





Abstract
Epidemiologists aim to identify modifiable causes of disease, this often being a prerequisite for the application of epidemiological findings in public health programmes, health service planning and clinical medicine. Despite successes in identifying causes, it is often claimed that there are missing additional causes for even reasonably well-understood conditions such as lung cancer and coronary heart disease. Several lines of evidence suggest that largely chance events, from the biographical down to the sub-cellular, contribute an important stochastic element to disease risk that is not epidemiologically tractable at the individual level. Epigenetic influences provide a fashionable contemporary explanation for such seemingly random processes. Chance events—such as a particular lifelong smoker living unharmed to 100 years—are averaged out at the group level. As a consequence population-level differences (for example, secular trends or differences between administrative areas) can be entirely explicable by causal factors that appear to account for only a small proportion of individual-level risk. In public health terms, a modifiable cause of the large majority of cases of a disease may have been identified, with a wild goose chase continuing in an attempt to discipline the random nature of the world with respect to which particular individuals will succumb. The quest for personalized medicine is a contemporary manifestation of this dream. An evolutionary explanation of why randomness exists in the development of organisms has long been articulated, in terms of offering a survival advantage in changing environments. Further, the basic notion that what is near-random at one level may be almost entirely predictable at a higher level is an emergent property of many systems, from particle physics to the social sciences. These considerations suggest that epidemiological approaches will remain fruitful as we enter the decade of the epigenome.
We cannot imagine these diseases, they are called idiopathic, spontaneous in origin, but we know instinctively there must be something more, some invisible weakness they are exploiting. It is impossible to think they fall at random, it is unbearable to think it.
James Salter, Light Years, 1975
Epidemiology is concerned with the identification of modifiable causes of disease, which is often a prerequisite for the application of epidemiological findings in public health programmes, health service planning and clinical medicine.1 Despite many successes, even with respect to the most celebrated—such as the identification of cigarette smoking as a major cause of lung cancer and other chronic diseases—it can appear that much remains to be done. Consider Winnie, lighting a cigarette from the candles on her centenary birthday cake, who, after 93 years of smoking, is not envisaging giving up the habit (Figure 1). Such people, who survive to a ripe old age despite transgressing every code of healthy living, loom large in the popular imagination2 and are reflected in the low positive predictive values and C statistics in many formal epidemiological prediction models. In general, epidemiologists do a rather poor job of predicting who is and who is not going to develop disease.

Winnie ain’t quitting now
This apparent failing of epidemiology has long been recognized. Writing about ischaemic heart disease (IHD) 40 years ago, Tom Meade and Ranjan Chakrabarti reported that ‘within any risk group, prediction is poor; it is not at present possible to express individual risk more precisely than as about a 1 in 6 chance of a hitherto healthy man developing clinical IHD in the next 5 years if he is at high risk’.3 This poor prediction of individual risk indicated that there was ‘a pressing need for prospective observational studies in which new risk factors are identified’.3 Many such calls followed over the succeeding decades, with funding applications often beginning with a statement that ‘identified risk factors account for only 30% of IHD risk’, before proposing the expensive exploration of novel putative causes of the disease.4 I have certainly promulgated such views in the (usually unsuccessful) pursuit of pounds or dollars, although the exact percentage of ‘explanation’ by established causes would fall and rise in relation to degree of desperation. The most feted contemporary candidate for better prediction is probably genetics. With the perception (in my view exaggerated) that genome-wide association studies (GWASs) have failed to deliver on initial expectations,5 the next phase of enhanced risk prediction will certainly shift to ‘epigenetics’6,7—the currently fashionable response to any question to which you do not know the answer.
As epidemiologists attempt to come to terms with ‘personalized medicine’ and individual risk prediction,8 they may want to consider how cognate disciplines concerned with individual trajectories address these issues. For example, in the study of criminality it has been suggested that ‘the concept of cause inevitably involves the concept of change within individual units’.9 One response to poor epidemiological prediction of individual outcomes is to consider that the framework of lifecourse epidemiology offers a solution: if only we collected better data on what happens to people from before birth and then throughout their lives we could better understand their ultimate fate. In a recent book entitled, ‘Epidemiological methods in lifecourse research’10 a chapter offers to tell us about ‘Measurement and design for life course studies of individual differences and development’.11 The suggestion is that the individual can indeed be the target of epidemiological understanding, and the lifecourse approach offers us a path towards this goal.
The issues that confront epidemiologists regarding understanding the trajectories and outcomes for individuals are ones that other disciplines also struggle with. For example, Stanley Lieberson and colleagues consider that their fellow sociologists are ‘barking up the wrong branch’12 when becoming involved in discussions of prediction of particular events. In this paper I attempt to bring together considerations from several fields of investigation—from behavioural genetics, genomics, epigenetics, evolutionary theory, epidemiology and public health—to illustrate why we should abandon any ambitions towards individualised prediction—as codified in personalized medicine, if we want to succeed as population health scientists. This involves initial discussion of issues—such as the notion of the non-shared environment—that may not be familiar to epidemiologists. However triangulation of the different disciplinary understandings is, I think, considerably more powerful than only engaging with the theoretical background of one approach, and I hope worth the effort.
Same origins, different outcomes
Lifecourse epidemiology has been defined as ‘the study of the effects on health and health-related outcomes of biological (including genetic), environmental and social exposures during gestation, infancy, childhood, adolescence, adulthood and across generations’.13 Family-level influences during gestation, infancy, childhood and adolescence are likely to be shared by siblings reared by the same parents, and are targets for epidemiological investigation. A large number of studies have, for example, examined the association of socio-economic circumstances in early life with later morbidity and mortality.14,15 The indicators used—such as occupational social class of the head of household—would generally be the same for different siblings from the same family. Exposures of this kind are, in the terminology popularized within behavioural genetics, shared (or common) environmental factors. It is therefore perhaps surprising that the groundbreaking 1987 paper by Robert Plomin and Denise Daniels,16 ‘Why are children in the same family so different from one another?’, recently reprinted with commentaries in the IJE,17–21 has apparently had little influence within epidemiology. The implication of the paper—which expanded upon an earlier analysis22—was that, genetics aside, siblings are little more similar than two randomly selected individuals of roughly the same age selected from the source population that the siblings originate from. This may be an intuitive observation for many people who have siblings themselves or have more than one child. Arising from the field of behavioural genetics, the paper focused on measures of child behaviour, personality, cognitive function and psychopathology, but, as Plomin points out, the same basic finding is observed for many physical health outcomes: obesity, cardiovascular disease, diabetes, peptic ulcers, many cancers, asthma, longevity and various biomarkers assayed in epidemiological studies.18 These findings come from studies of twins, adoptees and extended pedigrees, in which the variance in an outcome is partitioned into a genetic component, the contribution of common environment (i.e. that shared between people brought up in the same home environment) and the non-shared environment (i.e. exposures that are not correlated between people brought up in the same family). The shared environment—which is the domain of many of the exposures of interest to lifecourse epidemiologists—is reported to make at best small contributions to the variance of most outcomes. The non-shared environment—exposures which (genetic influences apart) show no greater concordance between siblings than between non-related individuals of a similar age from the same population—constitute by far the dominant class of non-genetic influences on most health and health-related outcomes (Box 1). Table 1 presents data from a large collaborative twin study of 11 cancer sites, with universally large non-shared environmental influences (58–82%), heritabilities in the range 21–42% (excluding uterine cancer, for which a value of 0% is reported) and smaller shared environmental effects, zero for four sites and ranging from 5% to 20% for the remainder.23 Many other diseases show a similar dominance of non-shared over shared environmental influences.18 Indeed, a greater non-shared than shared environmental component appears to apply to some,24–28 although not all,29 childhood-acquired infections and the diseases they cause. This is such a counter-intuitive observation that one commentator on an earlier draft of this paper used childhood infectious disease epidemiology as an example of a situation in which the shared environment must be dominant.
