





Abstract
The gilthead sea bream, Sparus aurata, is a coastal, commercially important fish. Contrasting results concerning the genetic structure of the species at different geographic scales have been reported. Here, an investigation is made into the population genetic structure of S. aurata along the coast of Italy, using samples analysed previously and material from new sampling sites (12) and using different microsatellite loci (10). One sample from the eastern Atlantic and three temporal replicates from one site were also included. The presence of a weak (overall FST = 0.0072), but significant, genetic population subdivision was detected by F-statistics. Temporal replicates indicate genetic data consistency over time. Isolation by distance between the Atlantic and the coast of Italy is suggested by a Mantel test. The distributional pattern of genetic variance obtained by analysis of molecular variation reflects the geographic sampling areas, but is only partially congruent with the results obtained with fewer sites and loci. The dispersal of passive eggs/larvae by the main currents appears to contribute to shaping the gene flow. Given the intensity of sea bream aquaculture activities in Italy, the possibility that aquaculture may have partially contributed to the population genetic pattern detected cannot be excluded.Franchini, P., Sola, L., Crosetti, D., Milana, V., and Rossi, A. R. 2012. Low levels of population genetic structure in the gilthead sea bream, Sparus aurata, along the coast of Italy. – ICES Journal of Marine Science, 69: 41–50.
Introduction
The levels of genetic differentiation or similarity inferred by neutral molecular markers represent a basic source of information for reconstructing the evolutionary history of a species and for depicting the actual situation in terms of genetic structure and gene flow. In marine environments, one would expect little or no genetic subdivision of a species into stocks or populations (Waples, 1998), but considerable evidence of population subdivision in marine fish species has been reported recently, even on a limited spatial scale (Hauser and Carvalho, 2008, and references therein). Moreover, in many marine fish species, the limited biological knowledge and lack of tracking experiments make it difficult to identify the mechanisms involved in the reproductive isolation. Often, it is also difficult to evaluate the extent to which genetic differentiation reflects either the influence of gene flow (past and present), genetic drift, or the degree of local adaptation (Grosberg and Cunningham, 2001). In addition, the consequences of anthropogenic practices in cultured species (e.g. stock enhancement and accidental escapes and/or gamete release from floating cages) can potentially alter the local gene pool composition and further complicate data interpretation (Perez-Enriquez et al., 2001).
The gilthead sea bream, Sparus aurata, is a marine coastal teleost that inhabits the Mediterranean Sea and the northeastern Atlantic Ocean. It is a sedentary fish that migrates seasonally towards and from brackish coastal lagoons and estuaries for feeding and reproductive purposes, respectively. It is a highly fecund, protandrous hermaphrodite, characterized by mass-spawning behaviour and a lengthy planktonic larval stage. It is an important species for capture fisheries and a key species for large-scale Mediterranean aquaculture owing to its excellent flesh quality and rapid growth. Aquaculture production of the species in 2008 reached >132 000 t, 90% of which was in the Mediterranean Sea (FAO, 2008), and was mainly from intensive farming in hatcheries or sea cages.
As a consequence of the rise of aquaculture production, research studies on the genome of S. aurata increased exponentially in the past decade. Linkage (Franch et al., 2006) and physical (Sarropoulou et al., 2007) maps became available and, as a first step for whole-genome sequencing, the first comparative BAC map has been produced (Kuhl et al., 2011), covering ∼75% of the sea bream genome. However, basic information on the biology of the species is still limited, and its genetic structure has been investigated only recently. The application of different nuclear molecular markers (allozymes, random amplified polymorphic DNAs, and microsatellites) at different geographic scales has provided ambiguous results, indicating either a weak genetic structure in the Atlantic Ocean and Mediterranean Sea (Alarcon et al., 2004; De Innocentiis et al., 2004; Rossi et al., 2006), or a strong genetic subdivision at short distances along the Tunisian coasts (Ben Slimen et al., 2004), or between the opposite coasts of the western Mediterranean (Chaoui et al., 2009). The low variability of the hypervariable domain I of the mitochondrial control region is inadequate to detect genetic divergence among populations (Alarcon et al., 2004). Therefore, the genetic structure is yet to be defined.
The main goal of the present study was to further the analysis of the genetic structure of S. aurata along the coast of Italy. For this purpose, we increased the number of microsatellite loci to ten on the same individuals (from six Italian localities and one in the Atlantic Ocean), with four loci previously investigated by our group (De Innocentiis et al., 2004), and we extended the analysis on fish from six additional sampling sites. Further statistical analyses were carried out using Bayesian and coalescent approaches in addition to the classical methods (e.g. F-statistics) applied in the previous studies with the same markers (Alarcon et al., 2004; De Innocentiis et al., 2004; Chaoui et al., 2009). In all, the allele size variation of ten polymorphic microsatellite loci was analysed in 15 gilthead sea bream samples (628 fish) from 12 sites along the coast of Italy and from 1 locality in the Atlantic Ocean. Three temporal replicates from one site were also included in the analysis.
In this way, we aimed to verify (i) whether the results obtained on the same samples with more loci were congruent with previous observations (De Innocentiis et al., 2004) and, therefore, how the number of loci could bias the results obtained, (ii) whether an increase in the number of sampling sites better resolves the genetic structure, and (iii) how many of the data were consistent over time, and hence whether temporal replicates from the same site gave homogeneous results.
Material and methods
Sampling and microsatellite genotyping
In all, 628 adult gilthead sea bream, ∼16–22 cm total length (corresponding to one year of age, mostly males), were collected from 12 locations along the Italian coasts in the central Mediterranean Sea and from 1 location in the Atlantic Ocean (Table 1, Figure 1). The site Lesina (Ad-L) was sampled in 3 years (Ad-L1, 2001; Ad-L2, 2007; Ad-L3, 2008) to investigate the temporal stability of genetic structure. For each fish, the distal portion of the pelvic fin was clipped and stored in 95% ethanol.
Summary of the 15 samples (from one Atlantic Ocean site and from 12 sites along the Italian coast and the Mediterranean Sea, with two additional temporal replicates from Lesina) of the gilthead sea bream (S. aurata).
| Sample ID | Sea/coast | Locality | Geographic coordinates | Sampling date | Number of individuals |
|---|---|---|---|---|---|
| At-A | Atlantic Ocean | Gulf of Cadice | 37°07′N 06°59′E | May 2001 | 48 |
| Mediterranean Sea | |||||
| Ad-U | Adriatic Sea | Umago | 45°25′N 13°30′E | January 2004 | 48 |
| Ad-L1 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2001 | 48 |
| Ad-L2 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2007 | 39 |
| Ad-L3 | Adriatic Sea | Lesina | 41°54′N 15°26′E | December 2008 | 31 |
| Ad-B | Adriatic Sea | Bisceglie | 41°15′N 16°32′E | June 2008 | 62 |
| Si-T | Sicily Channel | Trapani | 38°01′N 12°29′E | May 2008 | 42 |
| Ty-B | Tyrrhenian Sea | Bacoli | 40°48′N 14°06′E | May 1999 | 30 |
| Ty-S | Tyrrhenian Sea | Sabaudia | 41°17′N 13°00′E | October 2002 | 32 |
| Ty-Z | Tyrrhenian Sea | Anzio | 41°28′N 12°35′E | April 2008 | 48 |
| Li-G | Ligurian Sea | Genova | 44°22′N 08°53′E | May 2005 | 40 |
| Ty-T | Sardinian Coast | Tortolì | 39°54′N 09°42′E | November 2002 | 48 |
| Sa-M | Sardinian Coast | Muravera | 39°24′N 09°38′E | May 2004 | 32 |
| SC-G | Sardinian Coast | Santa Gilla | 39°11′N 09°05′E | May 1999 | 32 |
| SS-C | Sardinian Coast | Cabras | 39°49′N 08°30′E | November 2002 | 48 |
| Sample ID | Sea/coast | Locality | Geographic coordinates | Sampling date | Number of individuals |
|---|---|---|---|---|---|
| At-A | Atlantic Ocean | Gulf of Cadice | 37°07′N 06°59′E | May 2001 | 48 |
| Mediterranean Sea | |||||
| Ad-U | Adriatic Sea | Umago | 45°25′N 13°30′E | January 2004 | 48 |
| Ad-L1 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2001 | 48 |
| Ad-L2 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2007 | 39 |
| Ad-L3 | Adriatic Sea | Lesina | 41°54′N 15°26′E | December 2008 | 31 |
| Ad-B | Adriatic Sea | Bisceglie | 41°15′N 16°32′E | June 2008 | 62 |
| Si-T | Sicily Channel | Trapani | 38°01′N 12°29′E | May 2008 | 42 |
| Ty-B | Tyrrhenian Sea | Bacoli | 40°48′N 14°06′E | May 1999 | 30 |
| Ty-S | Tyrrhenian Sea | Sabaudia | 41°17′N 13°00′E | October 2002 | 32 |
| Ty-Z | Tyrrhenian Sea | Anzio | 41°28′N 12°35′E | April 2008 | 48 |
| Li-G | Ligurian Sea | Genova | 44°22′N 08°53′E | May 2005 | 40 |
| Ty-T | Sardinian Coast | Tortolì | 39°54′N 09°42′E | November 2002 | 48 |
| Sa-M | Sardinian Coast | Muravera | 39°24′N 09°38′E | May 2004 | 32 |
| SC-G | Sardinian Coast | Santa Gilla | 39°11′N 09°05′E | May 1999 | 32 |
| SS-C | Sardinian Coast | Cabras | 39°49′N 08°30′E | November 2002 | 48 |
Summary of the 15 samples (from one Atlantic Ocean site and from 12 sites along the Italian coast and the Mediterranean Sea, with two additional temporal replicates from Lesina) of the gilthead sea bream (S. aurata).
