fecal bacterial communities, which may vary in natural abundance and which would be prohibitive to examine with spread plating methods in the Splenda experiments,28 we developed a method herein referred to as “parallel lanes plating” to rapidly estimate total and relative bacterial abundances across numerous dilutions using a single agar plate. This method allows the plating of 8 to ten 10-fold dilutions for the simultaneous enumeration of CFU for bacterial subgroups exhibiting colonies of distinct morphological appearance, irrespective of their concurrent high or low abundances within the same sample. In brief, the method uses 10–20 μL of all 10-fold serial fecal dilutions in PBS, which are placed simultaneously as drops using a multichannel dispenser on 1 side of a tilted agar plate (>60o–80o angle). The inclination of the agar plate makes the drops slide downward over the agar surface, creating parallel lanes of spread solutions of up to 5-cm2 surface area, which allows the easy identification and enumeration of distinct bacterial colonies and inhibitory interactions across all fecal dilutions (0.5 × 10 cm) (Supplementary Fig. 1). Upon drying and 48 hours of aerobic and anaerobic incubation at 37oC, single colonies were enumerated relative to total CFUs of cultivable bacteria in the sample. Agars used included Brain Heart Infusion (BHI) or tryptic soy agar (TSA) supplemented with 5% sheep blood (BD, Downers Grove, IL, USA), Luria Brentani (LB; enterobacteria), de Mann Rogose Sharpe (lactobacilli), meat liver (fastidious anaerobes and sulphite reducers), and plain microbiological agar supplemented solely with either yeast extract, maltodextrin, or Splenda (at 3.5%). In selected media (TSA), purified colonies were Sanger sequenced following standard protocols. Specific Helicobacter primers were also used to semiquantify Helicobacter spp. in feces and tissue of selected animals using amplicon gel analysis (5’- B38 GCA TTT GAA ACT GTT ACT CTG; B39 CTG TTT TCA AGC TCC CCG AAG; C97 GCT ATG ACG GGT ATC C; C98 GAT TTT ACC CCT ACA CCA).29
16S rRNA Gene Microbiome Analysis
Microbiome analyses were conducted at CWRU using Ion Torrent protocols, which have been validated and described in detail.30, 31 To quantify bacterial changes associated with the 6-week supplementation of Splenda, we used pure DNA from end point fecal samples (day 42–47) to PCR-amplify the V4 region of the 16S rRNA gene in triplicate using primers Forward-S-D-Bact-0564-a-S-15 (5′-AYT GGG YDT AAA GNG) and Reverse-S-D-Bact-0785-b-A-18 (5′-TAC NVG GGT ATC TAA TCC), as previously reported.31 PCR products were then evaluated by electrophoresis in 2% agarose gel and purified with the Agencourt AMPure XP system. PCR amplicons were selected to obtain 400-bp length for library preparation and were sequenced using Ion Torrent reagents and a benchtop sequencer at CWRU.32, 33 For sequence analysis, the mothur package of algorithms (v1.32.1)34 and associated dependencies were used. As we previously reported,31 aligned paired-end reads aligned to Silva 16S rRNA reference database. Sequences that were >244 bp or <239 bp in length that contained any ambiguous base calls or long runs (>8 bp) of holopolymers or did not align with the correct region were removed. Chimeras were identified using uchime and were eliminated. CatchAll was used to assess species richness, while taxonomy assignment relied on the RDP taxonomy database (http://rdp.cme.msu.edu/index.jsp). Sequences were binned into operational taxon units (OTUs) at a 3% dissimilarity level. Instead of subsampling for normalization,31 we normalized the OTU tables by rescaling the abundances of all samples to the fecal sample having the lowest total sequence abundance in the study. R software was used to visualize the microbiome profile and compute univariate and multivariate statistics. Dendrograms were computed using Euclidean distances, and when distinct unsupervised hierarchical clusters were observed, we tested the mouse allocation using frequency statistics (Fisher exact). In this fashion, we determined whether the allocation of mice to the microbiome clusters was random or significantly linked to the study variables, especially mouse strain and treatment (Splenda or plain water).
Quantitative Polymerase Chain Reaction of Mucosa-Associated Bacteria
DNA was isolated from ileal tissue using the Roche High Pure PCR Template Prep Kit for genomic DNA isolation. qPCR was performed using 10 ng of DNA in all reactions and primers for Eubacteria35 or six 16S rRNA-specific sequences for Escherichia coli,36Bacteroides, Lactobacillus/Enterococcus, Eubacterium rectale/Clostridium coccoides (Erec), segmented filamentous bacteria (SFB), and mouse intestinal Bacteroides (MIB)37 in iTaq SYBR Green Supermix with ROX (BioRad) on an ABI prism 7900HT (ThermoFisher Scientific) using SDS2.4 software. Samples were run in triplicate. Because there is no proper widely accepted “reference control bacterial population marker” to normalize qPCR microbiome data, quantitative strain-specific 16S primer amplicon data were analyzed collectively using the raw qPCR-CT values as described earlier18 and multivariate statistics to visualize and quantify the overall impact of the AS supplementation on the mucosa-associated microbial composition in the ileum of mice.
Fluorescent In Situ Hybridization
To visualize the localization of bacteria carrying the malX gene, which encodes for maltodextrin-degrading enzymes,14, 38, 39 ileal tissues were fixed in methanol-based Carnoy’s fixative (60% absolute methanol, 30% glacial acetic acid, 10% chloroform), embedded in paraffin blocks, and sectioned. Five-µm sections were deparaffinized and hybridized with 250-ng E. coli-Cy3 probe (5’-Cy3-CAT CTT CAC AGC GAG TTC-3’), 500-ng MalX-Alexa488 probe (5’Alex488-ACG CGT TTC CTT TCG CAA-Alexa488-3’), and Eubacteria338-Alexa647 probe (5’Alexa647-GCT GCC TCC CGT AGG AGT-3’) or buffer-only controls in 20-mM Tris-HCL, 0.01% SDS, 0.9M NaCl at 46°C overnight.40 Slides were then rinsed twice with water, incubated 5 minutes in 20-mM Tris-HCL, 0.9M NaCl at 46°C, rinsed twice with water, dried 10 minutes at 46°C, and applied with Vectashield containing DAPI (Vector Labs) and coverslips. Imaging was acquired using a Leica TCS-SP spectral laser scanning confocal microscope equipped with a Q-Imaging Retiga EXi cooled CCD camera and Image ProPlus Capture and Analysis software (Media Cybernetics). Image z-stacks were collected every 0.49 μm spanning the full thickness of cells and exported for image analysis using Image ProPlus Capture and Analysis software (Media Cybernetics).