Effects of heritable and environmental factors on cancers at various sites, according to data from the Swedish, Danish and Finnish Twin Registries.23 Proportion of variance (95% CI)
| Site or type | Heritable factors | Shared environmental factors | Non-shared environmental factors |
|---|---|---|---|
| Stomach | 0.28 (0–0.51) | 0.10 (0–0.34) | 0.62 (0.49–0.76) |
| Colorectum | 0.35 (0.10–0.48) | 0.05 (0–0.23) | 0.60 (0.52–0.70) |
| Pancreas | 0.36 (0–0.53) | 0 (0–0.35) | 0.64 (0.47–0.86) |
| Lung | 0.26 (0–0.49) | 0.12 (0–0.34) | 0.62 (0.51–0.73) |
| Breast | 0.27 (0.04–0.41) | 0.06 (0–0.22) | 0.67 (0.59–0.76) |
| Cervix uteri | 0 (0–0.42) | 0.20 (0–0.35) | 0.80 (0.57–0.97) |
| Corpus uteri | 0 (0–0.35) | 0.17 (0–0.31) | 0.82 (0.64–0.98) |
| Ovary | 0.22 (0–0.41) | 0 (0–0.24) | 0.78 (0.59–0.99) |
| Prostate | 0.42 (0.29–0.50) | 0 (0–0.09) | 0.58 (0.50–0.67) |
| Bladder | 0.31 (0–0.45) | 0 (0–0.28) | 0.69 (0.53–0.86) |
| Leukaemia | 0.21 (0–0.54) | 0.12 (0–0.41) | 0.66 (0.45–0.88) |
| Site or type | Heritable factors | Shared environmental factors | Non-shared environmental factors |
|---|---|---|---|
| Stomach | 0.28 (0–0.51) | 0.10 (0–0.34) | 0.62 (0.49–0.76) |
| Colorectum | 0.35 (0.10–0.48) | 0.05 (0–0.23) | 0.60 (0.52–0.70) |
| Pancreas | 0.36 (0–0.53) | 0 (0–0.35) | 0.64 (0.47–0.86) |
| Lung | 0.26 (0–0.49) | 0.12 (0–0.34) | 0.62 (0.51–0.73) |
| Breast | 0.27 (0.04–0.41) | 0.06 (0–0.22) | 0.67 (0.59–0.76) |
| Cervix uteri | 0 (0–0.42) | 0.20 (0–0.35) | 0.80 (0.57–0.97) |
| Corpus uteri | 0 (0–0.35) | 0.17 (0–0.31) | 0.82 (0.64–0.98) |
| Ovary | 0.22 (0–0.41) | 0 (0–0.24) | 0.78 (0.59–0.99) |
| Prostate | 0.42 (0.29–0.50) | 0 (0–0.09) | 0.58 (0.50–0.67) |
| Bladder | 0.31 (0–0.45) | 0 (0–0.28) | 0.69 (0.53–0.86) |
| Leukaemia | 0.21 (0–0.54) | 0.12 (0–0.41) | 0.66 (0.45–0.88) |
Effects of heritable and environmental factors on cancers at various sites, according to data from the Swedish, Danish and Finnish Twin Registries.23 Proportion of variance (95% CI)
| Site or type | Heritable factors | Shared environmental factors | Non-shared environmental factors |
|---|---|---|---|
| Stomach | 0.28 (0–0.51) | 0.10 (0–0.34) | 0.62 (0.49–0.76) |
| Colorectum | 0.35 (0.10–0.48) | 0.05 (0–0.23) | 0.60 (0.52–0.70) |
| Pancreas | 0.36 (0–0.53) | 0 (0–0.35) | 0.64 (0.47–0.86) |
| Lung | 0.26 (0–0.49) | 0.12 (0–0.34) | 0.62 (0.51–0.73) |
| Breast | 0.27 (0.04–0.41) | 0.06 (0–0.22) | 0.67 (0.59–0.76) |
| Cervix uteri | 0 (0–0.42) | 0.20 (0–0.35) | 0.80 (0.57–0.97) |
| Corpus uteri | 0 (0–0.35) | 0.17 (0–0.31) | 0.82 (0.64–0.98) |
| Ovary | 0.22 (0–0.41) | 0 (0–0.24) | 0.78 (0.59–0.99) |
| Prostate | 0.42 (0.29–0.50) | 0 (0–0.09) | 0.58 (0.50–0.67) |
| Bladder | 0.31 (0–0.45) | 0 (0–0.28) | 0.69 (0.53–0.86) |
| Leukaemia | 0.21 (0–0.54) | 0.12 (0–0.41) | 0.66 (0.45–0.88) |
| Site or type | Heritable factors | Shared environmental factors | Non-shared environmental factors |
|---|---|---|---|
| Stomach | 0.28 (0–0.51) | 0.10 (0–0.34) | 0.62 (0.49–0.76) |
| Colorectum | 0.35 (0.10–0.48) | 0.05 (0–0.23) | 0.60 (0.52–0.70) |
| Pancreas | 0.36 (0–0.53) | 0 (0–0.35) | 0.64 (0.47–0.86) |
| Lung | 0.26 (0–0.49) | 0.12 (0–0.34) | 0.62 (0.51–0.73) |
| Breast | 0.27 (0.04–0.41) | 0.06 (0–0.22) | 0.67 (0.59–0.76) |
| Cervix uteri | 0 (0–0.42) | 0.20 (0–0.35) | 0.80 (0.57–0.97) |
| Corpus uteri | 0 (0–0.35) | 0.17 (0–0.31) | 0.82 (0.64–0.98) |
| Ovary | 0.22 (0–0.41) | 0 (0–0.24) | 0.78 (0.59–0.99) |
| Prostate | 0.42 (0.29–0.50) | 0 (0–0.09) | 0.58 (0.50–0.67) |
| Bladder | 0.31 (0–0.45) | 0 (0–0.28) | 0.69 (0.53–0.86) |
| Leukaemia | 0.21 (0–0.54) | 0.12 (0–0.41) | 0.66 (0.45–0.88) |
The terminology of shared and non-shared environments is not a familiar one within epidemiology, and such formulations are used in subtly different ways in the various social and behavioural sciences within which they have been evoked. In the context of classic twin studies, the shared environmental factors are those that make twins alike, and the non-shared environmental factors (sometimes referred to as the ‘unique environment’) are those that make twins different. In some contexts, what twins appear to share (for example, damp housing and mould on the walls) could either make them more similar—by providing a common exposure with an on-average main effect on a disease outcome—or more different, if a non-shared factor, such as cigarette smoking, strongly interacted with the shared exposure, leading to greatly divergent risks of disease in the presence of the shared exposure, but less divergence in its absence. With enough ingenuity it is possible to produce stories in which any exposure could either increase or decrease twin (or sibling) similarity. The mystery remains, however, that there appear to be a greater preponderance of difference-generating rather than similarity-generating exposures in the environments that twins (and other siblings) share.
The terms shared and non-shared environment will be used frequently in this paper. A variety of partially overlapping subcategorizations will also be encountered.
The shared environment may have both objective and effective aspects84; the effective element referring to how environments influence a particular person. In this sense the same objectively assessed exposure (e.g. emigration) may be experienced differently by two siblings, with one of them benefiting from the process and the other being adversely affected. The effective aspect of the shared environment could then act as a non-shared exposure, and be considered to generate non-shared effects from a shared exposure.
The non-shared environment has both systematic and non-systematic elements. Systematic differences are generally ones that can be more easily measured (and the term measured non-shared environment is used to refer to directly measured exposures as opposed to the quantitative estimation of non-shared environmental influences from behavioural genetics models). Systematic aspects of the non-shared environment include such factors as birth order, season of birth, sibling-sibling interactions, differential parental treatment and peer groups; factors that can systematically differ between siblings and may in principle be measured and studied. Non-systematic aspects include accidents, chance events and other life events that would be difficult to assess and analyse in most study settings.
Both shared and non-shared environmental effects may be either stable or unstable. Stable factors tend to track over time—such as smoking behaviour—whereas the existence and/or influence of unstable environmental factors changes to a considerable extent over time.
This basic conclusion seems to be that in the search for modifiable influences on disease the focus should be on factors that are unrelated to shared family background. This would appear to have important implications for epidemiology, as well as for social and behavioural sciences. However, as Neven Sesardic points out, even within behavioural genetics the central, rather momentous, finding regarding the apparently small or non-existent contribution of family background to child outcomes went under-appreciated; it was ‘an explosion without a bang’.19 Attempts at popularizing its message—such as Judith Rich Harris’ book The Nurture Assumption,30 which was headlined as saying parenting does not matter to children—may have simply increased the unwillingness of some researchers to come to terms with the key message regarding the importance of the non-shared environment (See Box 1).
For epidemiologists, the fact that the generally small shared environmental influences on many outcomes appeared to get even smaller (or disappear completely) with age—as is seen, for example, with respect to body mass index and obesity31—increases the relevance of the message, since later life health outcomes are often what we study. Yet, within epidemiology, the impact of this work has been minimal; of the 607 citations of the Plomin and Daniels paper on ISI Web of Science (as of May 2011), only a handful fall directly within the domain of epidemiology or population health. In the recent book, Family Matters: Designing, Analysing and Understanding Family-based Studies in Lifecourse Epidemiology,32 the issue is barely touched upon; the balanced one page it receives near the end of the 340-page book being perhaps too little, too late.33 Between-sibling studies as a way of controlling for potential confounding have been widely discussed within epidemiology, both in the book in question34 and elsewhere.35,36 Certainly, this is a useful method for taking into account shared aspects of the childhood environment. But if shared environment has little impact on many outcomes then, on the face of it, the approach might be missing the issue of real concern—the more important non-shared environmental factors. Despite this, the use of sibling controls sometimes appears to uncover substantial confounding. For example, maternal smoking during pregnancy was found in a large Swedish study to be associated with lower offspring IQ, even after adjustment for many potential confounding factors.37 In a between-sibs comparison, however, there was no association of maternal smoking with IQ of offspring, which the authors interpreted as indicating that the association seen for unrelated individuals was due to residual confounding. If shared environment is of such little importance, how can it generate meaningful confounding in epidemiological studies? We will return to this issue later.
Why are siblings so different?
Plomin and Daniels provided a catalogue of factors that could contribute to the large non-shared environmental effects impacting on many outcomes. An important concern was that in the statistical models used to estimate non-shared environmental effects, these usually come from subtraction: the non-shared environmental component being the remaining variance, after estimated genetic and shared environmental contributions have been taken into account. Measurement error would therefore appear as a non-shared environmental influence. A second possibility was that non-systematic aspects of the non-shared environment—essentially chance or stochastic events—could lead to children from the same family having very different trajectories throughout life. This was illustrated by the biography of Charles Darwin; if it were not for apparently chance events he would not have been present on the voyage of the Beagle, and we would probably be celebrating Alfred Russell Wallace as the founder of the theory of natural selection. Indeed, the narratives of people’s lives often emphasize serendipity and misfortune at crucial turning points that apparently had a major influence on their trajectories. The possibility that it was such non-shared ‘stochastic events that, when compounded over time, make children in the same family different in unpredictable ways’16 was, however, considered by Plomin and Daniels as ‘a gloomy prospect’ since it was ‘likely to prove a dead end for research’.16 Possible systematic sources of differences within families were considered a more promising avenue for future investigation.
Several categories of such systematic non-shared environmental influences were identified that could influence different outcomes in children from the same family. Some characteristics are clearly not shared by siblings, such as gender in gender-discordant sibships, birth order or season of birth. Sib–sib interactions generate different experiences for the participants involved, parental treatment of siblings may be more different than parents realize and there are extensive networks outside the family that provide unique experiences (in 1987 Plomin and Daniels mentioned peer groups, television and teachers;16 in 2011 the role of the internet, social networking and mobile communications in allowing one sibling to differentiate themselves from another might receive more emphasis).