| Sample ID | Sea/coast | Locality | Geographic coordinates | Sampling date | Number of individuals |
|---|---|---|---|---|---|
| At-A | Atlantic Ocean | Gulf of Cadice | 37°07′N 06°59′E | May 2001 | 48 |
| Mediterranean Sea | |||||
| Ad-U | Adriatic Sea | Umago | 45°25′N 13°30′E | January 2004 | 48 |
| Ad-L1 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2001 | 48 |
| Ad-L2 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2007 | 39 |
| Ad-L3 | Adriatic Sea | Lesina | 41°54′N 15°26′E | December 2008 | 31 |
| Ad-B | Adriatic Sea | Bisceglie | 41°15′N 16°32′E | June 2008 | 62 |
| Si-T | Sicily Channel | Trapani | 38°01′N 12°29′E | May 2008 | 42 |
| Ty-B | Tyrrhenian Sea | Bacoli | 40°48′N 14°06′E | May 1999 | 30 |
| Ty-S | Tyrrhenian Sea | Sabaudia | 41°17′N 13°00′E | October 2002 | 32 |
| Ty-Z | Tyrrhenian Sea | Anzio | 41°28′N 12°35′E | April 2008 | 48 |
| Li-G | Ligurian Sea | Genova | 44°22′N 08°53′E | May 2005 | 40 |
| Ty-T | Sardinian Coast | Tortolì | 39°54′N 09°42′E | November 2002 | 48 |
| Sa-M | Sardinian Coast | Muravera | 39°24′N 09°38′E | May 2004 | 32 |
| SC-G | Sardinian Coast | Santa Gilla | 39°11′N 09°05′E | May 1999 | 32 |
| SS-C | Sardinian Coast | Cabras | 39°49′N 08°30′E | November 2002 | 48 |
| Sample ID | Sea/coast | Locality | Geographic coordinates | Sampling date | Number of individuals |
|---|---|---|---|---|---|
| At-A | Atlantic Ocean | Gulf of Cadice | 37°07′N 06°59′E | May 2001 | 48 |
| Mediterranean Sea | |||||
| Ad-U | Adriatic Sea | Umago | 45°25′N 13°30′E | January 2004 | 48 |
| Ad-L1 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2001 | 48 |
| Ad-L2 | Adriatic Sea | Lesina | 41°54′N 15°26′E | November 2007 | 39 |
| Ad-L3 | Adriatic Sea | Lesina | 41°54′N 15°26′E | December 2008 | 31 |
| Ad-B | Adriatic Sea | Bisceglie | 41°15′N 16°32′E | June 2008 | 62 |
| Si-T | Sicily Channel | Trapani | 38°01′N 12°29′E | May 2008 | 42 |
| Ty-B | Tyrrhenian Sea | Bacoli | 40°48′N 14°06′E | May 1999 | 30 |
| Ty-S | Tyrrhenian Sea | Sabaudia | 41°17′N 13°00′E | October 2002 | 32 |
| Ty-Z | Tyrrhenian Sea | Anzio | 41°28′N 12°35′E | April 2008 | 48 |
| Li-G | Ligurian Sea | Genova | 44°22′N 08°53′E | May 2005 | 40 |
| Ty-T | Sardinian Coast | Tortolì | 39°54′N 09°42′E | November 2002 | 48 |
| Sa-M | Sardinian Coast | Muravera | 39°24′N 09°38′E | May 2004 | 32 |
| SC-G | Sardinian Coast | Santa Gilla | 39°11′N 09°05′E | May 1999 | 32 |
| SS-C | Sardinian Coast | Cabras | 39°49′N 08°30′E | November 2002 | 48 |
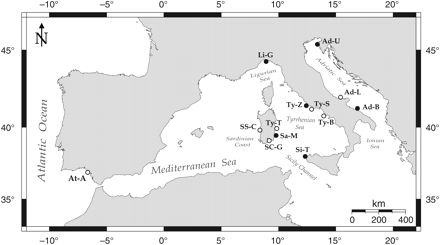
Sampling localities of S. aurata along the Italian coast, in the Mediterranean Sea, and in the Atlantic Ocean. Open circles, sites also examined by De Innocentiis et al. (2004); dots, additional sites examined in this study. Sampling localities are abbreviated as in Table 1.
Genomic DNA was obtained from fin clips using the salting-out extraction method described by Aljanabi and Martinez (1997), then used as a template in a polymerase chain reaction (PCR) for ten microsatellite loci: SaGT1, SaGT26, SaGT31, and SaGT32 (Batargias et al., 1999; previously scored in some populations by De Innocentiis et al., 2004); SauG46, SauD69, and SauK140 (Launey et al., 2003); and Pb-OVI-B2, Pb-OVI-D102, and Pb-OVI-D106 (Piñera et al., 2006). The PCRs were carried out in a thermocycler T1 (Biometra, Göttingen, Germany), applying the amplification conditions reported in the original studies for each primer pair. The forward primers for each locus were labelled with 5′-fluorescent dye (6-FAM, HEX, or TAMRA), and the amplified products were processed for polymorphism detection on an ABI 3730 automated sequencer. Allele sizes were screened using peak scanner v1.0 software (Applied Biosystems) by comparison with a genescan 500 ROX size standard. All steps, from amplification to genotyping, were repeated for some 4% of the total samples (25 fish) to verify repeatability in microsatellite scoring.
micro-checker v2.2.3 was used to detect null alleles and scoring errors (Van Oosterhout et al., 2004).
Genetic variability and differentiation
Deviation from Hardy–Weinberg (HW) expectations and linkage disequilibrium (LD) were tested for each locus and for each sample site, including the temporal replicates of Lesina (Ad-L), using genepop v3.4 (Raymond and Rousset, 1995). The significance levels of both analyses were calculated with the Markov chain Monte Carlo (MCMC) method using 10 000 permutations, 5000 dememorization steps, and 500 batches. Bonferroni correction (Rice, 1989) was used to determine the statistical significance for the HW tests, and a false discovery rate (FDR) approach was applied for the LD significance because of the large number of multiple tests involved (Benjamini and Hochberg, 1995).
The simulation-based program powsim v1.2 (Ryman and Palm, 2006) was used to estimate the statistical power of the dataset in detecting significant genetic differentiation. The use of powsim is recommended for population genetics studies when evaluating the hypothesis of genetic homogeneity. It can assess genetic homogeneity through optional combinations of the number of samples, sample sizes, and the number of loci, alleles, and allele frequencies for any hypothetical degree of true differentiation (quantified as FST). The allele frequency of the 15 samples examined (the 13 sampling sites and the temporal replicates from Lesina) was tested for genetic homogeneity for each locus separately and for all ten loci. The statistical power of the dataset was evaluated using Chi-squared and Fisher's exact tests. The simulations were performed using different combinations of Ne (effective population size) and t (time of divergence), leading to FST values in the range 0.005–0.010. Values of FST and Ne were selected according to real values detected in this study (see Results section). Estimates of the statistical α (type I) error were generated using samples drawn directly from the base population, omitting the drift steps (t = 0) leading to the absence of differentiation (FST = 0). The estimate of power was reported as the proportion of significant outcomes (p<0.05) after 1000 replicates.
The presence of the loci under selection was evaluated with a global outlier test adopting the coalescent approach developed by Beaumont and Nichols (1996), as implemented in the software lositan (Antao et al., 2008). Infinite alleles and stepwise mutation models were used. Additionally, the hierarchical method of Excoffier et al. (2009), based on the approach of Beaumont and Nichols (1996), was applied using the software arlequin v3.5 (Excoffier and Lischer, 2010). The latter method takes into account the hierarchical structure of the sample, wherein dispersal is likely to be more prevalent within the same geographic basins than between them [see analysis of molecular variation (AMOVA) structure hypotheses below]. The lositan and arlequin analyses were performed by running 50 000 simulations.