Statistical Analysis
In all experiments, mice were randomized using a systematic approach. We used optimal analytical and matching strategies to control for confounding bias, as implemented in interventional and observational studies.41 Further, blinding was enforced in experimental and analytical stages as Splenda supplementation or Helicobacter status was not obvious to handlers; noninformative codes were revealed after analysis. Both univariate and multivariate statistical analyses were conducted independently for each Splenda dosing. Parametric statistics (Student t tests and/or 1-way analysis of variance [ANOVA]) or their nonparametric alternatives were used to compare experimental data (ie, body weight, inflammatory scores, MPO activity). Metagenomic and microbiome bacterial abundance data (OTU taxonomic tables) were log transformed, normalized, and processed using R software or STATA. Multivariate Hotelling’s T-squared distribution statistics was used for the comprehensive analysis of bacterial qPCR tissue data. Data were presented as SD or 95% confidence intervals; significance was held at P ≤0.05. P values between 0.05 and 0.1 are also shown when appropriate.
RESULTS
The Gut Metagenome of Ileitis-Prone SAMP Mice Is Rich in Families of the Phylum Bacteroidetes
Prior to testing the effects of Splenda, we first examined the gut metagenome profile of the SAMP “experimental mouse” colony compared with the AKR, which have been maintained together in the same animal CWRU facility for more than a decade. Because each colony could have selected for their own microbiome, adapted to their health or diseased intestinal environment through the years, it was important to determine whether potential microbiome differences (“shifts/drifts”) could be attributable to the IBD susceptibility genotype and the progressive SAMP phenotype (mild inflammation in young; severe inflammation in adult). For this purpose, fecal samples from 6 experimental mice (3 males, 3 females; 6 cages) across 3 different age groups (7, 22, and 50 weeks) (see experimental design in Fig. 1A) were collected, processed, pooled for DNA extraction, and sequenced using metagenome MiSeq Illumina sequencing reagents. We used pooling to create a composite sample for each age group as a valid screening method to control for background individual and cage variability, because fecal pooling is a powerful and cost-effective approach to study the microbial phenotype of large animal populations (with pooling of 5–10 individual samples showing optimal performance).42, 43 At the phylum level, metagenomics revealed a significant increase in the Bacteroidetes phylum in SAMP mice. At the class level, within the Bacteroidetes, Sphingobacteriia and Bacteroidia were increased in SAMP compared with AKR mice (Figs 1B–E). Within these 2 classes, out of 7 possible in the phylum Bacteroidetes, which was the most abundant phylum in the study, it is remarkable that the most abundant species identified in the phylum belonged to 4 out of 6 possible genera in the class Bacteroidia (Prevotella, Alistipes, Parabacteroides, and Bacteroides; Order Bacteroidales). Collectively, Bacteroidia was most commonly altered in SAMP mice compared with only 1 genus increased out of 6 possible in Sphingobacteriia (1/6 vs 4/6; 1-tailed Fisher P = 0.045) (see species in Fig. 1E). This finding is experimentally relevant to CD because CD has been associated with Bacteroidetes-enriched dysbiosis (eg, Bacteroides and Prevotella genera in human gut mucosal samples).44
![Metagenomic analysis of pooled fecal matter reveals an abundance of Bacteroidetes and Helicobacteraceae in SAMP mice. A, En face stereomicroscopic images of healthy AKR ileal mucosa (top image) and SAMP cobblestone ileitis (bottom image); schematic illustration of metagenomic sampling design in SPF facilities (pooled feces in experiment 1 [summer] vs individual sampling in experiment 2 [8 months later, spring]). B, Multivariable unsupervised hierarchical analysis of metagenomic fecal bacterial abundance from 36 mice pooled as 6 mice per age group (7, 22, and 55 weeks old). Note clustering of AKR and SAMP samples as separate clades. Arrows illustrate distinct families, ranked by abundance across all samples. C, Compared with AKR, the SAMP metagenome have greater bacterial diversity (Shannon diversity). D, Boxplots reveal reduction of Firmicutes (Lactobacillaceae and Clostridiales) and expansion of Bacteroidetes families in SAMP (eg, Bacteroidaceae, Rickenellaceae, Sphingobacteriaceae). E, Circular diagram of relative taxonomic metagenomic abundance in phylogenetic tree format highlights significant enrichment of Helicobacteraceae (Proteobacteria phylum), and 4 of 6 known Bacteroidetes phylum families (Bacteroidaceae, Rickenellaceae, Porphyromonadaceae, and Prevotellaceae) in SAMP mice (1-tailed Fisher exact P = 0.046).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ibdjournal/24/5/10.1093_ibd_izy060/3/m_izy06001.jpeg?Expires=1716316517&Signature=GB8KInfRT5YYpLz-hFP5keQmQm1Es0yT0JZF~V~A1UDHSJ7M~ID0lUKovmrLHO35v4G5baBsIpbujoZKitlYlVUVp4LG6julzGCOGVxrorhABvnkG0rNoZqP6q~c98hnPjCNgaw4P66QXd2eVMVIeyOcrKyc1~aB5oUQFfGU~pyT3Yyo0BraL6PE7JhClUS9dLqyzvUx4J2s8A~O3fPmEn8Xzt3GgbVT76Q7FBKkHKL3qgkfcvn~AMVuJbPuaDmO5lotnfCLhT5a30tlW54yVgc5~AMiyJSebMPx0T6Chla9U3ksFREgh8BbI82hxPi7c30HQYAtbotv7Xz8w-jNBw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Metagenomic analysis of pooled fecal matter reveals an abundance of Bacteroidetes and Helicobacteraceae in SAMP mice. A, En face stereomicroscopic images of healthy AKR ileal mucosa (top image) and SAMP cobblestone ileitis (bottom image); schematic illustration of metagenomic sampling design in SPF facilities (pooled feces in experiment 1 [summer] vs individual sampling in experiment 2 [8 months later, spring]). B, Multivariable unsupervised hierarchical analysis of metagenomic fecal bacterial abundance from 36 mice pooled as 6 mice per age group (7, 22, and 55 weeks old). Note clustering of AKR and SAMP samples as separate clades. Arrows illustrate distinct families, ranked by abundance across all samples. C, Compared with AKR, the SAMP metagenome have greater bacterial diversity (Shannon diversity). D, Boxplots reveal reduction of Firmicutes (Lactobacillaceae and Clostridiales) and expansion of Bacteroidetes families in SAMP (eg, Bacteroidaceae, Rickenellaceae, Sphingobacteriaceae). E, Circular diagram of relative taxonomic metagenomic abundance in phylogenetic tree format highlights significant enrichment of Helicobacteraceae (Proteobacteria phylum), and 4 of 6 known Bacteroidetes phylum families (Bacteroidaceae, Rickenellaceae, Porphyromonadaceae, and Prevotellaceae) in SAMP mice (1-tailed Fisher exact P = 0.046).