An extensive research programme in the behavioural and social sciences consequent on the Plomin and Daniels review focused on the direct assessment of effects of the systematic aspect of the non-shared environment. Instruments were developed to collect detailed data on sibling-specific parenting practice, sib–sib interactions and the influence of schools and peer groups, and studies including more than one child per family were explicitly established to allow investigation of why siblings differ. However, a decade ago, a meta-analytical overview of such studies concluded that there was little direct evidence of important influences of specific non-shared environmental characteristics on behavioural and social outcomes mainly assessed during the first two decades of life.38 At best, only small proportions of the phenotypic variance attributed to the non-shared environment related to directly measured influences. The effects were rarely statistically robust and the median value of the proportion of variation accounted for was ∼3%. In the behavioural genetic studies, estimates of the proportion of the overall phenotypic variance accounted for by the non-shared environment are almost always over 50%, and often substantially so; similar findings apply to cancers (Table 1). There are more optimistic assessments of the current status of studies directly assessing the effects of non-shared environment,18,39 but in these the magnitude of the effects appears small. In an example presented in Plomin’s assessment of three decades of research on this issue18 non-shared aspects of maternal negativity does have a statistically robust association with offspring depressive symptoms, but accounts for only around 1% of the variance.40
In the epidemiological field there has been relatively little focused investigation of measured aspects of the non-shared environment on disease or disease-related phenomenon. The exception is mental health, particularly in early life, where at best small effects have been identified.18 Birth order has been strongly advocated as an important contributor in the psychological arena,41 but this does not stand up to scrutiny.42 In the health field associations are generally non-existent or small and when found often not robust, potentially reflecting confounding.43–48 Season or month of birth, which will generally differ between siblings, has been studied in relation to various—mainly psychiatric—health outcomes, and could reflect either biological processes (such as in-utero or early postnatal infections for seasonally variable infectious agents) or social processes, such as exact age at school entry and relative age within a school year. Effects are intriguing, but are variable, of low magnitude, and generally far from robust.49,50 The identified influence of particular aspects of the measured non-shared environment on health outcomes are, at best, weak in the medical field, and contrast with the large contribution that the non-shared environment appears to make in quantitative genetics analyses.
An issue with much epidemiological research is that adulthood environment is clearly of considerable potential importance. Similarities between siblings for adulthood environment will be less than for childhood environment. Much of the behavioural genetics literature is concerned with developmental outcomes assessed in childhood adolescence, or young adulthood. In these cases, the apparently small shared and large non-shared environmental components are seen during a period when siblings will usually have remained in the same household. For those health-related outcomes that have been assessed throughout life—such as obesity and body mass index—the non-shared environmental component is large from childhood to late adulthood, and the shared environmental contribution, evident in young childhood, declines to a small fraction by puberty, and remains either undetectable or small right through into old age.51 Systematic aspects of the non-shared environments of adults that have large effects on disease outcomes may await identification. However, the inability to identify such effects using intensive assessments of exposure and outcomes in childhood is sobering. Furthermore, in longitudinal twin studies, in which twin pairs have repeat assessments, the general finding is that the non-shared environmental variance at one age overlaps little with that at a later age—i.e. there appear to be unique and largely uncorrelated factors acting at different ages. For example, with respect to body mass index, the non-shared environmental components at age 20, 48, 57 and 63 years are largely uncorrelated with each other.52 This suggests that exposures contributing to non-shared environmental influences are often unsystematic and of a time- or context-dependent nature. Similar findings have emerged from studies of various other outcomes, with non-shared environmental influences contributing little, if anything, to tracking of phenotypes over time.53 A distinction can be drawn between the stable and unstable aspects of the non-shared environment, with studies tending to point to the latter as being of more statistical importance in terms of explaining variance in the distribution of disease risk. This is a crucial issue, since some environmental exposures which are partly non-shared in adulthood (such as cigarette smoking and occupational exposures) tend to track over time—and thus be stable components of the non-shared environment.
Currently, there is largely an absence of evidence—rather than evidence of absence—of directly assessed systematic non-shared environmental influences on health, and little active research in the biomedical field. However, as the phenotypic decomposition of variance shows similar patterns in the medical, behavioural and social domains, it seems prudent to assume that similar causal structures exist, and equivalent conclusions should be drawn: a large component of variation in health-related traits cannot be accounted for by measureable systematic aspects of the non-shared environment.
Why might the role of shared environment be under-estimated?
The contribution of the shared environment to outcomes may be being under-estimated by current approaches. In his IJE commentary Dalton Conley20 summarizes potential problems with the genetic models from which estimates of the contribution of the shared environment have been made, such as the assumption that twins have the same level of environmental sharing independent of zygosity. Conley,20 Turkheimer21 and Plomin18 all refer to the low proportion of the estimated heritability of many traits that can be accounted for by identified common genetic variants in GWASs, and the apparent mystery of the ‘missing heritability’,54 possibly indicating that heritability has been over-estimated by conventional twin studies. Many features of twin study analysis can be problematic. For example, twin study analysis often assumes that genetic contributions are additive, and that genetic dominance (in the classic Mendelian sense) or gene–gene interactions (epistasis) do not contribute to the genetic variance. Such an assumption can lead to under-estimation of the shared environmental component.55–57 Conversely, twin studies also assume no assortative mating (i.e. parents are no more genetically similar than if randomly sampled from the population) and no gene–environment covariation, both of which can lead to over-estimation of the shared environmental component.55 Different study designs for estimating components of phenotypic variation make different assumptions, however. Conventional twin studies, studies of twins reared apart, extended twin-family studies (in which other family members are included), other extended pedigree studies and adoption studies (including those in which there is quasi-random assignment of particular adoptees) generally come to the same basic conclusions about the relative magnitude of these components.58 All these designs have been applied to the study of body mass index and obesity, with the findings indicating roughly the same magnitude of heritability.55,59–64 This makes it less likely that these are seriously biased, because different biases would all have to generate the same effects, which is not a plausible scenario.
With respect to the ‘missing heritability’, to take the example of height—referred to by both Plomin18 and Turkheimer25—the estimate of the proportion of heritability explained by identified variants they give, of <5%, has already increased to >10%,65 and directly estimated heritability (relating phenotypic similarity to stochastic variation in the proportion of the genome shared between siblings) indicates similar heritabilities to those seen in twin studies.66 Genome-wide prediction using common genetic variation across the genome also points to the effects of measured genetic variation moving towards the expectation from conventional heritability estimates.67 Such data suggest there are large numbers of variants as yet not robustly characterized that are contributing to the heritability of height, with rare variants not identifiable through GWAS probably accounting for much of the remainder. For some diseases, a more considerable proportion of the heritability is already explained by common variants.68 In summary, it seems improbable that heritability has been substantially over-estimated at the expense of shared environment. The basic message that a larger non-shared than shared environmental component to phenotypic variance is the norm is unlikely to be overturned.
Shared environmental effects, although generally small, are more substantial for some outcomes, including musical ability69 and criminality in adolescents and young adults;70 respiratory syncytial virus infection,29 anti-social behaviour,53,71 mouth ulcers72 and physical activity73 in children and lung function in adults.74 Furthermore, findings with respect to shared environmental contributions have face validity. For example, in a twin study applying behavioural genetic variance decomposition to behaviours, dispositions and experiences, shared environmental effects were found for only 9 of the 33 factors investigated.75 However, they were identified for those aspects of life that would appear to depend on shared family characteristics, for example, for a child being read to by a parent, but not for the child reading books on their own. Similarly, the number of years a child had music lessons had a substantial shared environmental component, as might be expected as this will initially depend on the parents organizing such lessons. Continuing to play an instrument into adulthood, however did not have an identified shared environmental contribution. Strictness of parenting style and parental interest in school achievement also had shared environmental contributions, demonstrating that differences in perceptions and reporting styles of the twins do not prevent the identification of such effects. Together, this evidence makes it clear that the methods currently applied can identify the existence of shared environmental effects when they are present.
Shared environmental effects could be under-appreciated because of the limited range of shared environments in study samples, arising through both initial recruitment methods and sample attrition.76 Shared environmental influences on various outcomes have been found to be greater in high-risk families16,77: ones that often have low recruitment and retention rates in population-based studies. Measurement error in the classification of directly measured shared influences, in particular those that change over time, can lead to under-estimation of effects when they are directly studied.
Shared environmental influences within twin and related studies generally apply to infancy, childhood, adolescence and early adulthood and not to later adulthood experiences. Thus, they would encompass many of the aspects of the early life environment—from the antenatal period onwards—that are considered to be important potential contributors to adult disease within the developmental origins of health and disease (DOHaD) arena. Furthermore, many of the factors that are components of the shared environment are ones that are candidate influences on adulthood health—for example, housing conditions, characteristics of area of residence, environmental tobacco smoke exposure, socio-economic circumstances, disruptive social environments and other stressors. Their effects in adulthood would not be expected to be greater than their effects during the sensitive developmental periods of infancy, childhood, adolescence and young adulthood. Shared environment can be addressed through analysis of spousal similarities in health outcomes, as environments are shared to an extent by cohabiting couples, and these also yield what on the face of it are rather small effect estimates. For example, the cross-spousal correlation for body mass index does not change from when couples initially come together (reflecting assortative mating) over many years of them living together in an at least partially shared environment.61
Of most relevance to epidemiological approaches, however, is that models generally fix the shared environmental component to zero if it is not ‘statistically significantly’ different from zero. This is evident in Table 1; with respect to pancreatic cancer, for example, the shared environmental component is given as 0, with a 95% confidence interval (CI) 0–0.35 (i.e. the upper limit being 35% of phenotypic variance). In many cases, it is simply stated that these studies find no effect of shared environmental influences, even though the findings are compatible with quite substantial contributions, but these cannot be reliably estimated in the generally small samples available in twin and adoption studies. Thus, a twin study of aortic aneurysm reported that there was ‘no support for a role of shared environmental influences’,78 with the 95% CI around the effect estimate being 0–27%. A recent meta-analysis found that for various aspects of child and adolescent psychopathology, shared environment makes a non-negligible contribution in adequately powered analyses.79 The claims of there being ‘no shared environmental influence’, which are often made (Box 2), might more realistically be seen as an indication of inadequate sample size and the fetishization of ‘statistical significance’.80
In an entertaining paper, Eric Turkheimer proposed three laws of behavioural genetics, which can be modified for the health sciences as:
First Law: All human health and health-related traits are heritable.
