





Abstract
Cells adhere to each other and to the extracellular matrix (ECM) through protein molecules on the surface of the cells. The breaking and forming of adhesive bonds, a process critical in cancer invasion and metastasis, can be influenced by the mutation of cancer cells. In this paper, we develop a nonlocal mathematical model describing cancer cell invasion and movement as a result of integrin-controlled cell–cell adhesion and cell–matrix adhesion, for two cancer cell populations with different levels of mutation. The partial differential equations for cell dynamics are coupled with ordinary differential equations describing the ECM degradation and the production and decay of integrins. We use this model to investigate the role of cancer mutation on the possibility of cancer clonal competition with alternating dominance, or even competitive exclusion (phenomena observed experimentally). We discuss different possible cell aggregation patterns, as well as travelling wave patterns. In regard to the travelling waves, we investigate the effect of cancer mutation rate on the speed of cancer invasion.
1. Introduction
Normal cells proliferate, divide and die in a highly controlled manner. This proliferation requires mitogenic signals, which are transmitted into the cell by the transmembrane receptors and bind signalling molecules, i.e. diffusible growth factors, extracellular matrix (ECM) components and cell–cell adhesion molecules (Hanahan & Weinberg, 2000). When a cell divides, its DNA is copied by the two new cells. However, if the DNA is damaged or not copied correctly, the new damaged cells will either die or start to proliferate in an uncontrolled manner, creating a signalling of oncogenes that act by mimicking growth signalling (Hanahan & Weinberg, 2000), eventually leading to an abnormal mass of tissues from cells that differ in clinically important phenotypic features (Marusyk et al., 2012). More precisely, tumour formation is a result of clonal expansion driven by somatic mutation, developed by a single precursor (monoclonal) that undergoes genetic and biological changes (Khalique et al., 2007; Nowell, 1976). This transformation of normal cells to cancer cells is a multistep process described via the steps of hyperplasia, premalignant change and dysplasia (Beckmann et al., 1997). Cancer cells lose their ability to regulate genome stability which leads to further genetic changes and tumour development (Khalique et al., 2007). Over the last three decades it has been shown experimentally that tumours consist of heterogeneous populations of cells, which are the result of genetic instability (Stackpole, 1983; Khalique et al., 2007; Loeb & Loeb, 2000; Martelotto et al., 2014). Intra-tumour heterogeneity appears in almost all phenotypic cell features: from cell morphology, to gene expression, motility, proliferation, immunogenicity and metastatic potential (Nicholson, 1984, 1987; Marusyk & Polyak, 2010). It should be mentioned that also normal cells are heterogeneous for various characteristics (e.g. surface antigens). Nevertheless, cellular heterogeneity is more pronounced in malignant neoplasms (Nicholson, 1987). Since the characteristics of the most abundant cell types inside these heterogeneous tumours might not necessarily predict the properties of mixed populations (Marusyk et al., 2012), we need to gain a better understanding of the dynamics of tumours formed of different mutated cell types. In fact, experimental studies have shown complex interactions between clonal sub-populations: from stable coexistence to dominant behaviours (Schuh et al., 2012). For example, some studies have shown the possibility of having competitive exclusion of clonal cancer cell sub-populations in heterogeneous tumours (Leith et al., 1989; Schuh et al., 2012). Other studies have shown that some tumour clones can compete with alternating dominance (Keats et al., 2012). We emphasize that the clonal composition of heterogeneous tumours usually changes over time (Greaves & Maley, 2012), and hence we can expect to see different competition outcomes as time increases (e.g. from transient coexistence of multiple clones to long-term competitive exclusion). Moreover, in some cancer clones there is evidence of contact domination, i.e. the inhibition of growth in some clones is the result of cell–cell contact between various cell sub-populations (Aabo et al., 1994).