The interpopulation level of genetic differentiation was calculated through F-statistics (Weir and Cockerham, 1984) using fstat v2.9.3.2 (Goudet, 2001). The significance levels of FST overall loci and locus by locus for all populations were calculated with a randomization test, permuting alleles (Goudet et al., 1996). The significance of pairwise FST values was calculated with the MCMC method using 10 000 permutations, 5000 dememorization steps, and 500 batches. All analyses were carried out for each of the 15 samples examined, considering the three temporal replicates of Lesina as distinct. At the Lesina site, the variance-effective population size (NeV) from the three samples collected in 2001, 2007, and 2008 was estimated with the temporal method of Waples (1989) implemented in the program neestimator v1.3 (Peel et al., 2004) and with the pseudo-maximum-likelihood algorithm implemented in the program mlne v2.3 (Wang, 2001). As the three temporal samples consisted of fish ∼1-year old, the generation time (g) for the 2001, 2007, and 2008 replicates was set at 0, 6, and 7, respectively.
A hierarchical AMOVA, as implemented in arlequin, was performed to quantify the different genetic variance components (“among groups”, “among populations within groups”, “within populations”). In detail, the AMOVA was used to test the repartition of genetic variation in three different structure hypotheses: (i) the panmixia hypothesis, with a single group constituted by all 13 sampling sites; (ii) the 13 samples pooled in six groups, according to their geographic (sea/coast) origin (i.e. Atlantic Ocean, Adriatic Sea, Sicily Channel, eastern Tyrrhenian Sea, Ligurian Sea, and Sardinian coast); and (iii) the 13 samples pooled according to the five genetic assemblages proposed for gilthead sea bream by De Innocentiis et al. (2004). For this last hypothesis, two procedures were followed. In the first, we included only samples from the seven sites previously analysed by De Innocentiis et al. (2004). In the second, the complete sample set was included, considering samples from the Sicily Channel (Si-T) and from the Ligurian Sea (Li-G) as separate assemblages. The correlation between geographic and genetic distances was analysed through a Mantel test carried out by the program ntsyspc v2.20 (Rohlf, 2005). The value FST/(1 − FST) was compared with the natural logarithm of coastline distances.
Cluster analyses
Two programs that implement the Bayesian algorithms were used to infer the population structure in the sample; neither needs an a priori assumption of population subdivision to infer genetic structure in a genotype dataset: structure v2.3 (Pritchard et al., 2000) and structurama (Huelsenbeck and Andolfatto, 2007). structure was used to infer individual admixture and the number of populations (K) with the highest posterior probability. The “Admixture Model” and “Allele Frequencies Correlated” were selected because these parameters are recommended for detecting genetic structure when closely related populations are involved (Falush et al., 2003). Using the MCMC method, 20 replicates for each set value of K from 1 to 13 (the number of collection localities) with 1.5 × 106 iterations and a burn-in period of 1 × 105 iterations were performed. structurama, differently from structure, calculates the posterior probability distribution of K when it is treated as a random variable. A Bayesian MCMC algorithm that implements a Dirichlet process was assumed, and the analysis was carried out with 1.5 × 106 generations.
Gene flow
Gene flow was estimated with a coalescent method implemented in the migrate v3.03 program (Beerli and Felsenstein, 2001). The 13 samples were pooled according to structure (ii) tested by AMOVA (see above), i.e. according to the six different geographic sea/coasts to which they belonged. For each sample, the population size parameter Θ (Θ = 4Neµ, where Ne is the effective population size and µ the mutation rate) and the pairwise migration rates M (M = m/µ, where m is the migration rate per generation) were estimated using FST estimates and a UPGMA tree as starting parameters. The program conducted the analysis through a long chain of 1 × 106 recorded genealogies at a sampling increment of 50 iterations, after discarding the first 10 000 as burn-in for each locus. An adaptive heating scheme using four simultaneous Markov chains was implemented to increase the search efficiency.
Results
The genotypes for the ten microsatellite loci were determined for the 628 gilthead sea bream. The 25 fish amplified and scored twice produced identical results in each trial. It was not possible to score 2.5% of the genotypes unambiguously. The allele numbers (Supplementary Table S1) in all samples examined, the 13 sampling sites and the two additional temporal replicates of Lesina, ranged from 7 alleles for locus SaGT31 to 37 alleles for loci Pb-Ovi-D102 and SaGT1. A low variation in allele numbers among samples was observed (an average of 11.9 alleles for SC-G to 16.5 for SS-C), as also indicated by the average allele richness per sample (from 10.4 for SC-G to 11.5 for Ty-T).
After Bonferroni correction, only 3 of the 150 locus/population combinations showed a significant deviation from H–W expectation, all characterized by a heterozygote deficit (locus/population: Pb-Ovi-D106/Ad-B, SauD69/SC-G, and SaGT32/Si-T). LD was negligible for most samples except for population Ad-B (Adriatic Sea, Bisceglie). Indeed, after the application of the FDR approach, 19 of 45 pairs of loci showed genotyping linkage in this population. micro-checker did not indicate the presence of null alleles or scoring errors.
The powsim analysis revealed that the combination of microsatellite loci and sample sizes rendered a statistical power sufficient to detect a low level of differentiation. In fact, more than 92% (χ2) and 84% (Fisher's) of the tests where the Ne:t combination led to FST = 0.0010 and 100% (both χ2 and Fisher's) of the tests where Ne:t led to a FST value of ≥0.0025 were statistically significant (Table 2). The estimate of the statistical α (type I) error varied from 0.042 with the Fisher's exact test to 0.057 with a χ2 test.
POWSIM simulations to assess the statistical power of microsatellite loci to differentiate populations at varying levels of expected FST, showing the results of both χ2 and Fisher's exact tests for the proportion of simulations out of 1000 significant with a critical value of 0.05.
| Expected FST | Ne | t | χ2 test (%) | Fisher's test (%) |
|---|---|---|---|---|
| 0.0005 | 1 000/5 000/10 000 | 1/5/10 | 46.2/49.9/51.1 | 42.4/46.1/46.7 |
| 0.0010 | 1 000/5 000/10 000 | 2/10/20 | 92.3/94.1/96.0 | 86.8/84.7/91.2 |
| 0.0025 | 1 000/5 000/10 000 | 5/25/50 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0050 | 1 000/5 000/10 000 | 10/50/100 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0100 | 1 000/5 000/10 000 | 20/100/200 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| Expected FST | Ne | t | χ2 test (%) | Fisher's test (%) |
|---|---|---|---|---|
| 0.0005 | 1 000/5 000/10 000 | 1/5/10 | 46.2/49.9/51.1 | 42.4/46.1/46.7 |
| 0.0010 | 1 000/5 000/10 000 | 2/10/20 | 92.3/94.1/96.0 | 86.8/84.7/91.2 |
| 0.0025 | 1 000/5 000/10 000 | 5/25/50 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0050 | 1 000/5 000/10 000 | 10/50/100 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0100 | 1 000/5 000/10 000 | 20/100/200 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
The values of Ne used for the tests are based on the inferred Ne from this study (see Results section).
POWSIM simulations to assess the statistical power of microsatellite loci to differentiate populations at varying levels of expected FST, showing the results of both χ2 and Fisher's exact tests for the proportion of simulations out of 1000 significant with a critical value of 0.05.
| Expected FST | Ne | t | χ2 test (%) | Fisher's test (%) |
|---|---|---|---|---|
| 0.0005 | 1 000/5 000/10 000 | 1/5/10 | 46.2/49.9/51.1 | 42.4/46.1/46.7 |
| 0.0010 | 1 000/5 000/10 000 | 2/10/20 | 92.3/94.1/96.0 | 86.8/84.7/91.2 |
| 0.0025 | 1 000/5 000/10 000 | 5/25/50 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0050 | 1 000/5 000/10 000 | 10/50/100 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0100 | 1 000/5 000/10 000 | 20/100/200 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| Expected FST | Ne | t | χ2 test (%) | Fisher's test (%) |
|---|---|---|---|---|
| 0.0005 | 1 000/5 000/10 000 | 1/5/10 | 46.2/49.9/51.1 | 42.4/46.1/46.7 |
| 0.0010 | 1 000/5 000/10 000 | 2/10/20 | 92.3/94.1/96.0 | 86.8/84.7/91.2 |
| 0.0025 | 1 000/5 000/10 000 | 5/25/50 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0050 | 1 000/5 000/10 000 | 10/50/100 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
| 0.0100 | 1 000/5 000/10 000 | 20/100/200 | 100.0/100.0/100.0 | 100.0/100.0/100.0 |
The values of Ne used for the tests are based on the inferred Ne from this study (see Results section).
Locus-by-locus FST values overall populations ranged from 0.002 for locus SaGT31 to 0.038 for locus Pb-OVI-B2. The FST values were significant for all loci (Table 3). The global outlier test using all 15 populations detected directional selection at two loci (Pb-OVI-B2, p<0.001; SaGT26, p<0.05) using lositan under both infinite alleles and stepwise mutation models. arlequin identified only locus Pb-OVI-B2 as an outlier (p<0.001). To minimize the detection of false positives, we therefore considered only locus Pb-OVI-B2 to be under directional selection, because it was detected by both methods with high statistical support (p<0.001). For this reason, this locus was eliminated from subsequent analyses.