Second Law: The effect of being raised in the same family is smaller than the effect of genes.
Third Law: A substantial portion of the variation in health and health-related traits is not accounted for by the effects of genes or families.170
In many cases, reports have explicitly stated that there is no contribution of the shared environment to outcomes. For example, one representative study concludes that ‘In agreement with other twins studies of asthma and hay fever, no shared environmental influences were detected; in other words, factors related to home and family environment do not seem to contribute to the variance in asthma and hay fever liabilities’.171 Other studies—generally of children, adolescents or young adults, but some extending to later ages—have explicit statements of there being no shared environmental influences for amongst others, schizophrenia,172 bipolar affective disorder,173 blood pressure,174 aortic aneurysm,78 sleep characteristics,175 general cognitive ability,176 teacher-rated aggression,177 extraversion and neuroticism,178 atypical gender development in girls,179 stuttering,180 age at first sexual intercourse in young men,181 autism traits182 and aversion to new foods.183 In many of these cases confidence intervals around the ‘zero shared environmental influences’ are not provided, but when given they are often compatible with an effect magnitude that would be epidemiologically relevant.
Reasons for over-estimating or over-interpreting the non-shared environment
As already mentioned, measurement error in quantitative genetic models is generally categorized as being part of the non-shared environment and this will lead to over-estimation of this aspect of environmental influence on outcomes. Interaction between the non-shared environment and genotype if not modelled can lead to over-estimation of the non-shared environmental effects.81 However correlation between genetic variation and the non-shared environment if not modelled can lead to inflation of the additive genetic component and deflation of the non-shared environment estimates.81 Such correlation is likely to exist between genetic variation and the non-shared aspects of alcohol consumption, for example,82 and the same is likely to be the case for smoking.83 Finally, the effect of an apparently shared environmental factor could qualitatively differ according to characteristics of the siblings. Thus parental divorce can be considered to be a shared environmental influence (it will be reported as having happened for all siblings) and yet its effect on different offspring may be highly disparate, with some offspring suffering adverse consequences of family fracturing, whereas other offspring are benefited by escaping from a conflictive household environment. In this scenario there would be non-shared effects of the shared environment, with influences being due to ‘effective environments’ rather than ‘objective environments’.84 This could lead to an exposure having no detectable overall effect in a population, but still having a causal influence in particular cases.4
The ‘gloomy prospect’ after all?
The possibility that chance or stochastic events contribute to the large non-shared environmental component for many outcomes was not dismissed by Plomin and Daniels because of evidence against it, rather it was not considered a promising research topic.16 Indeed, in a reply to commentators on their original article, they stated that they ‘did not mean to minimize the possible importance of such events’ but that it ‘makes sense to start the search by looking for systematic sources of variance’.85 There is perhaps a reflection here of the story of the drunken man found searching for his keys under a street lamp, who when asked where he had dropped the keys gestured to a distant location, but said he was looking where the light was. If biographical narratives often hinge on chance events, why should the reasons behind the development of one particular case of disease be any less influenced by such events? Perhaps, like the poet Fausto Maijstral in Thomas Pynchon’s novel V, we need to begin ‘the process of learning life’s single lesson: that there is more accident to it than a man can ever admit to in a lifetime and stay sane’.86
The stochastic nature of phenotypic development is something we should not be surprised to encounter (Box 3). In his 1920 paper, ‘The relative importance of heredity and environment in determining the piebald pattern of guinea pigs’, Sewall Wright (Figure 2) presented a seminal path analysis (Figure 3), that has frequently been cited as a source of this particular statistical method.87 Wright observed that ‘nearly all tangible environmental conditions—feed, weather, health of dam, etc., are identical for litter mates’; in current terminology, they are part of the shared environment. Such factors were found to be of minor importance; instead, most of the non-genetic variance ‘must be due to irregularities in development due to the intangible sort of causes to which the word chance is applied’.87 Wright pointed out that measurement error could not be separated from this intangible variance, as is the case with non-shared environment in current parlance. In a later paper,88 Wright and his PhD student Herman Chase independently graded the guinea pig coat patterns, and demonstrated that measurement error was only a minor contributor (Figure 4). A summary table (Table 2) included a shared environmental influence on littermates—age of the mother—but the intangible variance dominated, with the estimate of the magnitude of this being similar to estimates seen for the contribution of the non-shared environment in relation to many human traits.16 In humans, of course, age of mother at conception could be a non-shared environmental factor influencing differences between siblings. In the inbred guinea pig strain, where genetic differences were minor, heredity was not an issue, and the intangible (‘non-shared environmental’) factors were even more dominant.

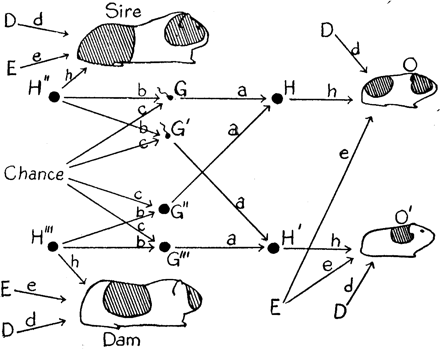
Random phenotypic variance? Sewall Wright's path analysis of the Piebald pattern in guinea pigs87

The ‘approximate analysis of the variance in the random bred stock and isogenic inbred strain’.88 Percentage of variance in coat pattern of guinea pigs attributable to different components
| Isogenic inbred strain | Random bred stock | |
|---|---|---|
| Heredity | 0 | 40 |
| Sex | 3 | 2 |
| Environment | ||
| Age of mother | 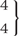 | |
| Other factors common to littermates | 6 | |
| Factors not common to littermates | 89 | 52 |
| 100 | 100 |
| Isogenic inbred strain | Random bred stock | |
|---|---|---|
| Heredity | 0 | 40 |
| Sex | 3 | 2 |
| Environment | ||
| Age of mother | | |
| Other factors common to littermates | 6 | |
| Factors not common to littermates | 89 | 52 |
| 100 | 100 |
The ‘approximate analysis of the variance in the random bred stock and isogenic inbred strain’.88 Percentage of variance in coat pattern of guinea pigs attributable to different components
| Isogenic inbred strain | Random bred stock | |
|---|---|---|
| Heredity | 0 | 40 |
| Sex | 3 | 2 |
| Environment | ||
| Age of mother | | |
| Other factors common to littermates | 6 | |
| Factors not common to littermates | 89 | 52 |
| 100 | 100 |
| Isogenic inbred strain | Random bred stock | |
|---|---|---|
| Heredity | 0 | 40 |
| Sex | 3 | 2 |
| Environment | ||
| Age of mother | | |
| Other factors common to littermates | 6 | |
| Factors not common to littermates | 89 | 52 |
| 100 | 100 |
Changes in obesity prevalence in adults by characteristics (per 100) US adults, by various characteristics151
| Characteristic | 1991 | 1998 | Difference | Increase (%) |
|---|---|---|---|---|
| Sex | ||||
| Men | 11.7 | 17.7 | 6.0 | 51.5 |
| Women | 12.2 | 18.1 | 5.9 | 47.4 |
| Age, years | ||||
| 18–29 | 7.1 | 12.1 | 5.0 | 69.9 |
| 30–39 | 11.3 | 16.9 | 5.6 | 49.5 |
| 40–49 | 15.8 | 21.2 | 5.4 | 34.3 |
| 50–59 | 16.1 | 23.8 | 7.7 | 47.9 |
| 60–69 | 14.7 | 21.3 | 6.6 | 44.9 |
| ≥70 | 11.4 | 14.6 | 3.2 | 28.6 |
| Race | ||||
| White | 11.3 | 16.6 | 5.3 | 47.3 |
| Black | 19.3 | 26.9 | 7.6 | 39.2 |
| Hispanic | 11.6 | 20.8 | 9.2 | 80.0 |
| Other | 7.3 | 11.9 | 4.6 | 62.0 |
| Education levels | ||||
| Less than high school | 16.5 | 24.1 | 7.6 | 46.0 |
| High school | 13.3 | 19.4 | 6.1 | 46.1 |
| Some college | 10.6 | 17.8 | 7.2 | 67.5 |
| College and further | 8.0 | 13.1 | 5.0 | 62.9 |
| Smoking status | ||||
| Never | 12.0 | 17.9 | 5.9 | 48.5 |
| Ex-smoker | 14.0 | 20.9 | 6.9 | 49.4 |
| Current | 9.9 | 14.8 | 4.9 | 50.3 |
| Characteristic | 1991 | 1998 | Difference | Increase (%) |
|---|---|---|---|---|
| Sex | ||||
| Men | 11.7 | 17.7 | 6.0 | 51.5 |
| Women | 12.2 | 18.1 | 5.9 | 47.4 |
| Age, years | ||||
| 18–29 | 7.1 | 12.1 | 5.0 | 69.9 |
| 30–39 | 11.3 | 16.9 | 5.6 | 49.5 |
| 40–49 | 15.8 | 21.2 | 5.4 | 34.3 |
| 50–59 | 16.1 | 23.8 | 7.7 | 47.9 |
| 60–69 | 14.7 | 21.3 | 6.6 | 44.9 |
| ≥70 | 11.4 | 14.6 | 3.2 | 28.6 |
| Race | ||||
| White | 11.3 | 16.6 | 5.3 | 47.3 |
| Black | 19.3 | 26.9 | 7.6 | 39.2 |
| Hispanic | 11.6 | 20.8 | 9.2 | 80.0 |
| Other | 7.3 | 11.9 | 4.6 | 62.0 |
| Education levels | ||||
| Less than high school | 16.5 | 24.1 | 7.6 | 46.0 |
| High school | 13.3 | 19.4 | 6.1 | 46.1 |
| Some college | 10.6 | 17.8 | 7.2 | 67.5 |
| College and further | 8.0 | 13.1 | 5.0 | 62.9 |
| Smoking status | ||||
| Never | 12.0 | 17.9 | 5.9 | 48.5 |
| Ex-smoker | 14.0 | 20.9 | 6.9 | 49.4 |
| Current | 9.9 | 14.8 | 4.9 | 50.3 |
Changes in obesity prevalence in adults by characteristics (per 100) US adults, by various characteristics151
| Characteristic | 1991 | 1998 | Difference | Increase (%) |
|---|---|---|---|---|
| Sex | ||||