The metastatic and invasive potential of heterogeneous tumours is influenced by the interactions amongst cells, and the interactions between cells and the ECM, via cell surface receptors and various cytokines and chemokines. A major group of cell surface receptors is represented by the integrins, which are involved in both cell–cell adhesion and cell–matrix adhesion (Weitzman et al., 1995). In particular, the successful colonization of new sites by cancer cells requires changes in integrin expression (Hanahan & Weinberg, 2000). Another group of molecules involved in cell–cell adhesion is represented by the cadherin families (Hanahan & Weinberg, 2000). The processes through which cells bind to each other and to ECM via these surface receptors, are responsible for tissue formation, stability and breakdown (Armstrong et al., 2006). In particular, to detach from the main aggregation/tissue, cells loose cell–cell adhesion and strengthen cell–matrix adhesion, which leads to ECM remodelling and degradation (with the help of enzymes called matrix metalloproteinases), thus helping cell migration through the ECM (Friedl & Wolf, 2003).
Over the last 20 years, mathematical models have been used intensively to try to gain a better understanding regarding the mechanisms behind cancer invasion and metastasis or the mechanisms behind the aggregation of other types of cells (see, e.g. Ambrosi & Preziosi (2002); Andasari & Chaplain (2012); Andasari et al. (2011); Anderson et al. (2000); Armstrong et al. (2006); Byrne & Preziosi (2003); Chaplain et al. (2011); Cristini et al. (2009); Deakin & Chaplain (2013); Domschke et al. (2014); Dyson et al. (2016); Enderling et al. (2006, 2010); Gerisch & Chaplain (2008); Green et al. (2010); Knútsdóttir et al. (2014); Mogilner & Edelstein-Keshet (1995); Mogilner et al. (1996); Painter et al. (2010); Sherratt et al. (2009); Painter et al. (2015) and many references therein). The early mathematical models were described by local systems of partial differential equations incorporating some generic chemotaxis or haptotaxis mechanisms (Gerisch & Chaplain, 2008; Painter, 2009), or incorporating implicit cell–cell interactions via tumour surface forces (Byrne & Chaplain, 1996). For example, Anderson et al. (2000) developed a local Partial Differential Equations (PDE) model of parabolic type for the invasion of cancer cells via cell–matrix interactions that lead to ECM degradation. Byrne & Chaplain (1996) modelled phenomenologically the influence of cell adhesion on tumour growth, by considering surface forces on tumour spheroids. As it became more clear that the movement in response to chemical/haptotactic gradients was facilitated by the binding and unbinding of cell surface molecules to other cells and to ECM, new mathematical models of parabolic type have been derived to describe these cell–cell and cell–matrix adhesion processes (Armstrong et al., 2006; Dyson et al., 2016; Gerisch & Chaplain, 2008; Gerisch & Painter, 2010; Green et al., 2010; Painter et al., 2015). Since these models incorporate the assumption that cells at position x bind/unbind to/from other cells at position x ± s (for some s > 0 within cells sensing radius), they are generally nonlocal. For example, Armstrong et al. (2006) focused on cell movements due to cell–cell adhesion, and introduced a nonlocal term that described the nature, the direction, as well as the strength of the adhesive forces between cancer cells. The authors also extended the nonlocal model to two populations that interact via adhesive forces (thus incorporating self-population and cross-population adhesion), and studied the effect of different adhesion strengths on the sorting or the mixing of cell populations. A similar two-population nonlocal model of parabolic type, which incorporated also cell proliferation and cell movement in response to cell ‘packing’, was described and investigated by Painter et al. (2015). The model in Armstrong et al. (2006) was further generalized by Gerisch & Chaplain (2008), Painter et al. (2010), Domschke et al. (2014) to include also cell-matrix interactions. While the majority of these models investigated cell–cell and cell–matrix adhesion in one cell population, a few models considered also multiple interacting cell populations (Domschke et al., 2014). In particular, Domschke et al. (2014) also assumed mutation between different cell populations. A different class of nonlocal models for cancer invasion has been developed in the context of the velocity-jump framework introduced in Othmer et al. (1988) and the kinetic models for active particles framework described, e.g. in Bellomo et al. (2010); see the transport models in Kelkel & Surulescu (2012), Lorenz & Surulescu (2014), Engwer et al. (2015, 2017), Hunt & Surulescu (2017).