Within the phylum Proteobacteria, analysis at the family level revealed a striking increase of Helicobacteraceae (C: Epsilonproteobacteria; O: Campylobacterales) in SAMP mice (Fig. 1B–E). However, as Helicobacteraceae (the sixth most abundant family in the study, out of 41) was already abundant in young SAMP mice (7 weeks of age), the findings suggested that Helicobacteraceae abundance was not due to the severity of ileitis, which is typically undetected at 3 weeks of age but affect >60% of ileum by 55 weeks of age. Because metagenomics is robustly based on the quantification of bacterial communities using single-copy gene data, results from pooling (n = 36) mice strongly indicated, for the first time, that the SAMP microbiome phenotype is rich in several Bacteriodetes families, and surprisingly dichotomous and rich in Helicobacteriaceae. Because Helicobacter has been considered both a confounding factor and a necessary organism in certain B6 mouse models of colitis, but not all,45 it was deemed necessary to confirm the findings with a new set of samples for individual (not pooled) metagenomics, especially as Bacteroidales and Helicobacteraceae are known inhabitants of the human intestine with the potential to become opportunistic pathogens, for example, Odoribacter splanchnicus (Fig. 1).46, 47
Virome Sequencing Reveals Absence of Norovirus in “Pooled Feces”
To further investigate the microbiome of the “pooled feces” experiment in our colony, fecal samples were also processed and shotgun-sequenced using a Roche 454-sequencing platform to screen for the presence of DNA and cDNA viral genomes, in addition to Illumina sequencing. Metagenomic examination of the sequences indicated the absence of norovirus in the pooled samples. Thus far, metagenomic analysis has indicated the absence of detectable dsDNA viruses in the pooled feces of young SAMP and AKR mice. PCR for norovirus and novel astroviruses were also negative, although testing of our SPF colony has been shown to be variably seropositive for norovirus over time. We recently reported a concurrent serological screening of adult SAMP mice reared under germ-free (GF) conditions, wherein SAMP exhibiting cobblestone ileitis had no seroconversion to norovirus for up to 62 weeks of age.26 Thus, serology indicated that SAMP ileitis occurred independently of norovirus, a virus needed to promote intestinal inflammation in ATG16L1 gene–dependent models using B6 mice.48, 49
Individual Metagenomics Illustrate Great Cage and Facility Variability of Helicobacter in Mice
To verify that Helicobacteraceae was stably abundant in SAMP mice over time, a second metagenomic analysis was conducted 8 months after the “pooled” metagenomics evaluation, but testing individual fecal samples from “breeder mice.” Paired male:female mice were sampled as clustered by cage to validate the reproducibility of our unsupervised metagenome hierarchical analysis used in the pooled metagenome analyses of “experimental mice.” Eighteen active 30-week-old breeders (1 male, 1 nonpregnant female/cage) were tested as representative because they would be the main source of gut commensal microbes inherited to the offspring “experimental mice” sampled earlier. To properly determine whether Helicobacteraceae was abnormal in SAMP, B6 breeders were also sampled, along with the original AKR vs SAMP colonies (Fig. 1A). To ensure compliance on scientific rigor and data reproducibility, SAMP and AKR mice were also sampled from an alternate colony (in facility B). Unexpectedly, metagenomic analysis (54.4 million raw reads; 20.9 million high-quality bacterial paired-end, 79 ± 5% unique) revealed major variability for Helicobacteraceae across the colonies and facilities, further challenging the relevance of Helicobacteraceae as causally associated with SAMP ileitis. This time, Helicobacter was absent in 3 cages of SAMP and B6 mice, but abundant in a cage of AKR in our facility A (Fig. 2A), which was opposite to concurrent findings in facility B. Analysis clearly showed that the ability to sequence Helicobacter in SPF mice was highly variable and dichotomous in metagenomics (either highly abundant or absent), likely due to seasonality or individual cage differences. Given the high variability, no absolute conclusion could be drawn regarding the potential causal or modulatory role of Helicobacter on SAMP cobblestone ileitis. However, the analysis (1) confirmed that Bacteroides are abundant in adult SAMP mice and (2) verified the optimal reproducibility of our metagenomic methods as pairs of breeders always clustered together (as expected within their cage assignment, despite notorious “mouse individualities”) using unsupervised clustered analysis (Fig. 2A–D).