| Men | 11.7 | 17.7 | 6.0 | 51.5 |
| Women | 12.2 | 18.1 | 5.9 | 47.4 |
| Age, years | ||||
| 18–29 | 7.1 | 12.1 | 5.0 | 69.9 |
| 30–39 | 11.3 | 16.9 | 5.6 | 49.5 |
| 40–49 | 15.8 | 21.2 | 5.4 | 34.3 |
| 50–59 | 16.1 | 23.8 | 7.7 | 47.9 |
| 60–69 | 14.7 | 21.3 | 6.6 | 44.9 |
| ≥70 | 11.4 | 14.6 | 3.2 | 28.6 |
| Race | ||||
| White | 11.3 | 16.6 | 5.3 | 47.3 |
| Black | 19.3 | 26.9 | 7.6 | 39.2 |
| Hispanic | 11.6 | 20.8 | 9.2 | 80.0 |
| Other | 7.3 | 11.9 | 4.6 | 62.0 |
| Education levels | ||||
| Less than high school | 16.5 | 24.1 | 7.6 | 46.0 |
| High school | 13.3 | 19.4 | 6.1 | 46.1 |
| Some college | 10.6 | 17.8 | 7.2 | 67.5 |
| College and further | 8.0 | 13.1 | 5.0 | 62.9 |
| Smoking status | ||||
| Never | 12.0 | 17.9 | 5.9 | 48.5 |
| Ex-smoker | 14.0 | 20.9 | 6.9 | 49.4 |
| Current | 9.9 | 14.8 | 4.9 | 50.3 |
| Characteristic | 1991 | 1998 | Difference | Increase (%) |
|---|---|---|---|---|
| Sex | ||||
| Men | 11.7 | 17.7 | 6.0 | 51.5 |
| Women | 12.2 | 18.1 | 5.9 | 47.4 |
| Age, years | ||||
| 18–29 | 7.1 | 12.1 | 5.0 | 69.9 |
| 30–39 | 11.3 | 16.9 | 5.6 | 49.5 |
| 40–49 | 15.8 | 21.2 | 5.4 | 34.3 |
| 50–59 | 16.1 | 23.8 | 7.7 | 47.9 |
| 60–69 | 14.7 | 21.3 | 6.6 | 44.9 |
| ≥70 | 11.4 | 14.6 | 3.2 | 28.6 |
| Race | ||||
| White | 11.3 | 16.6 | 5.3 | 47.3 |
| Black | 19.3 | 26.9 | 7.6 | 39.2 |
| Hispanic | 11.6 | 20.8 | 9.2 | 80.0 |
| Other | 7.3 | 11.9 | 4.6 | 62.0 |
| Education levels | ||||
| Less than high school | 16.5 | 24.1 | 7.6 | 46.0 |
| High school | 13.3 | 19.4 | 6.1 | 46.1 |
| Some college | 10.6 | 17.8 | 7.2 | 67.5 |
| College and further | 8.0 | 13.1 | 5.0 | 62.9 |
| Smoking status | ||||
| Never | 12.0 | 17.9 | 5.9 | 48.5 |
| Ex-smoker | 14.0 | 20.9 | 6.9 | 49.4 |
| Current | 9.9 | 14.8 | 4.9 | 50.3 |
The pioneering psychiatrist Henry Maudsley was present at a meeting of the newly founded Sociological Society in April 1904 when Sir Francis Galton talked on ‘Eugenics: its definition, scope and aims’,184 and considered how siblings ‘born of the same parents and brought up in the same surroundings’ could become so different. He concluded that in his opinion ‘we shall have to go far deeper down than we have been able to go by any present means of observation—to the germ-composing corpuscles, atoms, electrons, or whatever else there may be; and we shall find these subjected to subtle and most potent influences of mind and body during their formations and combinations, of which we yet know nothing and hardly realise the importance.’185 Here he is describing how heritability (‘germ-composing corpuscles’) and factors that would be subsumed under the non-shared environment could come together and that ‘in these potent factors the solution of the problem is to be found’.186 In his reply Galton was scathing.186 Referring to a discussion that included spoken or written contributions from George Bernard Shaw, H.G. Wells and William Bateson he declared himself unhappy with the quality of the debate, with two speakers that seemed to him ‘to be living forty years ago; they displayed so little knowledge of what has been done since’, others that ‘were really not acquainted with the facts, and they ought not to have spoken at all’.
Evidence of the importance of chance is abundant in the life histories of many creatures. In genetically identical Caenorhabditis elegans reared in the same environments there are large differences in age-related functional declines, attributable to purely stochastic events.89 In the case of genetically similar inbred laboratory rats, Klaus Gärtner noted the failure to materially reduce variance for a wide variety of phenotypes, despite several decades of standardizing the environment.90,91 Indeed, there was hardly any reduction in variance compared with that seen in wild living rats experiencing considerably more variable environments. The post-natal environment, controlled in these studies, seemed to have a limited effect on phenotypic variation. Embryo splitting and transfer experiments in rodents and cattle demonstrated that the prenatal environment was also not a major source of phenotypic variation.90,91 In genetically identical marbled crayfish raised in highly controlled environments considerable phenotypic differences emerge.94 These and numerous other examples from over nearly a century87,93–98 demonstrate the substantial contribution of what appear to be chance or stochastic events—which in the behavioural genetics field would fall into the category of non-shared environmental influences—on a wide range of outcomes. Finally, even in fully deterministic settings, it has been pointed out that non-linear aspects of autonomous epigenetic processes could generate phenotypic differences that cannot be attributed to either genetic or environmental influences; these have thus been termed ‘a third source of developmental difference’.99,100
As a thought experiment, imagine a lifecourse epidemiologist diligently recording every possible aspect of behaviour, environment and biomarker status of genetically identical marbled crayfish (Figure 5). These data are then used to predict outcomes with the usual epidemiological modelling approaches. How much of the variation in outcomes is our intrepid researcher going to be able to explain?

Variation of growth of genetically identical marbled crayfish in an aquarium: how well would epidemiologists be able to predict outcomes?94 Reproduced with permission from the Journal of Experimental Biology
Mechanisms of chance events: epigenetics to the rescue?
The chance events that contribute to disease aetiology can be analysed at many levels, from the social to the molecular. Consider Winnie (Figure 1); why has she managed to smoke for 93 years without developing lung cancer? Perhaps her genotype is particularly resilient in this regard? Or perhaps many years ago the postman called at one particular minute rather than another, and when she opened the door a blast of wind caused Winnie to cough, and through this dislodge a metaplastic cell from her alveoli? Individual biographies would involve a multitude of such events, and even the most enthusiastic lifecourse epidemiologist could not hope to capture them.101 Perhaps chance is an under-appreciated contributor to the epidemiology of disease.4,102,103
In model organism studies trait deviations due to developmental noise tend to be independent of one another,104 reflecting the generally non-stable characteristics of non-shared environmental influences on human traits discussed above. The stochastic nature of many subcellular processes related to gene expression that could influence development, phenotypic trajectory and disease have been extensively documented.93,97,105–107 Since influencing development and disease requires mitotic heritability of cellular phenotypes, it is unsurprising that epigenetic processes have come to the fore in this regard. For over 40 years Gilbert Gottlieb has stated that with respect to epigenetics ‘outcomes are probabilistic rather than predetermined’.108,109 The pioneering epigeneticist Robin Holliday points out that it is commonly stated that disease is either genetic or environmental, when in reality stochastic events are equally important.110 He goes on to consider that epigenetic defects are simply bad luck.110 Over the past decade the potential contribution of molecular epigenetic processes to stochastic phenotypic variation has been reiterated.97,111–118 Differences in epigenetic profiles between monozygotic twins119 and genetically identical animals92,94 have been presented, that could underlie other phenotypic differences, although the link between epigenotype and phenotype has not been reliably demonstrated. Mechanisms that are ultimately uncovered to explain phenotypic differences between genetically identical organisms would be classified as epigenetic according to many of the current definitions of epigenetics, thus there is an element of self-fulfilling prophecy in postulating that this will be the case. What is now required is concrete demonstrations of epigenotype/phenotype links that could account for much of the so-called non-shared environmental variance, and attempts to demonstrate the causal nature of such links120 (Box 4).
Epigenetics is an area of considerable current research interest, and also of increasingly high profile in the popular scientific literature.168 It is important to draw a distinction between mitotically stable epigenetic changes, which will underlie both normal development and disease within the life of an organism, and meiotically stable epigenetic changes, that can lead to intergenerational transmission of phenotypic dispositions. The former will almost inevitably be of importance in every aspect of development, including the development of disease. The latter, perhaps because of the frisson caused by the neo-Lamarckian heresy of inheritance of acquired characteristics169 has attracted considerable attention, and indeed in discussions of epigenetics it is this contested aspect that often attracts most attention. With regard to being involved in the genesis of phenotypic variation that is not dependent on genetic variation or the environment, intragenerational epigenetic processes are sufficient. Aspects of both shared and non-shared environment could, and indeed in many cases probably do, produce long-term phenotypic changes through the mediating role of epigenetic changes. Epigenetic mechanisms may then integrate the effects of the environment (both measurable and unmeasurable) and purely stochastic molecular events. In this interpretation, epigenetics is not an alternative to other accounts of how development occurs and disease arises, simply a description at one particular level of inherently multilevel processes.