We should emphasize here that all these nonlocal models for cell invasion are variations or generalizations of nonlocal models developed over the last two decades to describe the dynamics of cell populations (Edelstein-Keshet & Ermentrout, 1990), bacterial populations (Othmer et al., 1988; Xue et al., 2011; Perthame et al., 2016) or the dynamics of self-organized animal populations (Mogilner & Edelstein-Keshet, 1999; Topaz et al., 2006; Eftimie et al., 2007; Fetecau & Eftimie, 2010; Carrillo de la Plata et al., 2015; Fetecau, 2011). While the models describing collective cell movement are usually of parabolic type, the latest models for collective bacterial movement and collective animal movement are usually of hyperbolic/transport type.
In this study, we will investigate the role of cancer mutation on the possibility of clonal competition with alternating dominance or even competitive exclusion between two cancer cell sub-populations (as discussed before, these types of competition have been observed experimentally (Leith et al., 1989; Keats et al., 2012; Schuh et al., 2012)). To this end, we will introduce a nonlocal model for cell–cell and cell–matrix adhesion for two populations of cells (this model is a generalization of the models in Armstrong et al. (2006); Gerisch & Chaplain (2008); Painter et al. (2015)). However, in contrast to these previous models which are of parabolic type, here we consider a parabolic–hyperbolic model. More precisely, we assume that one ‘normal’ cancer cell population moves both randomly and in a directed manner in response to cell–cell and cell–matrix adhesive forces, while a second ‘mutant’ cancer cell population (i.e. a mutated clone) moves predominantly in a directed manner following cell–cell interactions (with cells from both populations) and cell–matrix interactions (as suggested by experimental observations in Goswami et al. (2005); Hagemann et al. (2005)). By incorporating this assumption, we aim to bring a more realistic approach to the models since the various intra-tumour cell sub-populations have been shown to be distinct not only in their adhesion capabilities but also in their motility and metastatic potential (Marusyk & Polyak, 2010). Moreover, we are interested in investigating the effect of mutation on the spreading speed of the mutated cancer cells, since the invasion levels of the mutant cancer cells are higher compared to those of ‘normal’ cancer cells (Chapman et al., 2014; Wojciechowska & Patton, 2015). Also, while previous nonlocal models have assumed constant adhesive interactions, here we will assume that these adhesive interactions depend on the level of integrins. Note that instead of assuming a kinetic (mesoscale) approach for cell-dependence on integrins as in Lorenz & Surulescu (2014), Engwer et al. (2015), Perthame et al. (2016), Engwer et al. (2017), Hunt & Surulescu (2017), here we follow a microscale–macroscale approach similar to Stinner et al. (2014, 2015, 2016) and Meral et al. (2015a,b) and couple the parabolic–hyperbolic model for spatial cell dynamics with an Ordinary Differential Equation (ODE) for integrins dynamics. We then investigate this complex multiscale model in terms of pattern formation, with particular attention being payed to the role of mutation rate on the coexistence (or not) of cell sub-populations that form stationary aggregations or travelling wave patterns.
The layout of this paper is as follows. In Section 2 we formulate a model of partial integro-differential equations for the dynamics of two cancer cell populations, coupled with ordinary differential equations describing ECM and integrins dynamics. In Section 3 we use linear stability analysis to investigate the ability of the model to form cell aggregations. In Section 4 we investigate numerically some types of spatio-temporal patterns exhibited by this nonlocal model. In Section 5 we investigate numerically and analytically travelling wave behaviours. We conclude in Section 6 with a summary and discussion of the results.