Locus-by-locus FST values for overall populations.
| Locus | FST | Outlier(L) | Outlier(A) |
|---|---|---|---|
| Pb-OVI-D106 | 0.013*** | NS | NS |
| SaUK140 | 0.003** | NS | NS |
| SaUG46 | 0.003** | NS | NS |
| Pb-OVI-D102 | 0.003* | NS | NS |
| Pb-OVI-B2 | 0.038*** | *** | *** |
| SaUD69 | 0.006*** | NS | NS |
| SaGT1 | 0.003** | NS | NS |
| SaGT26 | 0.021*** | * | NS |
| SaGT31 | 0.002* | NS | NS |
| SaGT32 | 0.007*** | NS | NS |
| Locus | FST | Outlier(L) | Outlier(A) |
|---|---|---|---|
| Pb-OVI-D106 | 0.013*** | NS | NS |
| SaUK140 | 0.003** | NS | NS |
| SaUG46 | 0.003** | NS | NS |
| Pb-OVI-D102 | 0.003* | NS | NS |
| Pb-OVI-B2 | 0.038*** | *** | *** |
| SaUD69 | 0.006*** | NS | NS |
| SaGT1 | 0.003** | NS | NS |
| SaGT26 | 0.021*** | * | NS |
| SaGT31 | 0.002* | NS | NS |
| SaGT32 | 0.007*** | NS | NS |
The columns Outlier(L) and Outlier(A) indicate the results of the tests for the detection of outlier loci performed by lositan and arlequin, respectively. NS (not significant) refers to loci where neutrality was found. Asterisks indicate the significance of the estimators.
*p<0.05.
**p<0.01.
***p<0.001.
Locus-by-locus FST values for overall populations.
| Locus | FST | Outlier(L) | Outlier(A) |
|---|---|---|---|
| Pb-OVI-D106 | 0.013*** | NS | NS |
| SaUK140 | 0.003** | NS | NS |
| SaUG46 | 0.003** | NS | NS |
| Pb-OVI-D102 | 0.003* | NS | NS |
| Pb-OVI-B2 | 0.038*** | *** | *** |
| SaUD69 | 0.006*** | NS | NS |
| SaGT1 | 0.003** | NS | NS |
| SaGT26 | 0.021*** | * | NS |
| SaGT31 | 0.002* | NS | NS |
| SaGT32 | 0.007*** | NS | NS |
| Locus | FST | Outlier(L) | Outlier(A) |
|---|---|---|---|
| Pb-OVI-D106 | 0.013*** | NS | NS |
| SaUK140 | 0.003** | NS | NS |
| SaUG46 | 0.003** | NS | NS |
| Pb-OVI-D102 | 0.003* | NS | NS |
| Pb-OVI-B2 | 0.038*** | *** | *** |
| SaUD69 | 0.006*** | NS | NS |
| SaGT1 | 0.003** | NS | NS |
| SaGT26 | 0.021*** | * | NS |
| SaGT31 | 0.002* | NS | NS |
| SaGT32 | 0.007*** | NS | NS |
The columns Outlier(L) and Outlier(A) indicate the results of the tests for the detection of outlier loci performed by lositan and arlequin, respectively. NS (not significant) refers to loci where neutrality was found. Asterisks indicate the significance of the estimators.
*p<0.05.
**p<0.01.
***p<0.001.
Multilocus FST estimates gave values of 0.0072 for all samples and of 0.0065 when only Mediterranean sites were included. When population pairs were analysed (Table 4), significant FST values were obtained in 32 of the 105 pairwise comparisons (Bonferroni correction applied). Most of the significant comparisons involved the Atlantic, At-A, and the Sicilian, Si-T, samples.
FST values for each pair of samples, with significant differences shown emboldened (105 multiple tests, corrected threshold via the Bonferroni procedure: p<0.0004).
| At-A | Ad-U | Ad-L 1 | Ad-L 2 | Ad-L 3 | Ad-B | Si-T | Ty-B | Ty-S | Ty-Z | Li-G | Ty-T | Sa-M | SC-G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ad-U | 0.0144 | – | ||||||||||||
| Ad-L1 | 0.0128 | 0.0055 | – | |||||||||||
| Ad-L2 | 0.0079 | 0.0025 | 0.0072 | – | ||||||||||
| Ad-L3 | 0.0092 | 0.0134 | 0.0067 | 0.0075 | – | |||||||||
| Ad-B | 0.0106 | 0.0069 | 0.0074 | 0.0054 | 0.0072 | – | ||||||||
| Si-T | 0.0120 | 0.0109 | 0.0128 | 0.0081 | 0.0091 | 0.0064 | – | |||||||
| Ty-B | 0.0164 | 0.0005 | 0.0062 | 0.0027 | 0.0100 | 0.0034 | 0.0086 | – | ||||||
| Ty-S | 0.0119 | 0.0027 | 0.0094 | 0.0032 | 0.0152 | 0.0029 | 0.0099 | −0.0012 | – | |||||
| Ty-Z | 0.0115 | 0.0050 | 0.0079 | 0.0038 | 0.0108 | 0.0052 | 0.0044 | 0.0024 | 0.0028 | – | ||||
| Li-G | 0.0129 | 0.0030 | 0.0072 | 0.0043 | 0.0139 | 0.0050 | 0.0097 | 0.0000 | −0.0022 | 0.0030 | – | |||
| Ty-T | 0.0088 | 0.0047 | 0.0080 | 0.0014 | 0.0086 | 0.0017 | 0.0073 | 0.0029 | 0.0007 | 0.0035 | 0.0030 | – | ||
| Sa-M | 0.0079 | 0.0104 | 0.0165 | 0.0040 | 0.0118 | 0.0042 | 0.0051 | 0.0056 | 0.0015 | 0.0046 | 0.0034 | 0.0034 | – | |
| SC-G | 0.0207 | 0.0139 | 0.0081 | 0.0139 | 0.0201 | 0.0108 | 0.0104 | 0.0101 | 0.0118 | 0.0106 | 0.0082 | 0.0099 | 0.0119 | – |
| SS-C | 0.0117 | 0.0047 | 0.0114 | 0.0033 | 0.0114 | 0.0055 | 0.0051 | 0.0053 | 0.0033 | 0.0025 | 0.0063 | 0.0038 | 0.0012 | 0.0162 |
| At-A | Ad-U | Ad-L 1 | Ad-L 2 | Ad-L 3 | Ad-B | Si-T | Ty-B | Ty-S | Ty-Z | Li-G | Ty-T | Sa-M | SC-G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ad-U | 0.0144 | – | ||||||||||||
| Ad-L1 | 0.0128 | 0.0055 | – | |||||||||||
| Ad-L2 | 0.0079 | 0.0025 | 0.0072 | – | ||||||||||
| Ad-L3 | 0.0092 | 0.0134 | 0.0067 | 0.0075 | – | |||||||||
| Ad-B | 0.0106 | 0.0069 | 0.0074 | 0.0054 | 0.0072 | – | ||||||||
| Si-T | 0.0120 | 0.0109 | 0.0128 | 0.0081 | 0.0091 | 0.0064 | – | |||||||
| Ty-B | 0.0164 | 0.0005 | 0.0062 | 0.0027 | 0.0100 | 0.0034 | 0.0086 | – | ||||||
| Ty-S | 0.0119 | 0.0027 | 0.0094 | 0.0032 | 0.0152 | 0.0029 | 0.0099 | −0.0012 | – | |||||
| Ty-Z | 0.0115 | 0.0050 | 0.0079 | 0.0038 | 0.0108 | 0.0052 | 0.0044 | 0.0024 | 0.0028 | – | ||||
| Li-G | 0.0129 | 0.0030 | 0.0072 | 0.0043 | 0.0139 | 0.0050 | 0.0097 | 0.0000 | −0.0022 | 0.0030 | – | |||
| Ty-T | 0.0088 | 0.0047 | 0.0080 | 0.0014 | 0.0086 | 0.0017 | 0.0073 | 0.0029 | 0.0007 | 0.0035 | 0.0030 | – | ||
| Sa-M | 0.0079 | 0.0104 | 0.0165 | 0.0040 | 0.0118 | 0.0042 | 0.0051 | 0.0056 | 0.0015 | 0.0046 | 0.0034 | 0.0034 | – | |
| SC-G | 0.0207 | 0.0139 | 0.0081 | 0.0139 | 0.0201 | 0.0108 | 0.0104 | 0.0101 | 0.0118 | 0.0106 | 0.0082 | 0.0099 | 0.0119 | – |
| SS-C | 0.0117 | 0.0047 | 0.0114 | 0.0033 | 0.0114 | 0.0055 | 0.0051 | 0.0053 | 0.0033 | 0.0025 | 0.0063 | 0.0038 | 0.0012 | 0.0162 |
FST values for each pair of samples, with significant differences shown emboldened (105 multiple tests, corrected threshold via the Bonferroni procedure: p<0.0004).