Chance encounters: the advantages of being random
If such a substantial role for chance exists in the emergence of phenotypic (including pathological) profiles, why is this? One possible answer, with a long pedigree,121–123 is that it provides for evolutionary bet-hedging.124 Fixed phenotypes may be tuned to a given environment, but in changing conditions a phenotype optimized for propagation in one situation may rapidly become suboptimal,125 a proposition supported by experimental evidence.126,127 Thus if variable phenotypes are produced from the same genotype, long-term survival of the lineage will be improved, an evolutionary version of the proverb ‘don’t put all your eggs in one basket’.124,128 Edward Miller suggests that large non-shared environmental influences have emerged to provide such a range of phenotypes, and relates this to Markowitz diversification in financial trading, in which holding broad portfolios of shares protects investors from the collapse of particular sectors of the market.129 Unsurprisingly, the extensive decades-old literature on this topic has more recently come to focus on epigenetic processes.111,114–116 A representative example comes from Gunter Vogt and colleagues. Reflecting on their demonstration of considerable phenotypic—including epigenetic—differences between genetically identical crayfish, they conclude that such variation may ‘act as a general evolution factor by contributing to the production of a broader range of phenotypes that may occupy different micro-niches’.94 The substantial non-shared environmental contribution to many outcomes could, therefore, include an element—perhaps substantial—of random phenotypic noise, consequent on stochastic epigenetic processes. At the molecular level, the potential existence of such processes has been observed within twin studies, with the formal demonstration of non-shared environmental contributions to epigenetic profiles130 and of substantial differences in epigenetic markers between monozygotic twins.119
Other mechanisms can also contribute to phenotypic diversity, including meiotic recombination and Mendelian assortment of genetic variants acting on highly polygenic traits, with such genetic variants having small individual effects. Mutation will also increase phenotypic variation. Sibling contrast effects—siblings becoming less similar than their genetic and shared environmental commonalities would suppose—could also provide for such evolutionary bet-hedging.129 Although evidence supporting such a process is sparse, it could lead to inflation of non-shared environmental influences and deflation of shared environment estimates from twin studies.
Evolutionary bet-hedging through random phenotypic noise can be seen as the other side of the coin to the effects of canalization. The latter process allows genetic variation to persist in a population without producing phenotypic effects, until environmental shocks produce decanalization.131 Together, these two apparently countervailing tendencies allow for the maintenance of genetic variation in a population, facilitating species survival during periods of ecological change. Random phenotypic variation can protect genotypes from elimination by selection during cycles of environmental change. Canalization, on the other hand, facilitates the accumulation of what has been termed cryptic genetic variation,131 maintaining within the population the genetic prerequisites for variable phenotypic responses to environmental change. As is generally the case, evolutionary explanations of biological (or social) processes need to be treated with caution.132 They are attractive propositions, however, and even when they have a long history—as with the notion that increased phenotypic variance (perhaps epigenetic in origin) is adaptive—can be greeted as though novel.133 Experimental studies of relevance to this hypothesis are appearing126,127 that will allow future evaluation of its importance.
A gloomy or a realistic prospect for epidemiology and public health?
Epidemiology and public health are population health sciences, but concern for the fate of individuals underlies attempts to control aggregate disease levels. Thus Geoffrey Rose started his seminal paper, ‘Sick individuals and sick populations’134 by saying that
In teaching epidemiology to medical students, I have often encouraged them to consider a question which I first heard enunciated by Roy Acheson: ‘Why did this patient get this disease at this time?’ It is an excellent starting-point, because students and doctors feel a natural concern for the problems of the individual. Indeed, the central ethos of medicine is seen as an acceptance of responsibility for sick individuals.134
Such a question reflects a long tradition in clinical medicine of emphasizing the need to understand the causes of specific events. In Claude Bernard’s An Introduction to the Study of Scientific Medicine (1865) a statistical average—such as the ratio of deaths to recoveries after surgery—is said to mean ‘literally nothing scientifically’, since it ‘gives us no certainty in performing the next operation’.135,136 For each patient who died ‘the cause of death was evidently something which was not found in the patient who recovered; this something we must determine, and then we can act on the phenomena or recognize and foresee them accurately’.135 Both prediction and understanding the causation of individual events are promised by what Bernard referred to as ‘scientific determinism’, the only route to useful knowledge. He went on to dismiss ‘the law of large numbers’ as ‘never teach[ing] us anything about any particular case’.135 Contemporary thought within many disciplines retains this notion. For example, a discussion of what underlies variation within plant clones argues against ‘positing probabilistic propensities governing the behaviour of the plant’; anyone doing so ‘is no biologist’ as an authentic biologist would indeed posit ‘hidden variables and seek evidence for them in more carefully constructed experiments. To do otherwise is to abdicate the scientist's self-appointed tasks.’137
Public health scientists can abdicate their responsibilities in this regard. For our purposes, it is immaterial whether there is true ontological indeterminacy—that events occur for which there is no immediate cause—or whether there is merely epistemological indeterminacy: that each and every aspect of life (from every single one of Winnie’s coughs down to each apparently stochastic subcellular molecular event) cannot be documented and known in an epidemiological context. Luckily, epidemiology is a group rather than individual level discipline,1 and it is at this level that knowledge is sought; thus averages are what we collect and estimate, even when using apparently individual-level data.
At around the same time as Bernard delineated what he considered to be the domain of scientific medicine a very different approach was advanced by Henry Thomas Buckle, in his ‘History of Civilization’.138 Anticipating Durkheim, Buckle reflected on the predictability of suicide rates within populations:
Whereas the exact motivations of the individual suicide are perhaps unknowable (Figure 6), the suicide rate of a population was a predictable phenomenon, and differences between populations were equally predictable. The fully probabilistic interpretation of the law of large numbers held by Simeon-Denis Poisson (holding that the underlying level varies, rather than Buckle’s view that there were ordained rates, with variation around these) accounts for why virtually random micro-level events come together to provide simple, understandable and statistically tractable higher-order regularities.139,140 Happily for epidemiologists, it is precisely these regularities that we deal with.In a given state of society, a certain number of persons must put an end to their own life. This is the general law; and the special question as to who shall commit the crime depends of course upon special laws; which, however, in their action, must obey the large social law to which they are all subordinate. And the power of the larger law is so irresistible, that neither love of life nor the fear of another world can avail anything towards even checking its operation.138

Sylvia Plath and Ted Hughes. In his poem about Plath's suicide, ‘Last letter’ Hughes wrote ‘what happened that night … is as unknown as if it never happened. What accumulation of your whole life, like effort unconscious, like birth pushing through the membrane of each slow second into the next, happened only as if it could not happen’207
Returning to Winnie (Figure 7), as she is part of the tail of a population distribution the existence of someone like her is inevitable. The problem is, of course, that it is not possible to know in advance who will be Winnie and who will be dead from smoking-related disease before their time. Most cases of lung cancer are attributable to smoking, but many smokers do not develop lung cancer. Thus, in the Whitehall Study of male civil servants in London cigarette smoking accounts for <10% of the variance (estimated as the pseudo-R2)141 in lung cancer mortality.102 At the population level, however, smoking accounts for virtually all of the variance—over 90% with respect to lung cancer mortality over time in the USA,142 and virtually all of the differences in rates between areas in Pennsylvania.143 It is in relation to this large contribution of smoking to the population burden of lung cancer that <10% of variance accounted for by cigarette smoking among individuals observed in prospective epidemiological studies, and the 12% shared environmental variance reported in Table 1, should be considered. The shared environmental component will in part reflect shared environmental differences in cigarette smoking initiation.144 The non-shared environmental component (62% of the variance in Table 1) will include the non-shared environmental influence on initiation, amount and persistence of smoking.144 However, as discussed earlier, stable aspects of the non-shared environment—which smoking would tend to be—are generally small contributors to the total non-shared environmental effect, and thus much of this will also reflect the substantial contribution of the kinds of chance events—from the sub-cellular to the biographical—discussed above. Richard Doll, reflecting on the 50th anniversary of the publication of his classic paper with Peter Armitage on the multi-stage theory of carcinogenesis145 considered that
‘whether an exposed subject does or does not develop a cancer is largely a matter of luck; bad luck if the several necessary changes all occur in the same stem cell when there are several thousand such cells at risk, good luck if they don't. Personally I find that makes good sense, but many people apparently do not’.146

Winnie: the tail of a distribution or a ‘black swan’?
In epidemiological studies, exposures and outcomes are assessed at a group level, even when we are apparently analysing individual-level data. In the Whitehall and other prospective studies, we estimate the relative risks as 10 or more for smoking and lung cancer risk,147 but these relative risks relate to groups of smokers compared with groups of non-smokers. Epidemiological inference is to the group, not to the individual.