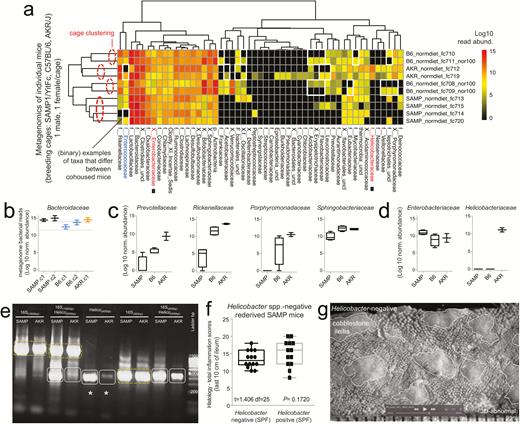
Metagenomic analysis reveals Helicobacter variability with minimal effect on SAMP ileitis. A, Individual fecal metagenomic analysis of breeder mice was performed 8 months after the previously pooled fecal sampling experiment, demonstrating bacterial profiles with reproducible cluster within pairs of breeders for each individual cage. Squares highlight absence of Helicobacteraceae in Helicobacteraceae-negative SAMP colony. When present, Helicobacteraceae was highly abundant in the feces of positive mice. Analysis highlights within-cage mouse microbiota individualities, eg, Enterococcaceae. B, Boxplots, showing averages for each cage, reveal high abundance of Bacteroidaceae in SAMP mice (compared with experiment 1 in Fig. 1D). C, The abundance of other Bacteroidetes is shown by boxplots. D, Compared with AKR and B6 mice, SAMP had a significantly higher level of Enterobacteracea (a family that contains Escherichia coli). Notice marked abundance of dichotomous variability of Helicobacteraceae in mice. E, Gel electrophoresis of PCR amplified target regions of Helicobacter spp. within the 16S rRNA gene from fecal DNA of random mice form our SAMP and AKR colony, 4 months after the individual fecal metagenomic experiment. Notice high band intensity in SAMP (asterisks). Universal and PCR-specific primers were used to generate full or partial gene amplicons (1500 or 400 bp). F, Histological inflammation scores of distal ileum from rederived Helicobacter-negative SAMP compared with that of the SAMP colony show no differences. Notice wider variability in Helicobacter-positive mice. G, Representative snapshot of videostereomicroscopy shows unchanged morphological appearance of ileal mucosal surface in Helicobacter-negative SAMP mice. Scale = 500 um.
The Presence of Helicobacteraceae Does Not Alter Cobblestone Lesion Progression in SAMP Ileitis
Helicobacter spp. specific 16S-primer amplification of fecal bacterial DNA confirmed that both mouse lines (SAMP and AKR) in facility A harbored the bacterium in their feces and that SAMP could have more DNA copy numbers than AKR (Fig. 2E). Because the initial pooled metagenomic analysis indicated a strong signature for Helicobacter spp. in SAMP, we then created a new SAMP colony by rederivation and colonization with a Helicobacter-negative mouse microbiota at the National Institutes of Health. Following rederivation, breeding pairs were transported and housed in our facility for phenotype testing. SM analysis of the small intestines of mice at 30 weeks of age revealed a persistence of the 3D cobblestone lesion architecture in the inflamed ileum, whereas histological analysis over time of this colony revealed that Helicobacter was not necessary for the disease to occur or histologically worsen (the study of immunophenotyping differences is in progress and will be reported separately) (Fig. 2F).
The SAMP Ileitis Is Not Transmissible or Preventable During Long-term Cohabitation With Healthy Mice
Previous studies using DSS-induced colitic mice and cohousing suggested that acute colitis is “transmissible” from affected B6 mice to unaffected mice by exposure to their gut microbiota.50 By using cohousing, others have reported that the microbiota of NOD2 knockout B6 mice transmit colitogenic properties to B6 mice, primarily via changes in Butyrivibrio, Lachnobacterium spp., and in Bacteroides (above shown to be also increased in SAMP).51 In contrast, we recently determined that the same NOD2 mutation in SAMP unexpectedly improved, rather than exacerbated, the cobblestone ileitis (50% better at 30 weeks old) and reduced the severity of DSS colitis.52 In contrary, we found no evidence to suggest that the anti-inflammatory benefit of the NOD2 mutation was due to a given microbiome profile based on fecal metatranscriptomic (bacterial gene expression mRNA) and 16S microbiome analyses. Although the concept of transmissible colitis seemed relevant to the genetics of B6 mice, the genomic differences in our SAMP model and its spontaneous progressive ileitis phenotype21 prompted us to determine whether microbiome exposure of young 3-week-old healthy AKR mice during cohousing could protect young 3-week-old SAMP mice from developing ileitis, and whether disease could be transmissible from SAMP to AKR in a long-term experiment. The experiment was conducted for 6 months with 14 mice. 3D-SMAPgut analyses showed that the 3D ileitis phenotype was not transmissible to AKR mice and that SAMP ileitis was not prevented during cohabitation with AKR. Culture analysis (anaerobes and enterobacterial fecal enumeration) indicated that although the total bacterial load was similar for AKR and SAMP cohoused mice, SAMP favored gut enterobacterial growth (Supplementary Fig. 1).
Together, the aforementioned analyses indicate that SAMP ileitis (1) had a Bacteroidetes-rich microbiome phenotype, (2) had increased likelihood to favor enterobacterial growth, (3) could develop cobblestone lesions unaffected by the presence of Helicobacter and norovirus in the gut, and (4) was not transmissible to AKR by long-term cohousing. Crossover experimentation with fecal transplantation from SAMP into GF-AKR mice, and vice versa, was considered to verify that SAMP ileitis was not microbiome-transmissible, but the experiment was not possible due to the lack of available GF-AKR mice at the time. Due to animal welfare concerns (SAMP-AKR mouse fights, with SAMP hierarchical dominance in cohousing experiments), we determined that Splenda experiments could not be conducted cohousing adult AKR and SAMP mice.