| At-A | Ad-U | Ad-L 1 | Ad-L 2 | Ad-L 3 | Ad-B | Si-T | Ty-B | Ty-S | Ty-Z | Li-G | Ty-T | Sa-M | SC-G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ad-U | 0.0144 | – | ||||||||||||
| Ad-L1 | 0.0128 | 0.0055 | – | |||||||||||
| Ad-L2 | 0.0079 | 0.0025 | 0.0072 | – | ||||||||||
| Ad-L3 | 0.0092 | 0.0134 | 0.0067 | 0.0075 | – | |||||||||
| Ad-B | 0.0106 | 0.0069 | 0.0074 | 0.0054 | 0.0072 | – | ||||||||
| Si-T | 0.0120 | 0.0109 | 0.0128 | 0.0081 | 0.0091 | 0.0064 | – | |||||||
| Ty-B | 0.0164 | 0.0005 | 0.0062 | 0.0027 | 0.0100 | 0.0034 | 0.0086 | – | ||||||
| Ty-S | 0.0119 | 0.0027 | 0.0094 | 0.0032 | 0.0152 | 0.0029 | 0.0099 | −0.0012 | – | |||||
| Ty-Z | 0.0115 | 0.0050 | 0.0079 | 0.0038 | 0.0108 | 0.0052 | 0.0044 | 0.0024 | 0.0028 | – | ||||
| Li-G | 0.0129 | 0.0030 | 0.0072 | 0.0043 | 0.0139 | 0.0050 | 0.0097 | 0.0000 | −0.0022 | 0.0030 | – | |||
| Ty-T | 0.0088 | 0.0047 | 0.0080 | 0.0014 | 0.0086 | 0.0017 | 0.0073 | 0.0029 | 0.0007 | 0.0035 | 0.0030 | – | ||
| Sa-M | 0.0079 | 0.0104 | 0.0165 | 0.0040 | 0.0118 | 0.0042 | 0.0051 | 0.0056 | 0.0015 | 0.0046 | 0.0034 | 0.0034 | – | |
| SC-G | 0.0207 | 0.0139 | 0.0081 | 0.0139 | 0.0201 | 0.0108 | 0.0104 | 0.0101 | 0.0118 | 0.0106 | 0.0082 | 0.0099 | 0.0119 | – |
| SS-C | 0.0117 | 0.0047 | 0.0114 | 0.0033 | 0.0114 | 0.0055 | 0.0051 | 0.0053 | 0.0033 | 0.0025 | 0.0063 | 0.0038 | 0.0012 | 0.0162 |
| At-A | Ad-U | Ad-L 1 | Ad-L 2 | Ad-L 3 | Ad-B | Si-T | Ty-B | Ty-S | Ty-Z | Li-G | Ty-T | Sa-M | SC-G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ad-U | 0.0144 | – | ||||||||||||
| Ad-L1 | 0.0128 | 0.0055 | – | |||||||||||
| Ad-L2 | 0.0079 | 0.0025 | 0.0072 | – | ||||||||||
| Ad-L3 | 0.0092 | 0.0134 | 0.0067 | 0.0075 | – | |||||||||
| Ad-B | 0.0106 | 0.0069 | 0.0074 | 0.0054 | 0.0072 | – | ||||||||
| Si-T | 0.0120 | 0.0109 | 0.0128 | 0.0081 | 0.0091 | 0.0064 | – | |||||||
| Ty-B | 0.0164 | 0.0005 | 0.0062 | 0.0027 | 0.0100 | 0.0034 | 0.0086 | – | ||||||
| Ty-S | 0.0119 | 0.0027 | 0.0094 | 0.0032 | 0.0152 | 0.0029 | 0.0099 | −0.0012 | – | |||||
| Ty-Z | 0.0115 | 0.0050 | 0.0079 | 0.0038 | 0.0108 | 0.0052 | 0.0044 | 0.0024 | 0.0028 | – | ||||
| Li-G | 0.0129 | 0.0030 | 0.0072 | 0.0043 | 0.0139 | 0.0050 | 0.0097 | 0.0000 | −0.0022 | 0.0030 | – | |||
| Ty-T | 0.0088 | 0.0047 | 0.0080 | 0.0014 | 0.0086 | 0.0017 | 0.0073 | 0.0029 | 0.0007 | 0.0035 | 0.0030 | – | ||
| Sa-M | 0.0079 | 0.0104 | 0.0165 | 0.0040 | 0.0118 | 0.0042 | 0.0051 | 0.0056 | 0.0015 | 0.0046 | 0.0034 | 0.0034 | – | |
| SC-G | 0.0207 | 0.0139 | 0.0081 | 0.0139 | 0.0201 | 0.0108 | 0.0104 | 0.0101 | 0.0118 | 0.0106 | 0.0082 | 0.0099 | 0.0119 | – |
| SS-C | 0.0117 | 0.0047 | 0.0114 | 0.0033 | 0.0114 | 0.0055 | 0.0051 | 0.0053 | 0.0033 | 0.0025 | 0.0063 | 0.0038 | 0.0012 | 0.0162 |
Analyses of the three temporal replicates collected in Lesina (Ad-L1, Ad-L2, and Ad-L3) revealed homogeneity among them. Indeed, expected and observed heterozygosity, the absence of LD at all loci combinations, and non-significant pairwise FST values were observed. Moreover, homogeneity in effective size was detected in pairwise comparisons (Table 5). The Waples moment method (Waples, 1989) gave a wider range of mean Ne estimates than the mlne method (Wang, 2001), but the two methods yielded overlapping 95% confidence intervals (CIs).
Temporal estimates of Ne calculated from the microsatellite loci for both the Waples moment and mlne methods, with associated 95% confidence intervals.
| Temporal comparison | Years | Waples moment Ne | −95% | +95% | MLNE Ne | −95% | +95% |
|---|---|---|---|---|---|---|---|
| Ad-L1 vs. Ad-L2 | 2001–2007 | 374.3 | 184.0 | 1454.8 | 243.3 | 53.9 | 10 000 |
| Ad-L1 vs. Ad-L3 | 2001–2008 | 652.1 | 229.8 | ∞ | 202.4 | 44.1 | 10 000 |
| Ad-L2 vs. Ad-L3 | 2007/2008 | 104.9 | 27.6 | 1349.2 | 177.1 | 33.9 | 10 000 |
| Temporal comparison | Years | Waples moment Ne | −95% | +95% | MLNE Ne | −95% | +95% |
|---|---|---|---|---|---|---|---|
| Ad-L1 vs. Ad-L2 | 2001–2007 | 374.3 | 184.0 | 1454.8 | 243.3 | 53.9 | 10 000 |
| Ad-L1 vs. Ad-L3 | 2001–2008 | 652.1 | 229.8 | ∞ | 202.4 | 44.1 | 10 000 |
| Ad-L2 vs. Ad-L3 | 2007/2008 | 104.9 | 27.6 | 1349.2 | 177.1 | 33.9 | 10 000 |
Temporal estimates of Ne calculated from the microsatellite loci for both the Waples moment and mlne methods, with associated 95% confidence intervals.
| Temporal comparison | Years | Waples moment Ne | −95% | +95% | MLNE Ne | −95% | +95% |
|---|---|---|---|---|---|---|---|
| Ad-L1 vs. Ad-L2 | 2001–2007 | 374.3 | 184.0 | 1454.8 | 243.3 | 53.9 | 10 000 |
| Ad-L1 vs. Ad-L3 | 2001–2008 | 652.1 | 229.8 | ∞ | 202.4 | 44.1 | 10 000 |
| Ad-L2 vs. Ad-L3 | 2007/2008 | 104.9 | 27.6 | 1349.2 | 177.1 | 33.9 | 10 000 |
| Temporal comparison | Years | Waples moment Ne | −95% | +95% | MLNE Ne | −95% | +95% |
|---|---|---|---|---|---|---|---|
| Ad-L1 vs. Ad-L2 | 2001–2007 | 374.3 | 184.0 | 1454.8 | 243.3 | 53.9 | 10 000 |
| Ad-L1 vs. Ad-L3 | 2001–2008 | 652.1 | 229.8 | ∞ | 202.4 | 44.1 | 10 000 |
| Ad-L2 vs. Ad-L3 | 2007/2008 | 104.9 | 27.6 | 1349.2 | 177.1 | 33.9 | 10 000 |
In light of these results, to have data comparable with those from previous investigations (De Innocentiis et al., 2004) and to ensure sample-size homogeneity, only the temporal replicate Ad-L1, the same used by De Innocentiis et al. (2004), was included in further analyses of genetic structure and gene flow.
The AMOVA (Table 6) showed significant genetic differentiation within the whole sample set when the panmixia hypothesis was tested, although most genetic variation was explained by the “within populations” hierarchical level (99.59%). When genetic structuring of geographic source areas was tested (six groups: Atlantic Ocean, Adriatic Sea, Sicily Channel, eastern Tyrrhenian Sea, Ligurian Sea, and Sardinian coast), microsatellite loci explained significant molecular variance at the “among groups” level and non-significant variance at the “among populations within groups” level. The hypothesis that genetic variation was structured according to the genetic assemblages proposed by De Innocentiis et al. (2004) was not supported by the AMOVA, either when only the same individuals from seven sampling localities (five groups hypothesis) therein analysed were considered or when the complete sample set (5 + 2 groups) was included.