These reflections will be unexceptional to epidemiologists, as they merely illustrate a key point made by Geoffrey Rose in his contributions to the theoretical basis of population health148,149—that the determinants of the incidence rate experienced by a population may explain little of the variation in risk between individuals within the population. Accounting for incidence differs from understanding particular incidents. Consider obesity in this regard;150 its prevalence has increased dramatically over the past few decades, yet estimates of the shared environmental contributions to obesity are small. Clearly germline genetic variation in the population has not changed dramatically to produce this increase in obesity. However, as Table 3 demonstrates, the prevalence of obesity has increased in both genders, all ages, all ethnic and socio-economic groups, and in both smokers and non-smokers.151 The most likely reason for this is that there has been an across the board shift in the ratio of energy intake to energy expenditure. Study designs utilized to estimate heritability cannot pick this up—twins, for example, are perfectly matched by birth cohort.150 Thus, although energy balance may underlie the burden of obesity in a population—and behind this, the social organization of food production, distribution and promotion, together with policies influencing transportation, urban planning and leisure opportunities—the determinants of who, against this background, is obese within a population could be largely dependent on a combination of genetic factors and chance. The basic principle—that different factors may underlie variation within a population and variations over time or between populations—can be found in the writings of such disparate figures as R.A. Fisher,152 Sewall Wright153,154 and Richard Lewontin,155 although with greatly varying emphasis. Epidemiologists and other population health scientists have drawn out the implications of this well-established and non-controversial insight in ways that link it with classical epidemiological reasoning (Box 5).156–158
‘It is desirable that … loose phrases about the ‘percentage of causation,’ which obscure the essential distinction between the individual and the population, should be carefully avoided’ RA Fisher, Transactions of the Royal Society of Edinburgh, 1918.152
The relationship between explanation of variance within a population and identification of the causes of events or outcomes has a fraught and contested history.58,187 The furore over ‘The Bell Curve’,188 a polemical work on apparent population-level differences in abilities, is one high-profile example. This controversy exists despite some of the basic propositions being accepted by most commentators. Even Francis Galton—the sometime bogeyman of the eugenics movement—wrote ‘Nature prevails enormously over nurture when the differences of nature do not exceed what is commonly to be found among persons of the same rank of society and in the same country’.189 In other words, the contribution of genetic inheritance to differences within a population is large when there is limited environmental variation between people within a particular context. If the context were broadened, the contribution of such environmental factors would be greater. Heritability is not a fixed characteristic, nor does high heritability within a particular situation indicate that environmental change cannot lead to dramatic modification of outcomes. Height—the topic of much of Galton’s own work—is both highly heritable and highly malleable, as changes over time in height make clear.190 Wilhelm Johannsen, the coiner of the term ‘gene’ recognized that in a genetically highly homogeneous group ‘hereditary may be vanishingly small within the pure line’,191 and that in this situation ‘all the variations are consequently purely somatic and therefore non-heritable’.191 Conversely, in a highly standardized environment, the contribution of genetic factors will be increased. It is traditional in epidemiological and related fields to hark back to such trusted thought experiments as how phenylketonuria (PKU) would be expressed against the background of different levels of phenylalanine intake within populations, to demonstrate that the same outcome can be 100% heritable and 100% environmental in different contexts.5,192–197 The point is well made that the presence of a clear genetic predisposition does not mean that environmental change cannot have major effects on disease risk. Perhaps reflecting the contested nature of this area, however, public health academics are sometimes asymmetrical in their reasoning, and after having presented the clear example of PKU they then claim that secular trends and migrant studies—with their unambiguous demonstrations of environmental influences on disease—provide arguments against strong genetic predisposition to common disease.5 This is equivalent to saying that the clear demonstration that genetic lesions underlie PKU in permissive environments argues against any major environmental contribution to PKU.
A second popular thought experiment relates to the possession of two eyes or two legs. The reason humans are almost always born with two of each is genetically determined. However, within a population the trait would not be highly heritable—and certainly not 100% heritable—with loss of a leg or eye generally reflecting accidental events. The distinction between explaining individual trajectories (genes are responsible for the development of two eyes and two legs) and variation in a population is clear, and reflects the distinction between ‘who?’ (why does one person have a disorder or problem rather than another?) and ‘how many’ (what proportion of the population are affected?) questions.198 A distinction between historical origins of explanations of variance in populations (with R.A. Fisher as the exemplar) and of development (with Lancelot Hogben as the advocate) has been highlighted by James Tabery.199,200 This distinction has sometimes been misunderstood, however, as indicating that claims for the ontological status of particular gene by environment interactions should not be judged within the usual framework of scientific scrutiny: consistency of effects and replication. Thus, celebrated apparent gene by environment interactions (with no main genetic effects), such as that between the serotonin transporter gene (5-HTTLPR) and stressful life events in relation to the risk of depression,201 which essentially fail such tests202,203 are said to be being misjudged by the application of statistical evaluation, and should instead be considered as part of the phenomenology of the biology of development.204 Rather than this being the case it may be that inappropriate initial claims are made regarding the existence of group-level processes (an exposure meaningfully categorizable at the population level interacting with a single genetic variant with no appreciable main effect on the outcome) followed by an unwillingness to allow such claims to be evaluated within the framework appropriate for group-level effects. Instead, we may imagine that there are a myriad of almost unimaginable higher order interactions—combinations of unique environments interacting with combinations of genetic variants, which themselves show epistasis—with only a single individual who bears these exposure combinations. Although these may (and in my view almost certainly do) have important influences on individual trajectories, we do not have (and will never have) the tools to identify them. This should not surprise us given the difficulty in disciplining variation during the laboratory study of such processes in highly controlled mouse experiments, for example.205
This paper has focused on the role of ‘the gloomy prospect’ within epidemiology and public health, but similar considerations apply within many other disciplines and discourses. Within sociology, for example, the perhaps under-appreciated role of chance has been emphasised,206 illustrated with entertaining examples from the sporting world. A striking example of what is known as Stein's paradox in statistics is that within-season prediction of the end of season batting averages for particular baseball players is generally better if strongly weighted towards the average of all players at that stage in the season.207 The best guess at what will happen to an individual can often be made by largely discounting individual characteristics. The popular recognition of the importance of chance in people's lives164 can also influence response to cultural artefacts. Thus in films, novels or plays explanation of events is often near-deterministic, which in certain circumstances appears satisfying. Consider Alfred Hitchcock's film Marnie. The behaviour of the eponymous character—fear of thunderstorms, the colour red and men, together with her thieving and frigidity—is all explained at the end of the film by a particular event occurring when Marnie was six. She discovered her prostitute mother with a client during a thunderstorm and ended up killing him (in a cinematic shock of bright red blood) with a poker. Everything seamlessly rolled on from this event. In crime stories this is often what the reader wants. As Stephen Kern entertainingly demonstrates208 the range of causal models in such narratives has a similar range to epidemiology—from the long-arm of early life (or prenatal) events through to primarily psychological and social causation. Outside of murder novels, however, the factitious nature of such explanations can be entirely unsatisfactory. The apparent reality of the well-told narrative appears unreal precisely because everything is tied up and explained—a notion that has resonance with David Shield's literary manifesto Reality Hunger.209 To take one example, the clunking plots of the novels of Ian McEwan—Saturday for example—revolve around such faux ‘explanations’. The work of McEwan—and similar purveyors of book club fare, such as Jonathan Franzen—appear, paradoxically, much less true than such novels as Laurence Sterne's Tristram Shandy, Macado de Assis' Epitaph of a Small Winner, Blaise Cendrars' Moravagine or Alasdair Gray's Lanark, which are apparently not seeking such realism. In these works explanations, when offered, become things to be explained, and the often random nature of the world as codified in people's experience is respected.
Poor individual prediction does not just apply to human lives. Each earthquake surprises us, although we know very well in which parts of the world they are likely to occur. The science of earthquake prediction is certainly one that has had to embrace the gloomy prospect.210 Historical occurrences are of an essentially individual nature, with both chance and potentially understandable causes playing a role.211 Attempting a confident causal narrative in the absence of group level data from multiple but equivalent events is analogous to providing a complete description of why Winnie didn't develop smoking related disease whilst another particular individual did. Evolution is another one-off, in which much appears random and even detecting correlations between major environmental change and the speed of evolutionary change has proved difficult.212 Of course if we could run historical or evolutionary processes repeatedly, there would have been different trajectories and outcomes on each occasion. Some regularities would also appear analogous to the group-level differences in disease incidence rates seen in relation to group-level exposure differences. Chance (at one level) and near necessity (at another) may be the only certainty in attempting to understand epidemiological—and many other—processes.
Rose illustrated this point with the thought experiment of a population in which all the individuals smoke 20 cigarettes a day, in which ‘clinical, case–control and cohort studies alike would lead us to conclude that lung cancer was a genetic disease; and in one sense that would be true, since if everyone is exposed to the necessary agent, then the distribution of cases is wholly determined by individual susceptibility’.134 I would contend that the role of chance events, in addition to genetic variation, in influencing who would develop lung cancer in this setting should be added here.
We can now reflect again on Table 1, where it is suggested that the components of variance for lung cancer are 26% heritable, 12% shared environment and 62% non-shared environment. These figures are entirely compatible with smoking being far and away the preeminent tangible environmental cause of lung cancer, and responsible for the incidence rate of lung cancer in any population. The heritability of lung cancer will in part reflect the heritability of smoking behaviours.144 Indeed, the first molecular genetic variation identified in hypothesis-free genome wide association studies of lung cancer was in a gene related to nicotine reception and smoking behaviour.159–161 This association of a genetic variant linked to a modifiable exposure (in this case cigarette smoking) with lung cancer constitutes evidence within the Mendelian randomization framework162 that cigarette smoking causes lung cancer. We clearly do not require such confirmation in this case. However if the link between smoking and lung cancer had not already been established identification of such germline genetic influences would have pointed epidemiologists in the right direction. Interaction between genetic variation and the non-shared environment—in this case the non-shared aspect of smoking behaviour—is classified within the genetic variance in quantitative genetic models.56 Such interactions may not be insubstantial and could be informative with respect to the casual nature of environmental factors.167 The link between smoking-associated genetic variation and lung cancer illustrates the potential of hypothesis-free identification of causal relationships within observational data.163 Importantly, a genetic variant that itself accounts for a very limited proportion of the heritability of lung cancer—which in turn is a modest proportion of the overall variance in lung cancer risk—can provide robust causal evidence about a modifiable risk factor that the large majority of lung cancer cases can be attributed to. Indeed, such genetic variants can provide randomized evidence in situations where randomized controlled trials may not be possible.
As might be expected, there is a more substantial shared environmental contribution to initiation of cigarette smoking—generally occurring in adolescence, when siblings are residing in a common home—than to smoking persistence (which can stretch into much later adulthood) and amount.144 The modest shared component of variance in lung cancer risk indexed in Table 1 will relate, in part at least, to an exposure that accounts for most of the burden of lung cancer in a population. The very substantial non-shared environmental component will contain some non-shared contribution to smoking behaviour (such as peer group influences), together with random events occurring within, or to, particular individuals. From an epidemiological or public health perspective the relatively small shared environmental and individual molecular genetic contributions to lung cancer risk can be very informative about what underlies the vast majority of all of the disease in a population. The large non-shared environmental component, on the other hand, is much less informative in this regard.