Splenda Does Not Alter the SAMP Cobblestone Ileitis Phenotype but Increases Tissue MPO Activity
Due to the difficulties in cohousing AKR and SAMP mice and in controlling for the observed metagenome cage variability, we implemented the IsPreFeH protocol18, 25 among all animals52 subjected to the Splenda experiments, which was followed by individual animal housing and Splenda supplementation for 42–47 days. Although low-dose supplementation resulted in transient body weight reduction during supplementation (generalized linear regression [GLM] P < 0.0001), no changes were significant at higher doses. In addition, although fecal cultures showed that Splenda promotes enterobacterial and maltodextrin-utilizing bacterial growth in SAMP, no evidence of systemic effects was present from culture of spleens, serum TNF levels, or blood glucose tolerance tests. Compared with the total number of anaerobes (TSA) and lactobacilli (MRS agar), the number of enterobacteria (LB) was significantly increased in Splenda-supplemented SAMP mice. No differences were seen at 1, 3, or 14 days after supplementation (Fig. 3A–G). Unexpectedly, SMAPgut and histological analyses of the ileum showed that Splenda did not augment the ileal inflammation scores, the percentage of SM-abnormal mucosa, or the 3D-morphological features of cobblestone lesions in CD-prone (SAMP) mice at any of the 3 doses tested. More importantly, supplementation did not cause either ileitis or colitis in healthy (AKR) mice. In contrast, a well-validated quantitative enzymatic activity assay showed that the amount of MPO reactivity in the ileum of SAMP mice treated with the Splenda FDA maximum approved dose (3.5 mg/mL) was 2.7-times higher compared with SAMP mice drinking plain water (219.1 vs 81.8 U/g, t test P = 0.022). Controlling for multiple variables, analysis of ileal tissues of SAMP mice in experiment 3 (high-dose, 35 mg/mL) confirmed that increased increments of MPO activity occurred only in Splenda-treated SAMP (2-way ANOVA F2,27 = 4.08, P = 0.055, controlling for significant interaction between Splenda and organ [colon vs ileum], P = 0.0515). Interestingly, the same doses of Splenda had no effect on ileal MPO activity in AKR mice (ie, experiment 3, 2-way ANOVA F2,21 = 0.09, P > 0.76). The MPO activity in the colon was lower compared with the ileum (2-way ANOVAs for AKR and SAMP, P = 0.012 and P < 0.0001), which was unaltered by Splenda in both mouse lines (Fig. 4A–E). These results suggested that Splenda promoted the increase of intestinal MPO tissue reactivity (“biochemical inflammation”) only in the gut wall of mice prone to IBD, without inducing major (noticeable histologic) microscopic changes associated with active inflammation. This finding is remarkable considering that subclinical inflammation (ie, increased MPO and other inflammatory biomarkers, with no evidence of histological abnormalities) has recently been observed in healthy twins of monozygotic and dizygotic discordant pairs with IBD compared with healthy nontwin relatives. This further supports the concept that a combination of genetic and environmental factors predisposes individuals to biochemical (reactivity) inflammation in IBD.53
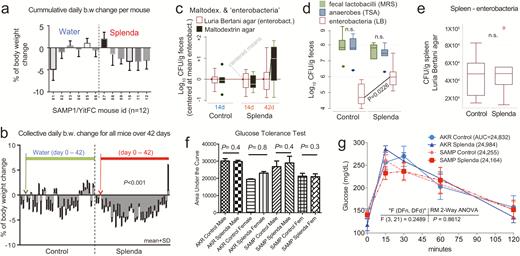
Effects of Splenda on body weight, fecal bacteria, and glucose tolerance in SAMP mice. A, Mean body weight change in 12 SAMP mice (individually caged, 7 days of adaptation, more than 42 days of supplementation of drinking water with and without low-dose Splenda). B, Daily group average of body weight change over time; low Splenda dose. C, Bacterial enumeration from feces using standard LB agar (used for enterobacteria) and in-house “maltodextrin agar.” Notice maltodextrin agar yielded an increasing bacterial count trend toward the end of study in the Splenda group (GLM P > 0.05; relative to centered log-transformed data for LB agar). Supplementary Figure 1 illustrates other in-house agars, yeast extract agar, and Splenda agar yielding similar trends, compared with LB agar. D, Total number of anaerobes (TSA), lactobacilli (MRS agar), and enterobacteria (LB) after 42 days of Splenda supplementation in SAMP mice (unpaired t test, n = 6/group). E, The total number of enterobacteria in the spleen suggests that Splenda had no systemic bacteremic effect. F, Glucose tolerance test on day 40 with animals with Splenda supplementation in experiment 2 (high FDA-approved dose). Notice the lack of significant effect across experimental mice. G, Glucose tolerance test curves illustrated as mean ± SD. Univariate analysis across time points showed no differences due to Splenda. Abbreviation: AUC, area under the curve (n = 6 mice/group).
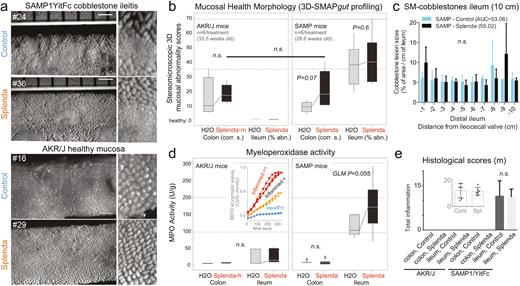
Splenda has no effect on 3D-SM or histological scores but promotes tissue MPO activity in mice with ileitis. A, En face stereomicroscopic images of AKR and SAMP ileal mucosa after 47 days of supplementation of Splenda at the maximum FDA-approved dose (experiment 2, medium dose, AKR vs SAMP mice, n = 6/group, supplementation started at 25.2 ± 2.4 weeks of age). Notice similar 3D stereoenterotype for SAMP cobblestone ileitis and the healthy mucosa in AKR mice. B, Cumulative 3D-SMAPgut scores on mucosal surface morphology for the colon (corrugation score/cm) and ileum (percentage of abnormal mucosa). Despite the positive trend (upward lines connecting means), there was no statistical difference between groups in all 3 Splenda experiments, irrespective of dose. C, Average size for average cobblestone for each centimeter of the distal ileum. D, MPO activity from colon and ileum tissue at the end of the experiment. Notice that increased MPO occurred only in mice with ileitis. Inset: Example of MPO activity for 3 tissues with different MPO reactivities. Notice the reproducibility among triplicates (Supplementary Table 1). E, Histological scores of the colon and ileum. Notice that there were no differences between groups.