AMOVA hierarchical analysis examining the partitioning of genetic variance at three hierarchical levels (“among groups”, “among populations within groups”, and “within populations”) in different hypothesized structures (see text), with significant genetic variations indicated by emboldened p-values.
| Structure tested | Degree of freedom | Variation (%) | F-statistic | p-value |
|---|---|---|---|---|
| One group | ||||
| Among populations | 12 | 0.75 | FST = 0.0034 | 0.00 |
| Within populations | 1 103 | 99.25 | ||
| Six groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Si-T) vs.(Ty-B, Ty-S, Ty-Z) vs. (Li-G) vs. (Ty-T, Sa-M, SC-G, SS-C) | ||||
| Among groups | 5 | 0.45 | FCT = 0.0045 | 0.03 |
| Among populations within groups | 7 | −0.04 | FSC = −0.0004 | 0.51 |
| Within populations | 1 103 | 99.59 | FST = 0.0040 | 0.00 |
| Five groups (structure tested by De Innocentiis et al., 2004): (At-A) vs. (Ad-L) vs. (Ty-B, Ty-S, Ty-T) vs. (SC-G) vs. (SS-C) | ||||
| Among groups | 4 | 0.09 | FCT = 0.0009 | 0.43 |
| Among populations within groups | 2 | 0.11 | FSC = 0.0011 | 0.41 |
| Within populations | 565 | 99.80 | FST = 0.0020 | 0.01 |
| 5 + 2 groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Ty-B, Ty-S, Ty-T, Ty-Z, Sa-M) vs. (SC-G) vs. (SS-C) vs. (Si-T) vs. (Li-G) | ||||
| Among groups | 6 | −0.13 | FCT = −0.0012 | 0.64 |
| Among populations within groups | 6 | 0.45 | FSC = 0.0044 | 0.00 |
| Within populations | 1 103 | 99.68 | FST = 0.0031 | 0.00 |
| Structure tested | Degree of freedom | Variation (%) | F-statistic | p-value |
|---|---|---|---|---|
| One group | ||||
| Among populations | 12 | 0.75 | FST = 0.0034 | 0.00 |
| Within populations | 1 103 | 99.25 | ||
| Six groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Si-T) vs.(Ty-B, Ty-S, Ty-Z) vs. (Li-G) vs. (Ty-T, Sa-M, SC-G, SS-C) | ||||
| Among groups | 5 | 0.45 | FCT = 0.0045 | 0.03 |
| Among populations within groups | 7 | −0.04 | FSC = −0.0004 | 0.51 |
| Within populations | 1 103 | 99.59 | FST = 0.0040 | 0.00 |
| Five groups (structure tested by De Innocentiis et al., 2004): (At-A) vs. (Ad-L) vs. (Ty-B, Ty-S, Ty-T) vs. (SC-G) vs. (SS-C) | ||||
| Among groups | 4 | 0.09 | FCT = 0.0009 | 0.43 |
| Among populations within groups | 2 | 0.11 | FSC = 0.0011 | 0.41 |
| Within populations | 565 | 99.80 | FST = 0.0020 | 0.01 |
| 5 + 2 groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Ty-B, Ty-S, Ty-T, Ty-Z, Sa-M) vs. (SC-G) vs. (SS-C) vs. (Si-T) vs. (Li-G) | ||||
| Among groups | 6 | −0.13 | FCT = −0.0012 | 0.64 |
| Among populations within groups | 6 | 0.45 | FSC = 0.0044 | 0.00 |
| Within populations | 1 103 | 99.68 | FST = 0.0031 | 0.00 |
AMOVA hierarchical analysis examining the partitioning of genetic variance at three hierarchical levels (“among groups”, “among populations within groups”, and “within populations”) in different hypothesized structures (see text), with significant genetic variations indicated by emboldened p-values.
| Structure tested | Degree of freedom | Variation (%) | F-statistic | p-value |
|---|---|---|---|---|
| One group | ||||
| Among populations | 12 | 0.75 | FST = 0.0034 | 0.00 |
| Within populations | 1 103 | 99.25 | ||
| Six groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Si-T) vs.(Ty-B, Ty-S, Ty-Z) vs. (Li-G) vs. (Ty-T, Sa-M, SC-G, SS-C) | ||||
| Among groups | 5 | 0.45 | FCT = 0.0045 | 0.03 |
| Among populations within groups | 7 | −0.04 | FSC = −0.0004 | 0.51 |
| Within populations | 1 103 | 99.59 | FST = 0.0040 | 0.00 |
| Five groups (structure tested by De Innocentiis et al., 2004): (At-A) vs. (Ad-L) vs. (Ty-B, Ty-S, Ty-T) vs. (SC-G) vs. (SS-C) | ||||
| Among groups | 4 | 0.09 | FCT = 0.0009 | 0.43 |
| Among populations within groups | 2 | 0.11 | FSC = 0.0011 | 0.41 |
| Within populations | 565 | 99.80 | FST = 0.0020 | 0.01 |
| 5 + 2 groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Ty-B, Ty-S, Ty-T, Ty-Z, Sa-M) vs. (SC-G) vs. (SS-C) vs. (Si-T) vs. (Li-G) | ||||
| Among groups | 6 | −0.13 | FCT = −0.0012 | 0.64 |
| Among populations within groups | 6 | 0.45 | FSC = 0.0044 | 0.00 |
| Within populations | 1 103 | 99.68 | FST = 0.0031 | 0.00 |
| Structure tested | Degree of freedom | Variation (%) | F-statistic | p-value |
|---|---|---|---|---|
| One group | ||||
| Among populations | 12 | 0.75 | FST = 0.0034 | 0.00 |
| Within populations | 1 103 | 99.25 | ||
| Six groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Si-T) vs.(Ty-B, Ty-S, Ty-Z) vs. (Li-G) vs. (Ty-T, Sa-M, SC-G, SS-C) | ||||
| Among groups | 5 | 0.45 | FCT = 0.0045 | 0.03 |
| Among populations within groups | 7 | −0.04 | FSC = −0.0004 | 0.51 |
| Within populations | 1 103 | 99.59 | FST = 0.0040 | 0.00 |
| Five groups (structure tested by De Innocentiis et al., 2004): (At-A) vs. (Ad-L) vs. (Ty-B, Ty-S, Ty-T) vs. (SC-G) vs. (SS-C) | ||||
| Among groups | 4 | 0.09 | FCT = 0.0009 | 0.43 |
| Among populations within groups | 2 | 0.11 | FSC = 0.0011 | 0.41 |
| Within populations | 565 | 99.80 | FST = 0.0020 | 0.01 |
| 5 + 2 groups: (At-A) vs. (Ad-U, Ad-L, Ad-B) vs. (Ty-B, Ty-S, Ty-T, Ty-Z, Sa-M) vs. (SC-G) vs. (SS-C) vs. (Si-T) vs. (Li-G) | ||||
| Among groups | 6 | −0.13 | FCT = −0.0012 | 0.64 |
| Among populations within groups | 6 | 0.45 | FSC = 0.0044 | 0.00 |
| Within populations | 1 103 | 99.68 | FST = 0.0031 | 0.00 |
The Mantel test applied to all sample sites revealed the presence of correlation between genetic, expressed as FST, and geographic distances (r = 0.37, p = 0.046; Figure 2). Excluding the Atlantic population (At-A) from the analyses, however, there was no correlation within the Mediterranean Sea populations (r = 0.12, p = 0.193).
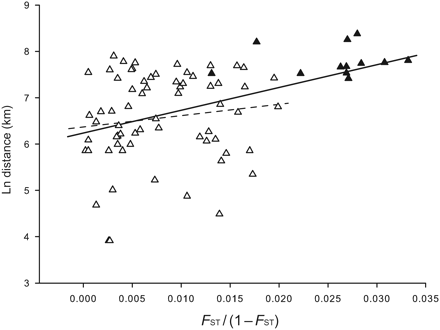
Genetic distances in gilthead sea bream populations inferred from multilocus estimates of FST plotted against geographic distance. The pairwise comparisons involving samples from the Mediterranean Sea and the Atlantic Ocean are indicated by solid triangles and those involving exclusively samples from the Mediterranean Sea by open triangles. Solid regression lines interpolate comparisons for all samples and dashed regression lines for Mediterranean Sea samples.
Clustering analyses by structure identified the greatest posterior probability value at K = 1 (a single genetic cluster) of the 13 tested K (Figure 3). This is supported by the symmetric proportion of the sample assigned to all populations (P = 1/K) in which individuals showed comparable assignment probabilities for all different inferred K clusters. The greatest partition probability was found at K = 1 (P = 0.99) also by structurama, where the K number of populations was not specified a priori.