2. A mathematical model of two cancer cell sub-populations
2.1. Derivation of the model
Cancer invasion is a complex process involving cell–cell interactions, and interactions between cells and non-cellular components (Calvo & Sahai, 2011). To develop our model we consider two populations of cancer cells, a ‘normal’ (original clone) cancer population and a ‘mutant’ (descendant clone) population, which interact with each other as well as with the ECM via long-range adhesive and repulsive forces (Deman et al., 1976; Geiger, 1991); see Fig. 1.
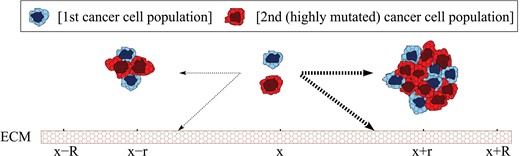
A caricature illustration of movement decisions made by cells at x, following interactions with neighbouring cells at x − r and x + r, and with the ECM. For very strong attraction (represented here by thicker arrows), cells move towards larger cell aggregations.
Let |$\varOmega \subset \mathbb {R}^{d} $| denote a bounded spatial domain (here we consider only d = 1; for a 2D version of the model see Appendix A), with periodic boundary conditions. Let |$I_{T} = \left [0,\infty \right )$| be the time interval. Denote by |$u_{1}\left (x,t\right )$| the density of ‘normal’ cancer cells at position x and time t, and by |$u_{2}\left (x,t\right )$| the density of ‘mutant’ cancer cells at position x and time t. We also denote by |$f\left (x, t\right )$| the ECM density, and by |$c\left (x, t\right )$| the density of integrin receptors on the surface of cancer cells (receptors involved in cell–cell and cell–matrix interactions). For compact notation, we define the vectors |$\underline {u}\left (x,t\right ) =\left ( u_{1}\left (x,t\right ), u_{2}\left (x,t\right )\right )^{\top }$| and |$\underline {\upsilon }\left (x,t\right ) =\left (\underline {u}\left (x,t\right ), f\left (x,t\right )\right )^{\top }$|.
Note that with this choice of boundary condition, the nonlocal terms are wrapped around the domain. We will return to this aspect in Section 4.
Note that equations (2.13)–(2.16) describe a ‘microscale–macroscale’ model, where the spatially-averaged microscale integrin dynamics (described by an ODE) is coupled with the spatial dynamics of cancer cells (described by nonlocal PDEs). As mentioned above, this modelling approach follows various other recent studies on cancer cell–matrix adhesion via integrins (Stinner et al., 2014, 2015, 2016; Meral et al., 2015a,b). Moreover, the coupling of PDEs for cell movement with ODEs for the dynamics of cell receptors has been considered not only for cancer cells but also for other types of cells, such as ‘Dictyostelium’ cells (Höfer et al., 1995a,b). However, the past 10–15 years have seen the development of structured cell population models (described by ‘mesoscale’ models) where the macroscopic cell dynamics is dependent on a microscopic variable that characterizes the cells at molecular level (Erban & Othmer, 2004, 2005; Xue et al., 2011; Lorenz & Surulescu, 2014; Engwer et al., 2015; Perthame et al., 2016; Engwer et al., 2017; Hunt & Surulescu, 2017). In Appendix B we present a modified mesoscale version of model (2.13)–(2.16) where we assume that the two cancer cell populations are structured by an internal variable that describes the integrin level. (Here we use the terminology of ‘mesoscale’ and ‘microscale–macroscale’ models in the same spirit as Stinner et al. (2014), Lorenz & Surulescu (2014) and Hunt & Surulescu (2017).)
2.2. Non-dimensionalization of the model
From now on, we will always refer to the non-dimensional quantities. In the next two sections, we discuss the dynamics of model (2.22).
3. Linear stability analysis of the model
Next, we investigate the conditions under which the two populations of cancer cells form aggregations. We first calculate the spatially homogeneous steady states of the model, and then apply standard linear stability analysis to investigate the conditions (for parameter values) under which the two cancer cell populations can form aggregations.