Splenda Promotes Gut Microbiome Dysbiosis and Proteobacteria in SAMP and AKR Mice
Differences in fecal bacterial composition were assessed using 16S microbiome analysis (1.42 million V4 16S rRNA gene sequences from 20 mice; 70,879 reads per sample passed quality control filters; 99.36% had a Blast hit match in the 16S database). Corroborating the culture findings from SAMP mice treated with Splenda, analysis revealed that the most consistent effect that Splenda supplementation had on the mouse microbiome was a significantly widespread promotion of bacterial species across the 5 microbial classes identified within the Proteobacteria phylum (Alphaproteobacteria, Betaproteobacteria, Epsilonproteobacteria, Deltaproteobacteria, and Gammaproteobacteria). Comparison of proteobacterial mean abundances between AKR and SAMP indicates that SAMP were likely to have more Proteobacteria in the control (6/6; 1-tail sign P = 0.016) (see culture agars from cohouse AKR-SAMP mice in Supplementary Fig. 1E) and Splenda groups (5/6; 1-tail sign P = 0.109) (Fig. 5A–D). Comparatively, microbiome analysis also showed that subtle differences exist between AKR and SAMP mice, which clusters them separately as mouse strains. In addition, Splenda altered the gut microbiome by reducing some other phyla, while no significant effect was observed in Bacteroidetes or Firmicutes (eg, lactobacilli and clostridia), which also clustered the mice according to the dietary impact on their gut microbes (Fig. 5A–D). Culture data from Splenda-treated SAMP compared with mice drinking water showed that Splenda had no effect on the counts of lactobacilli (MRS agar), total bacteria (TSA), or anaerobic clostridial species (MRS, TSA and meat liver agars; GLM of normalized log10 adjusted P > 0.2), whereas it promoted the remarkable growth of Escherichia coli in the feces of mice. Of interest, E. coli overgrowth apparently occurred at the expense of displacing, at least, cultivable Streptococcus-like organisms, which were evident in the feces of mice drinking only water (Fig. 6). Because several lines of evidence indicated that Splenda promotes MPO tissue reactivity and the overgrowth of E. coli (Gammaproteobacteria) in intestinal content (ie, feces), we then used qPCR and fluorescence in situ hybridization (FISH) analysis to determine whether ileal tissue from SAMP mice treated with Splenda would have mucosal-associated dysbiosis and increased bacterial infiltration into deep intestinal layers.
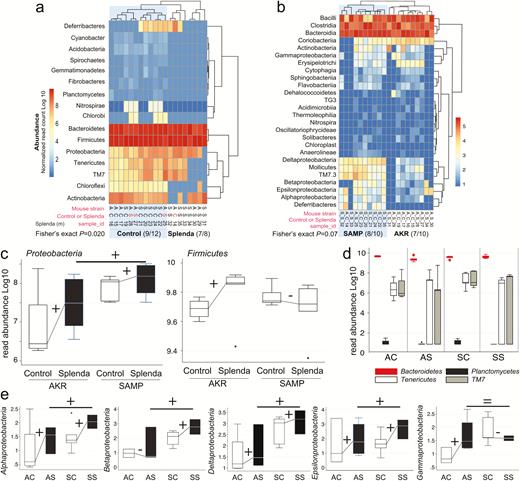
Splenda promotes gut dysbiosis characterized by enrichment of Proteobacteria in mice. A, Phylum analysis. 16S rRNA gene copy microbiome abundance normalized and presented as an unsupervised clustered hierarchical heat map that illustrates a significant effect attributable to Splenda (increase in Proteobacteria and reduction of other phyla including Chloroflexi; P = 0.02). Note the high relative abundance of Bacteroidetes with respect to Firmicutes. Notice that when present several proteobacterial classes contribute to microbiome separation between SAMP and AKR (P = 0.07). Notice highly abundant Bacteroidia, Bacilli, and Clostridia cluster at the top of the panel. C, Boxplot illustrates the effect of Splenda on phylum Proteobacteria, compared with Firmicutes. Lines connecting normalized averages indicate positive trends. D, Boxplot illustrates high Bacteroidetes abundance and the comparative reduction of other phyla in Splenda-treated mice. E, Bacterial abundance across all 5 Proteobacteria classes detected in the study. Sign binomial statistics of means in log10 scale suggests that Splenda promotes a positive effect (including (C); 10/12 were positive, 2/12 were negative, 1-tail sign P = 0.019). Abbreviations, C, control water; diet S, Splenda; mouse A, AKR/J; S, SAMP. B, Class analysis.
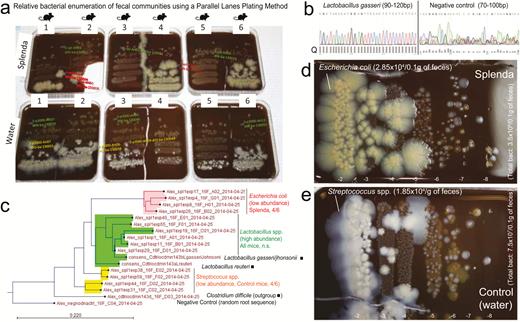
Splenda promotes the replacement of Streptococcus spp. with E. coli in the intestinal tract of SAMP mice. A, “Parallel Lanes Plating” Method. Photograph of BHI agar plates supplemented with 5% sheep defibrinated blood inoculated with 10-fold serial dilutions of feces from SAMP mice using a parallel minidrop slide and lanes method developed for tracking and relative enumeration of complex bacterial communities (Supplementary Fig. 1). Mice were caged individually and exposed to a composite of bedding and feces (IsPreFeH) prior to receiving Splenda at a low dose (1.5 mg/mL) or water for 42 days. Representative colonies comprising all possible morphologies in each agar plate were selected for purification and Sanger sequencing for speciation (from high dilutions, green labels; and low dilutions, pink or red labels). Notice the colony morphology (large, thick, spreading) of 4/6 mice in the Splenda group is different from that of 4/6 control mice (whitish, smaller). B, Sanger sequence chromatograph. Single-colony PCR revealed Lactobacillus gasseri (umbonate, brown) as the most common abundant bacteria in mice, unaffected by Splenda supplementation (see the panels below). Q, quality of consensus sequence. C, Phylogenetic analysis of 16S rRNA paired-end consensus sequences revealed that the whitish colonies in the control (water) group were closely related to Streptococcus spp., while bacteria in Splenda mice were E. coli. D and E, Close-up of colony morphologies on BHI agar after 5 days of aerobic incubation. Notice the “parallel lanes method” of 2 mice representing the Splenda and control groups. Negative numbers indicate 10-fold dilution factor.