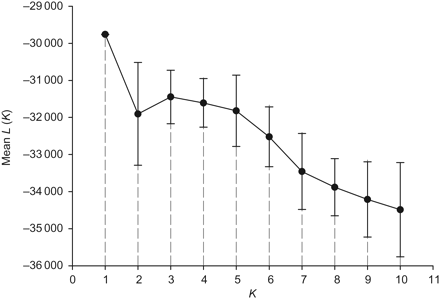
Number of gilthead sea bream populations with the greatest posterior probability expressed as the mean likelihood (L) over 20 runs for each of the 13 inferred K (the highest K value was set as the number of sampling sites).
The estimates of the population size parameter (Θ), the migration rate (M), and the effective population size (Ne), calculated assuming a microsatellite mutation rate of 10−4 per locus per generation (Whittaker et al., 2003), are listed in Table 7. Ne values ranged from 2450 gilthead sea bream for the Sicily Channel to 8025 for the Ligurian Sea (Table 7). Gene flow between many of the assemblages is limited, but in some cases, the values of M indicated high outcoming and low incoming gene flow (or vice versa; Table 7). The asymmetric gene flow is confirmed by the one-way ANOVA applied to test the null hypothesis of equal immigration rate among samples (p<0.01). The greatest difference in gene-flow direction was in comparisons involving the Adriatic Sea, which estimated substantial gene flow towards all other Mediterranean sites and low incoming flow from all of them. For the Atlantic site, low gene flow in both directions was observed with all the Mediterranean sites, except the consistent incoming gene flow from the Sardinian coast (M = 42.11).
Bayesian inference estimates of mean values of the population size parameter Θ (emboldened on the diagonal) and the scaled migration rate, M (reported as the mean M value) for the Mediterranean (Adriatic Sea: Ad-U, Ad-L, Ad-B; Sicily Channel: Si-T; Tyrrhenian Sea: Ty-B, Ty-S, Ty-Z; Ligurian Sea: Li-G; Sardinian Coast: Ty-T, Sa-M, SC-G, SS-C) and the Atlantic Ocean assemblages identified by AMOVA.
| Site | Atlantic Ocean | Mediterranean Sea | Ne | ||||
|---|---|---|---|---|---|---|---|
| Adriatic Sea | Sicily Channel | Tyrrhenian Sea | Ligurian Sea | Sardinian Coast | |||
| Atlantic Ocean | 1.46 | 5.67 | 5.43 | 8.06 | 5.29 | 42.11 | 3 650 |
| Mediterranean Sea | |||||||
| Adriatic Sea | 5.13 | 1.58 | 5.21 | 6.32 | 5.05 | 13.15 | 3 950 |
| Sicily Channel | 9.78 | 51.32 | 0.98 | 32.23 | 14.20 | 59.93 | 2 450 |
| Tyrrhenian Sea | 8.56 | 32.90 | 11.84 | 1.76 | 7.25 | 43.01 | 4 400 |
| Ligurian Sea | 7.01 | 22.22 | 5.91 | 69.38 | 3.21 | 61.37 | 8 025 |
| Sardinian Coast | 9.19 | 19.92 | 8.55 | 42.06 | 4.95 | 2.12 | 5 300 |
| Site | Atlantic Ocean | Mediterranean Sea | Ne | ||||
|---|---|---|---|---|---|---|---|
| Adriatic Sea | Sicily Channel | Tyrrhenian Sea | Ligurian Sea | Sardinian Coast | |||
| Atlantic Ocean | 1.46 | 5.67 | 5.43 | 8.06 | 5.29 | 42.11 | 3 650 |
| Mediterranean Sea | |||||||
| Adriatic Sea | 5.13 | 1.58 | 5.21 | 6.32 | 5.05 | 13.15 | 3 950 |
| Sicily Channel | 9.78 | 51.32 | 0.98 | 32.23 | 14.20 | 59.93 | 2 450 |
| Tyrrhenian Sea | 8.56 | 32.90 | 11.84 | 1.76 | 7.25 | 43.01 | 4 400 |
| Ligurian Sea | 7.01 | 22.22 | 5.91 | 69.38 | 3.21 | 61.37 | 8 025 |
| Sardinian Coast | 9.19 | 19.92 | 8.55 | 42.06 | 4.95 | 2.12 | 5 300 |
Rows and columns represent recipient and donor populations, respectively. Ne was calculated assuming µ = 10−4 per locus per generation.
Bayesian inference estimates of mean values of the population size parameter Θ (emboldened on the diagonal) and the scaled migration rate, M (reported as the mean M value) for the Mediterranean (Adriatic Sea: Ad-U, Ad-L, Ad-B; Sicily Channel: Si-T; Tyrrhenian Sea: Ty-B, Ty-S, Ty-Z; Ligurian Sea: Li-G; Sardinian Coast: Ty-T, Sa-M, SC-G, SS-C) and the Atlantic Ocean assemblages identified by AMOVA.
| Site | Atlantic Ocean | Mediterranean Sea | Ne | ||||
|---|---|---|---|---|---|---|---|
| Adriatic Sea | Sicily Channel | Tyrrhenian Sea | Ligurian Sea | Sardinian Coast | |||
| Atlantic Ocean | 1.46 | 5.67 | 5.43 | 8.06 | 5.29 | 42.11 | 3 650 |
| Mediterranean Sea | |||||||
| Adriatic Sea | 5.13 | 1.58 | 5.21 | 6.32 | 5.05 | 13.15 | 3 950 |
| Sicily Channel | 9.78 | 51.32 | 0.98 | 32.23 | 14.20 | 59.93 | 2 450 |
| Tyrrhenian Sea | 8.56 | 32.90 | 11.84 | 1.76 | 7.25 | 43.01 | 4 400 |
| Ligurian Sea | 7.01 | 22.22 | 5.91 | 69.38 | 3.21 | 61.37 | 8 025 |
| Sardinian Coast | 9.19 | 19.92 | 8.55 | 42.06 | 4.95 | 2.12 | 5 300 |
| Site | Atlantic Ocean | Mediterranean Sea | Ne | ||||
|---|---|---|---|---|---|---|---|
| Adriatic Sea | Sicily Channel | Tyrrhenian Sea | Ligurian Sea | Sardinian Coast | |||
| Atlantic Ocean | 1.46 | 5.67 | 5.43 | 8.06 | 5.29 | 42.11 | 3 650 |
| Mediterranean Sea | |||||||
| Adriatic Sea | 5.13 | 1.58 | 5.21 | 6.32 | 5.05 | 13.15 | 3 950 |
| Sicily Channel | 9.78 | 51.32 | 0.98 | 32.23 | 14.20 | 59.93 | 2 450 |
| Tyrrhenian Sea | 8.56 | 32.90 | 11.84 | 1.76 | 7.25 | 43.01 | 4 400 |
| Ligurian Sea | 7.01 | 22.22 | 5.91 | 69.38 | 3.21 | 61.37 | 8 025 |
| Sardinian Coast | 9.19 | 19.92 | 8.55 | 42.06 | 4.95 | 2.12 | 5 300 |
Rows and columns represent recipient and donor populations, respectively. Ne was calculated assuming µ = 10−4 per locus per generation.
Discussion
The results of the study revealed a high level of genetic variability in gilthead sea bream. Allelic polymorphism and expected heterozygosity values are comparable with those observed in S. aurata and other sparids (Batargias et al., 1999; Perez-Enriquez et al., 2001; Jeong et al., 2003; Alarcon et al., 2004; De Innocentiis et al., 2004; Chaoui et al., 2009) as well as in other marine and anadromous teleosts (DeWoody and Avise, 2000).
Physical linkage of loci appears to be unlikely because a substantial level of disequilibrium was only recorded in 1 population of the 15 analysed. The disequilibrium observed at the Adriatic site Bisceglie (Ad-B) may indicate population subdivision as a result of the mixing of distinct populations/broodstocks/generations (Hartl and Clark, 1997). Indeed, the Bisceglie coast is characterized by massive presence of floating cages for aquaculture, and frequent escapes have been documented (CoProMar, pers. comm.). As cultured sea bream show reduced variability (Karaiskou et al., 2009; Porta et al., 2010; Loukovitis et al., 2011) that could also have been obtained by allochthonous breeders (Porta et al., 2010), their accidental release may have altered allelic frequencies of the local wild genetic assemblage. However, the homogeneity detected among the three temporal replicates from Lesina supports previous evidence, based on a lower number of microsatellite loci (Rossi et al., 2009), excluding chaotic variation in gene frequencies over time in gilthead sea bream when human intervention is absent.
Population structure
The powsim simulations, carried out with different combinations of effective population size (Ne) and time (t) since divergence, demonstrated that the microsatellite loci screened in this study provided sufficient statistical power to detect a low level of genetic structure (FST = 0.001) in the sample set.