Since all eigenvalues are negative, the state (0, 1, 0, p2/q) is always linearly stable to homogeneous perturbations.
Cellular aggregations form when the steady states |$(u_{1}^{*},u_{2}^{*},f^{*},c^{*})$| are unstable to spatial perturbations, and for this to happen we require the maximum real part of eigenvalues of the Jacobian matrix of system (3.6) to be positive, for at least one discrete value of k > 0.
- For the state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 0, f^{\ast }, 0\right )$| we obtain the eigenvalues(3.7)\begin{align} \lambda_{1}=0, \;\;\; \lambda_{2}=-k^{2}D-M+r_{1}, \;\;\; \lambda_{3}=r_{2}>0, \;\;\; \lambda_{4}=-q<0. \end{align}
Thus, the steady state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 0, f^{\ast }, 0\right )$| is unstable.
- For the state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 1, 0, p_{2}/q\right )$| we obtain the eigenvalueswhere |$Y\left (k\right )=\frac {2k}{R}\hat {K}^{s}\left (k\right )$|. Recall that in the absence of diffusion and advection, the steady state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 1, 0, p_{2}/q\right )$| is linearly stable. Thus, for aggregation to form, we require this state to be unstable to spatially inhomogeneous perturbations (Benson et al., 1993), i.e. the maximum real part of the eigenvalues to be greater than zero. Since λ1, λ2, λ4 < 0, the steady state |$\left (0, 1, 0, p_{2}/q\right )$| is unstable if the following dispersion relation holds:(3.8)\begin{align} \lambda_{1}=-\beta <0, \quad \lambda_{2}=-k^{2}D-M<0, \quad \lambda_{3}=S_{2}\left(c^{\ast}\right)Y\left(k\right)-r_{2}, \quad \lambda_{4}=-q<0, \end{align}(3.9)\begin{align} Re\left(\lambda_{3}\left(k\right)\right)=Re\left(-r_{2}+\frac{2ks^{\ast}_{2}}{R}\left(1+\tanh\left(a_{2}\frac{p_{2}}{q}\right)\right)\hat{K}^{s}\left(k\right)\right)>0. \end{align}
An example of such dispersion relation is shown in Fig. 2. Note the steady-state/steady-state mode interaction that occurs when we increase the parameters qa and qr (i.e. the strength of attractive and repulsive cell–cell interactions). There is a range of k −values for which |$Re\left (\lambda _{3}\left (k\right )\right )$| is positive, and thus aggregations can arise from spatial perturbations of the steady state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 1, 0, p_{2}/q\right )$|. This means that the steady state |$\left (0, 1, 0, p_{2}/q\right )$|, which is linearly stable for the homogeneous system, is destabilized due to the spatial dynamics (diffusion and advection). Setting diffusion coefficient zero we observe that there is still a range of k −values for which |$Re\left (\lambda _{3}\left (k\right )\right )>0$|, implying the adhesion-driven instability.
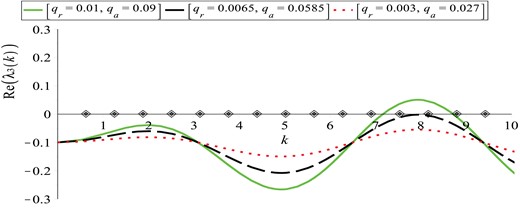
Plot of the dispersion relation (3.12) for the steady state |$\left (0, 1, 0, p_{2}/q\right )$|. The curves represent Re(λ3(k)) for: (a) qr = 0.01, qa = 0.09 (solid green curve); (b) qr = 0.0065, qa = 0.0585 (dashed black curve); (c) qr = 0.003, qa = 0.027 (dotted red curve); the rest of the model parameters are given in Table 2. The diamonds on the x-axis represent the discrete wave numbers |$k_{j}=2\pi j/L, j=1, 2, \dots $|. The two critical wave numbers that become unstable at the same time (giving rise to steady-state/steady-state mode interactions) are k12 and k13.