These considerations also address the apparent paradox, mentioned above, regarding the use of sibling controls in epidemiological studies. The relatively small shared environmental effects can generate associations through residual confounding that are of the order of magnitude of many epidemiological associations, although in terms of variance explained for the outcome the effects are small. This is because such shared environmental factors can be strongly related to the exposure under consideration. In the example discussed previously of maternal smoking during pregnancy, this could be very strongly related to family-level socio-economic circumstances and parental education. Confounding by parental genetic factors may also occur, and this could generate or contribute to the observed associations. Confounding by these family—level socio-economic or genetic factors will be taken into account in a between—siblings analysis. When the substantial non-shared environmental influences are largely due to chance events they will be unrelated to exposures under investigation, and will not lead to systematic confounding. Thus despite the often large apparent effects of the non-shared environment, such contributions will generally not be a source of systematic confounding in epidemiological studies.
Lay and professional epidemiology: catching up with common sense?
In the first sustained presentation on the importance of the non-shared environment,22 Rowe and Plomin noted that after the birth of a second child parents are often struck by how different their two children are, despite upbringing being in common. In relation to health, non-professional understanding of causes of disease regularly identify the role of chance (or fate)164 and heritable factors165 as being of considerable importance. Indeed I have to confess that when I was involved in a cross-disciplinary project exploring the construction of models of disease causation held by the general public—which we referred to as ‘lay epidemiology’2—I was disappointed that, for the public at large, there appeared to be a concentration on such apparently individual factors as inheritance and fate, rather than my preferred model of the socio-political determinants of health.166 The cognitive mapping of the lay epidemiology of disease is predicated on observing individual cases among relatives, friends, acquaintances and public figures, and as such the essential unpredictability of these events fully supports the popular analysis. At a group level, the underlying social causes of IHD could be social and political structure, sequentially mediated through free trade in toxic microenvironments, in health-related behaviours, and in elevated body mass index, blood pressure, serum cholesterol, glucose and insulin. At an individual level, it is mostly genes and chance.
Learning to live with randomness: reaffirming the role of epidemiology in the decade of the epigenome
Epidemiologists have survived the decade of the genome relatively unscathed, content to be phenotypers for their genetic colleagues, and accept the redefinition of authorship on >100 ‘author’ papers reporting tiny relative risks associated with common genetic variants. As we enter the next decade—clearly with the epigenome to the fore—how should we understand our role? One perhaps counter-intuitive way is to embrace the findings of quantitative genetics and realize they actually enhance the importance of the insights that epidemiology brings. First, most traits have a non-trivial genetic component. This is good news: it means that genetic variants can be utilized as instrumental variables for the near-alchemic act of turning observational into experimental data, and allow the strengthening of causal inference with respect to environmentally modifiable exposures, in the absence of randomized trials.162,167 Indeed, we might even enter the age of hypothesis-free causality.163 Second, exposures that affect disease risk at a group level may have small effects in quantitative genetics terms (‘variance explained’), but they are both something that public health policy can do something about and they can account for the large majority of the cases of disease in a population. Third, unstable aspects of the non-shared environment in childhood and adulthood probably largely consist of chance events, about which we can do nothing. We should be happy that their random nature means they are not systematically related to the things we are interested in—and are therefore not confounders. Stable aspects of the non-shared environment, whilst in terms of ‘variance explained’ appearing small, are more promising as indicators of potential levers for health improvement. Finally, in terms of public health policy, we should target the modifiable causes of disease that heritability and shared environment tell us about. This must be at a group level, however, and we should do so without pretending to understand individual-level risk (Box 6), or misrepresent population level data (smokers die earlier on average) as individual level events (each smoker shortens her or his life). If we pretend the latter then every Winnie (Figure 7) is a ‘black swan’, the existence of whom proves that not all swans are white. Health promotion approaches that have less coherent views on disease causation than those popularly held are bound to be unsuccessful.2 Chance leads to averages being the only tractable variables in many situations, and this is why epidemiology makes sense as a science. We should embrace the effects of chance, rather than pretend to be able to discipline them.
Personalized medicine has been promoted as a way of improving therapeutic effectiveness, by targeting treatments to the characteristics of individual patients. The figure215 presents trajectories of severity of depression over time for 20 participants with the same initial diagnosis treated with the same anti-depressant in one arm of a randomized controlled trial, GENDEP,216 the aim of which was described at its inception as being to revolutionize the treatment of depression … [and] … to make it easier for doctors to decide which antidepressant will be most likely to work for a given depressed person.217 The trajectories in the figure have been used to illustrate the potential to identify gene by environment interactions, or phenotypic sub-groups that would respond differentially to particular treatments.215 The lines of evidence that have been reviewed in this paper suggest that this may be an over-ambitious aim, with the trajectories reflecting ontologically or epistemologically stochastic events rather than epidemiologically tractable ones. Indeed, in the event the GENDEP study failed to identify any robust genetic influences on treatment response, or sub-categorizations of depression that reliably predicted outcome.218,219
Now let us consider the most celebrated cases of supposedly personalized medicine. These include the identification of genetic variants related to adverse responses to the drugs abacavir, the statins and flucloxacillin,8,220,221 genetic variants related to the appropriate dose of drugs such as warfarin and clopidogrel,8 or the identification of sub-groups of patients with leukaemia or breast cancer who respond to particular treatments.8 These findings do not in reality relate to individual patients; rather the data have been produced with respect to (and can only be appropriately applied to) particular groups of patients. Consider statin myopathy, a condition that occurred in around 1% of participants in the SEARCH randomized controlled trial.221 A common variant in the gene SLCO1B1 was strongly associated with risk (odds ratio for myopathy 4.5 in heterozygotes and 16.9 in risk allele homozygotes). However over a quarter of the population are carriers of the risk variant, and any treatment implications apply to the large groups defined by such carriage. In the case of the use of imatinib in leukaemia the personalization of treatment relates to identification of the sub-group of leukaemias that fall into the chronic myelogenous leukemia category. Similarly trastuzumab (Herceptin) is appropriate treatment for the quarter to a third of breast cancer patients whose tumours express the growth factor receptor HER2. In all these cases treatments are not personalized; rather they are stratified—hence the adoption of the term stratified medicine rather than personalized medicine by many authorities.
In the case of prevention rather than therapeutics an analogous situation is encountered. With respect to coronary heart disease (CHD), for example, individually targeted health promotion aimed at risk factor management (such as smoking cessation) has had very disappointing results.222 Conversely, population-level data demonstrate substantial and rapid reductions in smoking levels and CHD rates over time.223 Population aggregate data present a very different picture regarding the preventability of CHD than data on individuals suggests. Epidemiological reasoning would have led us to anticipate that group-level processes require group-level analysis and group-level solutions. As with therapeutics, stratified rather than personalized approaches are what is required.
A major component of inter-individual differences in risk of disease is accounted for by events that are not epidemiologically tractable, including stochastic events ranging across the sub-cellular and cellular level, to chance biographical events and idiosyncratic gene by environment interactions. These will fall into the ‘non-shared environmental’ component of variance identified in behavioural genetic analyses. Even if their causal contribution could be identified, there would often be no implications for disease prevention, as such events do not generally provide targets for intervention.
At the level of populations, rather than individuals, a large proportion of cases of disease will often be attributable to modifiable influences that only account for a small proportion of inter-individual variation in risk. These may be elements of ‘the shared environment’ in childhood or the adulthood equivalent of such group-level exposures.
Epidemiology is a group-level discipline. As Jerry Morris stated in his seminal ‘The Uses of Epidemiology’ over 50 years ago, ‘the unit of study in epidemiology is the population or group, not the individual’.1 Epidemiology relates to incidence rather than particular incidents.
Ecological studies directly address causes of population disease burden but are subject to many well-known biases, and aetiological hypotheses they support require testing in different study designs. However, the fact that we collect data at the level of the individual does not detract from the fact that in most situations we can only make inferences to groups, and not to individuals.
Genetic variants are borne by individuals but, like other exposures in an epidemiological context, must usually be analysed at a group level.
We should not conflate individual- and group-level explanation. In an insightful paper David Coggon and Chris Martyn4 convincingly present the case for the highly stochastic nature of disease causation. However, they consider that substantial variation between populations in disease rates, or rapid changes in incidence over time, provide an exception to this rule. In fact chance processes at an individual level together with almost entirely explicable group level differences are in no way contradictory, indeed should be expected.
The substantial stochastic element in disease causation and treatment response suggests that fully personalized medicine is an unlikely scenario. Indeed the move from personalized to stratified medicine reflects the fact that in most situations group-level rather than purely individual data contribute to appropriate treatment decisions, and provide the empirical basis for evidence – based medicine and best practice treatment guidelines. The tension between more reliable estimates based on larger groups and the essentially individual nature of medical encounters is a long running one,213 highlighting the importance and difficulty of identifying the smallest coherent groups for which reliable treatment effects can be estimated.
Acknowledgements
Thanks to Ezra Susser for comments on an earlier draft of this article. Ezra reminds me that when we first met—longer ago than I care to remember—one of our first topics of conversation was why siblings were so different, and the implications of this for epidemiology. We have intermittently continued this conversation ever since. Martin Shipley kindly provided me with estimates of variance from the Whitehall Study many years ago; these have finally been used here. Thanks also to Yoav Ben-Shlomo, Jenny Donovan, Shah Ebrahim, Dave Evans, Sam Harper, Nancy Krieger, Debbie Lawlor, Dave Leon, John Lynch, Nick Martin, Marcus Munafo, Neil Pearce, Robert Plomin, Caroline Relton, Benjamin Rokholm, Sharon Schwartz, Marc Schuckit, Thorkild Sørenson and Nic Timpson for helpful comments on earlier drafts.
Conflict of interest: None declared.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}