Splenda Causes Distinct Ileal Tissue Microbiota With Increased Bacterial Malx Gene in SAMP Mice
Multivariate quantitative analysis of DNA copy numbers in ileal (full thickness) tissue samples from SAMP mice using 7 specific primers for various bacterial families/species revealed that supplemented mice had a distinct (dysbiotic) microbiome profile when compared with untreated animals (see enrichment of eubacteria, lactobacilli, and the contribution of E. coli illustrated as 2-dimensional vectors in Fig. 7A). Although SAMP ileitis has been known to be a highly segmental disease of the small intestine, blinded FISH analysis of SAMP mice confirmed that the ilea of Splenda-treated SAMP mice had increased numbers and larger clusters of bacteria within the villi compared with untreated SAMP and AKR mice. These bacterial clusters positively hybridized with an E. coli probe and a probe to malX, a gene that encodes a maltodextrin-binding protein of the maltose/maltodextrin metabolism system.54 Of translational value, the presence of FISH-positive clusters was almost imperceptible in ileitis-resistant AKR mice in both groups, with and without the sucralose/maltodextrin (Splenda) supplementation (Fig. 7B and C). This observation further supports that sucralose/maltodextrin have a dysbiotic effect in ileitis-prone SAMP mice, but not in healthy AKR mice.
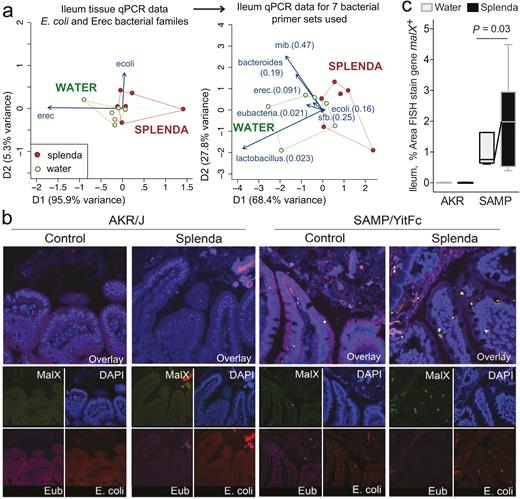
Bacterial qPCR and FISH staining of ileal tissue illustrates distinct microbiota and increased invasive malX+ bacteria (E. coli) in Splenda-supplemented SAMP but not AKR mice. A, Multivariate analysis of DNA qPCR data from ileum tissues of SAMP mice after 42 days of supplementation. Notice the display of mice (points) and the vector influence of the variables (arrows) on the overall matrix data variability (D1 and D2) for the E. coli and Erec primers, and for all the 7 primer sets used in this study. Hotelling’s T-test P values are in parentheses. Notice the separation of the 2 clusters (water, Splenda). B, Ileal sections from SAMP mice supplemented with 3.5% Splenda for 42 days (Splenda) or nontreated control mice (Water) were hybridized with probes to Eubacteria (purple), E. coli (red), and malX (maldodextrin, green), a component of the maltose/maltodextrin metabolism system. Cell nuclei are visualized with DAPI (blue). Images shown are representative of analyses performed in 5 mice per group. Notice the presence of E. coli in both the epithelial layer and the subepithelial lamina propria tissue (villi), and the large bacterial clusters in the lamina propria of SAMP mice on the Splenda panels. C, Percentage of area that stained positive for the malX gene probe (pixels, malX+ area/total tissue area*100). Unpaired t test statistics. A minimum of 4 fields were analyzed/sampled using ImageProPlus v7 software (AKR control, n = 4; AKR Splenda, n = 6; SAMP control, n = 6; SAMP Splenda, n = 10).
Together, our data indicate that sucralose/maltodextrin daily supplementation in the water (for 6 weeks) promotes bacterial dysbiosis with proliferation of Proteobacteria species, including E. coli, and increased bacterial invasiveness into villi tissue, which may in turn increase the MPO reactivity of ileal tissues during the course of murine cobblestone CD-like ileitis.
DISCUSSION
CD is a chronic relapsing-remitting IBD with a tendency to develop highly distinctive fecal microbiota disturbances (dysbiosis) compared with other forms of IBD.55 Characteristically, CD has abundant Bacteroidetes and reduced presence of Firmicutes.56, 57 A recent meta-analysis of raw sequencing data from IBD microbiome studies, together with DGGE studies,56, 57 indicated that some CD patients may not have a high abundance of fecal Bacteroidetes, suggesting that either there are 2 types of CD patients based on the presence of this phylum or there is large data variability. Our own studies support the role of Bacteroidetes in CD, as we recently discovered Bacteroidetes-enriched intramural cavernous fistulous lesions in the severely inflamed bowel samples removed from patients who underwent surgery for CD.18 For the first time, we presented data indicating that the SAMP ileitis phenotype is enriched (and not deficient) in Bacteroidetes, making this model microbiologically relevant to conduct dietary studies in a Bacteroidetes-rich context, or under germ-free conditions. The metagenome analysis also showed a large variability for Helicobacteraceae. Although we assessed the relevance of this genus experimentally by re-deriving the SAMP mouse colony free of Helicobacter, it is important to highlight the inherently difficulty in achieving the composition of a natural microbial community with a single absent organism in the re-deriviation process. Despite this inherent difficulty, the presence of SAMP ileitis in animals lacking Helicobacter indicates that the role of this genus as necessary for inducing ileitis in SAMP is minimal. Mono-strain association studies are currently examining the potentially negligible modulatory effect of this genus on local and systemic immunity in the development of SAMP ileitis.