Note that model (2.22) does not exhibit Hopf bifurcations (i.e. |$Re\left (\lambda \left (k\right )\right )=0, Im\left (\lambda \left (k\right )\right )\neq 0$|). Therefore, although aggregations will develop, there will be no travelling patterns that arise via Hopf bifurcations.
3.1. Aggregation with different types of kernel
In Mogilner & Edelstein-Keshet (1999) it was shown that even kernels give rise to group drift, while odd kernels have a greater effect in regions where the distribution of the density is uneven (e.g. the edges of the population), leading to stationary groups. Considering the importance of the symmetry of the interaction kernels, we next discuss the cases of even and odd kernels, and compare these results with our previous results where we used a translated Gaussian kernel (see equation (3.10)), to investigate the possibility of cell aggregations to form from spatial perturbation of the steady state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 1, 0, p_{2}/q\right )$|.
Aggregation with even kernel
We consider the situation that the interaction kernel is an even function with respect to the y-axis. We choose the following Gaussian kernel (Mogilner & Edelstein-Keshet, 1999):where qa, qr, ma and mr have the same meaning as in equation (3.10). The Fourier sine transform of the above even kernel is zero: |$\hat {K}^{s}\left (k\right )=0$|. We know (see equation (3.8)) that the Jacobian matrix of system (3.6) has three negative eigenvalues λ1, 2, 4 < 0. For the even kernel considered here we also have(3.13)\begin{align} K\left(x\right):=\frac{q_{a}}{\sqrt{2\pi {m^{2}_{a}}}}\exp\left(-x^{2}/2{m^{2}_{a}}\right)-\frac{q_{r}}{\sqrt{2\pi {m^{2}_{r}}}}\exp\left(-x^{2}/2{m^{2}_{r}}\right), \end{align}(3.14)\begin{align} \lambda_{3}\left(k\right)&=-r_{2}<0. \end{align}Therefore, the steady state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 1, 0, p_{2}/q\right )$| is stable, and no aggregations will form.
Aggregation with an odd kernel
We investigate now the stability of |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 1, 0, p_{2}/q\right )$| when we consider an odd kernel with respect to the origin. To this end, we choosewhere qa, qr, ma and mr have the same meaning as in equation (3.10). Then the Fourier sine transform of the kernel (3.15) is(3.15)\begin{align} K\left(x\right):=\frac{q_{a}x}{\sqrt{2\pi {m^{2}_{a}}}}\exp\left(-x^{2}/2{m^{2}_{a}}\right)-\frac{q_{r}x}{\sqrt{2\pi {m^{2}_{r}}}}\exp\left(-x^{2}/2{m^{2}_{r}}\right), \end{align}(3.16)\begin{align} \hat{K}^{s}\left(k\right)=q_{a}k{m_{a}^{2}}{\exp}(-(km_{a})^{2}/2)-q_{r}k{m_{r}^{2}}{\exp}(-(km_{r})^{2}/2). \end{align}As before, the Jacobian matrix of system (3.6) has three negative eigenvalues: λ1, 2, 4 < 0. The stability of the state |$\left (0, 1, 0, p_{2}/q\right )$| is given by the sign of(3.17)\begin{align} Re\left(\lambda_{3}\left(k\right)\right)=-r_{2}+\frac{2k^{2}s^{\ast}_{2}}{R}\left(1+\tanh\left(a_{2}\frac{p_{2}}{q}\right)\right)\left(q_{a}{m_{a}^{2}}e^{-\frac{\left(km_{a}\right)^{2}}{2}}-q_{r}{m_{r}^{2}}e^{-\frac{\left(km_{r}\right)^{2}}{2}}\right). \end{align}In Fig. 3 we plot |$Re(\lambda _{3}\left (k\right ))$| against the wave number k for this steady state. We observe that for values of qa greater than those in the translated Gaussian case (see Fig. 2), there is a range of k–values for which |$Re\left (\lambda _{3}\left (k\right )\right )$| is positive, thus the steady state |$\left (0, 1, 0, p_{2}/q\right )$| can become unstable, and spatial aggregations could arise.