The FST estimates here are lower than those obtained with the same markers by both De Innocentiis et al. (2004) and Alarcon et al. (2004), but do indicate a weak but still well-defined genetic structure along the Italian coast. However, the Bayesian analyses depict a different scenario and lean towards the lack of any population subdivision. Hence, the questions arise whether, in a situation of weak genetic structuring, the F-statistics are more sensitive than the Bayesian methods or, alternatively, whether they overestimate the real genetic differentiation between groups of individual fish. It is difficult to distinguish between these two possibilities. Bayesian clustering methods have the unchallenged advantage of requiring just the individual genotype and no reference to sample origin or a priori grouping of individuals in populations (Pritchard et al., 2000). Nevertheless, Latch et al. (2006) demonstrated that their performances depend on the levels of population differentiation. Indeed, in the presence of low FST values (≤0.02), different programs applying these methods not only fail to identify the real number of subpopulations, but also provide false certainty (extremely high probability values) regarding the number of inferred K. The overall FST value (0.0072) estimated here is an order of magnitude lower than the FST threshold indicated by Latch et al. (2006). Hence, the single panmictic population suggested by the Bayesian clustering methods appears to be incorrectly identified, and the real genetic structure of gilthead sea bream is more reliably reflected by the results based on FST estimates, i.e. a (weak) population subdivision in the species.
Comparison of the Italian and Atlantic samples indicates that the latter comprise the most divergent population, consistent with previous observations by De Innocentiis et al. (2004). When applied to the complete dataset, the Mantel test performed using the pairwise FST values provides a significant correlation with geographic distance. However, no correlation was found when At-A is excluded from the analysis. This could be explained considering an isolation-by-distance model between the Atlantic Ocean and the Italian coast, a model not identifiable in the study of De Innocentiis et al. (2004) carried out with four of the ten loci analysed here.
The present study was carried out with an increased number of microsatellite loci (10 vs. 4) and has a greater spatial coverage of sampling (12 vs. 6 sites) along the Italian coast, compared with the previous study of De Innocentiis et al. (2004). The patterns of subdivision of the two studies are only partially congruent. AMOVA, based on four loci, identified four subpopulations (Sardinian Sea, Sardinian Channel, Tyrrhenian Sea, and Adriatic Sea) from six sites (De Innocentiis et al., 2004). However, the same structure was not statistically significant when AMOVA was applied to the same fish from the same six sites based on a total of ten loci. The AMOVA applied to the complete dataset identified five assemblages along the Italian coast (in addition to the sixth Atlantic assemblage) and that all samples from Sardinia, including those from Ty-T previously (De Innocentiis et al., 2004) included in the Tyrrhenian group, gather within the same assemblage (Sardinian coast). Therefore, for gilthead sea bream, data from a reduced number of microsatellite loci, although sensitive enough to detect a weak genetic structure, appear not to be representative of the real molecular variance within and between populations.
The increase in sampling coverage here better resolved genetic structure, which is likely to have been shaped by marine currents (see below) and allows us to conclude that at the medium geographic scale, genetic subdivision along the Italian coast is weak.
Framing these data into results obtained for gilthead sea bream from other sites of the species' distribution, the strong genetic subdivision found at short distances along the Tunisian coasts (Ben Slimen et al., 2004) or between the French and the Algerian coasts (Chaoui et al., 2009) is intriguing. Ben Slimen et al. (2004) used different markers, allozymes, to analyse genetic differentiation. Notwithstanding, allozymes have been used previously on some of the samples analysed here (Rossi et al., 2006) and provided results consistent with results obtained using microsatellites. Chaoui et al. (2009) used microsatellites and random amplified polymorphic DNA and found an overall FST value of 0.069, indicating strong genetic differentiation between the two western banks of the Mediterranean Sea.
Gene flow
Data on migration rates were obtained on a non-homogenous sample size collected over a long timespan, so the results of this analysis have to be taken with caution. Nevertheless, according to the M values calculated, the estimated gene flow between the six assemblages of gilthead sea bream are of the same order of magnitude as those observed in pelagic fish such as sardine, Sardina pilchardus (Gonzalez and Zardoya, 2007), and bigeye tuna, Thunnus obesus (Gonzalez et al., 2008). The estimates indicate migration between the selected assemblages, and in some cases, it is asymmetrical.
Along the Italian coast, the pattern of gene flow appears to be compatible with the main currents characterizing water circulation (Figure 4). As an example, the outgoing gene flow from the Adriatic Sea mirrors the direction of the currents that move water towards the western Mediterranean, where the other four samples are located. At the same time, it is not surprising that no immigration is directed from these towards the Adriatic Sea, which mainly receives water from the eastern Mediterranean, where no gilthead sea bream samples were collected. In a similar way, the asymmetry of gene flow could be explained between gilthead sea bream from the Tyrrhenian and Ligurian seas.
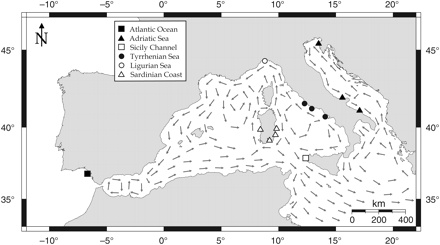
Representation of the main sea currents in the central and western Mediterranean Sea. Sampling sites symbols refer to the six assemblages identified by AMOVA.
Unfortunately, little is known about the biology of the gilthead sea bream, and the dispersal capability or territoriality of juvenile/adult individuals cannot be quantified because of the lack of tracking experiments. For this reason, it is not possible to estimate the contribution of adult gilthead sea bream active migration, if any, to the observed pattern of gene flow. However, the congruence between migration estimates and marine currents within the Mediterranean Sea suggests a key role for passive dispersal of eggs and larvae in shaping gene flow, compatible with the long-lasting larval stage of the gilthead sea bream (∼50 d). Similar results of a principal contribution of larval dispersal on gene flow were recently observed in another coastal sparid, Chrysoblephus laticeps (Teske et al., 2010). Water circulation and larval dispersal, however, does not allow direct interpretation of either the migration pattern between the Atlantic and the Sardinian assemblages or the genetic subdivision found along the Tunisian coast by Ben Slimen et al. (2004) or between the French and Algerian coasts by Chaoui et al. (2009).
The effective population size estimated by the coalescent method gave high mean values of T, comparable with the recently published estimates of Ne for Adriatic Sea wild samples (Loukovitis et al., 2011). However, when we tried to infer Ne from the three Lesina temporal replicates, we obtained smaller mean values than those estimated with the coalescent method. Nevertheless, the few fish in each temporal sample and the short period between replicates might have affected the estimation of Ne in temporal methods, as also revealed by the large CIs (Luikart and Cornuet, 1999).
Management implications and perspectives
Sparus aurata is an important aquaculture species throughout its distribution. Its culture has increased exponentially in the past 25 years (FAO, 2008). Intensification of rearing in the absence of an appropriate control of broodstock origin and history, associated with the extensive exchange of breeders and fry between hatcheries, may already have caused a mixture of different genetic pools. In addition to restocking with hatchery fry in natural lagoons, an input of farmed fish into the wild could have been caused by accidental releases from sea cages. Indeed, in cases of accidental escapes, massive numbers of fish enter the wild. Moreover, the longer culture period adopted in the past decade to address the market requirement for larger fish caused the release of gametes by gilthead sea bream spawners from the cages to the marine system (Dimitriou et al., 2007). The impact of these activities on wild gilthead sea bream populations remains to be quantified (Sola et al., 2007). However, it is worth emphasizing that, given the intensity of sea bream aquaculture activity in Italy, aquaculture may already have partially contributed to wild population homogenization, i.e. aquaculture may be the factor that best explains current population patterns. However, all studies carried out so far on the population genetics of the species still cover a limited area of its distribution. Further studies need to be carried out at different geographic scales and over time to provide a more general picture, to permit distinction between micro-evolutive processes and domestication effects. In this sense, S. aurata may become a model species for evaluating the genetic impact of aquaculture on wild populations.
Finally, it needs to be emphasized that this study was performed applying neutral genetic markers, as in most population genetics studies involving marine fish in recent years. As gene flow is expected to hamper adaptive population divergence, molecular markers with presumably neutral functional meaning indicate that local adaptations may be rare or absent in marine fish. There is now increasing interest in studying adaptive genetic variation (Luikart et al., 2003; Nielsen et al., 2009) and in identifying molecular genetic markers under selection (e.g. Joost et al., 2007). For this purpose, genome-wide datasets, which can improve the inference of population parameters and provide a better understanding of adaptive evolution (Luikart et al., 2003), are suggested for future study.
Supplementary material
Acknowledgements
Financial support for this work was provided by the Italian Ministry of Instruction, University and Research (MIUR), and POR Puglia. We thank all those who helped in the collection of gilthead sea bream: A. Santulli (University of Palermo), M. Rampacci (A.Ge.I., Rome), M. Vacchi (ISPRA, Genoa), R. Vaccarella (Provincia di Bari), and E. Rambaldi (COIPA, Rome).
References
Author notes
Present address: Lehrstuhl für Zoologie und Evolutionsbiologie, Department of Biology, University of Konstanz, Universitätstrasse 10, 78457 Konstanz, Germany.
Handling editor: Bill Turrell

{kind=link}
{kind=link}
{kind=link}
{kind=link}