Because current microbiome approaches are not quantitative with respect to the net fecal matter biomass content (eg, changes in bacteria taxa per gram of feces, and variable limit of detection thresholds Fig. 5)58 and because changes in fecal microbiota relative composition may not necessarily imply changes in the gut wall, we conducted complementary microbial enumeration–based methods on fecal samples (Fig. 6) and then qPCR and FISH assays in ileal tissues (Fig. 7), and we confirmed that AS supplementation had a modulatory effect on the mouse microbiome composition of SAMP ileal tissues. Because the effect of Splenda on MPO activity was exclusive to ileitis-prone SAMP mice, and not control IBD-free mice, it is reasonable to assume that CD patients may have increased MPO-inflammation susceptibility if sucralose/maltodextrin-containing foods were chronically consumed, even at low concentrations, as a regular habit. Similarly, it is also reasonable to highlight that increased biochemical MPO activity could put CD patients at risk of having exaggerated inflammation if other circumstances trigger mucosal inflammation, for instance, during unexpected food-borne bacterial superinfections that further recruit MPO-containing leukocytes to the intestinal tract.59, 60 Increases in the relative abundance of Proteobacteria, as identified in our study due to Splenda, have been reported with various types of intestinal tract diseases in diverse species, including humans. Proteobacteria are a major phylum of gram-negative bacteria that includes a variety of pathogens common to humans and animals, including Vibrio, Salmonella, Yersinia, Helicobacter, and Escherichia spp. As a pathological feature, most Proteobacteria, being gram-negative, have an outer membrane composed of lipopolysaccharides (LPS), which are potent triggers of innate immunity, with local and systemic responses, depending on the LPS dose that is proportional to the rate of bacterial growth. In this study, we found no clinically or tumor necrosis factor–based evidence of systemic inflammation induced by Splenda. However, we found microbiological and pathological evidence that Splenda promotes increased local MPO activity, bacterial penetration of the intestinal epithelium, and an increased predominance of E. coli. Together, our findings further support the role of dysbiotic Proteobacteria expansion as a microbial signature of intestinal and epithelial dysfunction.61, 62
The MPO enzyme produced by macrophages and neutrophils evolutionarily plays an essential role in degrading invading pathogens as one of the earliest lines of defense in innate immunity. However, the induced MPO-derived oxygen radicals cascade (which causes reactive oxydative stress), in turn, catalyzes reactions that can lead to modifications of proteins and destruction of the extracellular matrix that may exaggerate inflammation. Therefore, excessive MPO activity with sucralose/maltodextrin supplementation, together with the detection of increased bacterial fecal abundance and penetration of E. coli in the gut wall, could result in subclinical exaggeration of ileitis in CD. As IBD patients believed that “sugary foods” might be a culprit contributing to the increased severity of their symptoms, with epidemiological evidence supporting their clinical deterioration, our AKR mouse data indicate that it is reasonable to consider that healthy individuals should not be worried or expect to develop CD ileitis (or MPO hyper-reactivity) if they consume Splenda. However, IBD-free “healthy” consumers of sucralose and maltodextrin should be aware that proliferation of Proteobacteria is one of the features expected with AS-induced dysbiosis, which may functionally63 modulate other conditions not considered in our study. Our study only assessed 6 weeks of sweetener supplementation, but a chronic increase of reactive oxidative stress in inflamed tissues could also carry a higher risk for DNA oxidation and tissue damage, which have long been considered predisposing mechanisms for IBD-associated cancer and other serious complications.64–66
It has been almost a century since the introduction of noncaloric AS in our diets, which are consumed by ~30% of adult Americans. Of interest, some analyses have suggested that there may be a direct temporal correlation between the incidence of IBD and the amount of AS sold in various countries.67 Although other major shifts have occurred in human nutrition, it is also important to highlight that obesity and diabetes appear to parallel the increased incidence of IBD.16 Additionally, the term “Western diets” implies a proven shift of the gut microbiota that enhances the susceptibility to adherent-invasive E. coli infections and intestinal inflammation in mice.68 In this study, we report similar findings due solely to the administration of a minor component of the diet (an artificial sweetener based on a common combination of sucralose and maltodextrin in retail food markets), suggesting that several dietary habits or additives may lead to similar microbiota alterations. For instance, diet emulsifiers used as food additives have also been shown recently to alter the gut microbiota and promote colitis in mice.69 Our results provide experimental data to help inform the mechanisms of IBD modulation by the human diet, and guide the dietary habits of susceptible individuals. In addition to illustrating the experimental role of a sucralose-maltodextrin-based artificial sweetener (relevant to similar products on the market) in promoting intestinal dysbiosis and MPO activity, our studies also indicate that it might be possible to measure Proteobacteria and MPO activity as simultaneous fecal biomarkers in patients to monitor their gut’s (disease/health) adjustment to their diets. Monitoring MPO and Proteobacteria in humans could help identify individual factors that trigger patient IBD susceptibility to seemingly unharmful dietary habits.
SUPPLEMENTARY DATA
Supplementary data are available at Inflammatory Bowel Diseases online.
Supported by: This work was partially supported by National Institutes of Health Grants DK091222, DK055812, and DK042191 to F.C. and by a Career Development Award from the Crohn’s & Colitis Foundation to A.R.P. C.M. received support from grant 01DK082437, and K.P.N. from the Howard Hughes Medical Institute “Med into Grad” Initiative. Special acknowledgement goes to the CCF Imaging Core Leica SP5 Confocal Multi-photon Microscope Shared Instrument Grant 1S10RR026820-01, and the Mouse Models Core and the Histology/Imaging Core of the NIH P30 Silvio O. Conte Cleveland Digestive Disease Research Core Center (DK097948).
Author contributions: A.R.P. and F.C. designed the study. A.H., L.K., C.H., and P.M. assisted with laboratory and mouse experiments. S.K.D. and M.H.M. rederived and provided Helicobacter-negative mice. T.T.P. provided mice and samples from alternate mouse facility B. S.I. and A.R.P. developed and validated the “Parallel Lane Plating” culture strategy. A.R.P., A.H., G.M., M.R., and S.I. led microbiome and microbiology analysis. C.M. and K.P.N. performed qPCR and FISH assays. C.M.C. and L.K. conducted glucose tolerance test assays. F.C., T.T.P., S.I., and M.L. commented on and edited the manuscript. All authors read and approved the manuscript.
Conflicts of interest: All authors declare no competing interests.
ACKNOWLEDGMENTS
We thank John D. Ward and Lindsey N. Kaydo for their technical support and Dr. Wei Xin for the histological scoring of ileitis severity. ARP is an Assistant Professor of Medicine at CWRU School of Medicine. Metagenomic sequencing was conducted in the laboratory of Dr. Skip Virgin at Washington University, School of Medicine, St. Louis, MO.
Raw sequencing data files will be available upon request.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}