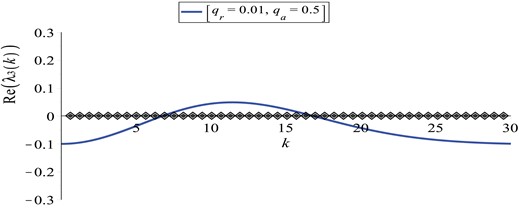
Plot of the dispersion relation (3.17) for the steady state |$\left (u_{1}^{\ast }, u_{2}^{\ast }, f^{\ast }, c^{\ast }\right )=\left (0, 1, 0, p_{2}/q\right )$|. Here, qa = 0.5. The rest of the model parameters are given in Table 2. The solid curve represents the real part of λ3, while the diamonds represent the discrete wave numbers |$k_{j}=2\pi \frac {j}{L}, j=0, 1, 2, \dots $|.
4. Numerical results
Stationary aggregation patterns. To check the validity of our results obtained via linear stability analysis (see Fig. 2), we first run simulations for small random perturbations of the spatially homogeneous steady state |$\left (u^{\ast }_{1}, u^{\ast }_{2}, f^{\ast }, c^{\ast }\right )=(0, 1, 0, p_{2}/q)$|, and for qa = 0.09 and qr = 0.01. The results, presented in Fig. 4, show 12–13 stationary pulses (corresponding to critical wave numbers k12 − k13). The spread of cancer cells over the whole domain (panels (a), (b)) leads to the degradation of ECM (panel (c)). The integrin density (panel (d)) follows the patterns in the density of cancer cells. We observe here a coexistence between a low u1 population and a high u2 population.
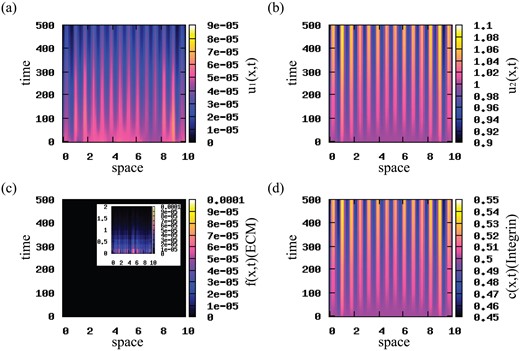
Patterns exhibited by model (2.22) for small random perturbations of the steady state |$\left (0, 1, 0, p_{2}/q\right )$|, corresponding to the dispersion relation shown in Fig. 2. Here, we use the cell–cell interaction kernel given by relation (3.10), and the model parameters given in Table 2. (a) Density of u1 cell population; (b) density of u2 (highly mutated) cell population; (c) ECM density f; (d) integrin density c. Note the formation of 12 or 13 peaks (if counting separately the peaks on the periodic boundaries) of high cell density corresponding to critical wave numbers k12/k13.
To check the effect of initial data on model dynamics, in Fig. 5 we investigate the behaviour of model (2.22) when we change the initial conditions to a rectangular pulse describing an initial aggregation of tumour cells. The magnitudes of attractive-repulsive interactions (and all other parameter values) are the same as in Fig. 4. The short-term results in Fig. 5(a)–(d) show a travelling pulse behaviour exhibited by the u2 population that moves away from population u1 (which is mostly stationary—although even this population spreads a bit). This faster spreading of the u2 cells compared to the u1 cells is consistent with experimental observations (Chapman et al., 2014; Wojciechowska & Patton, 2015). The movement of u2 population eventually stops near the boundary (due to the periodic boundary conditions, the left-moving and right-moving subgroups can sense each other across the boundary). The fast-moving u2 cells degrade the ECM (panel (c)). We also note the high density of integrins associated with the u1 population (panel (d)). The long-term results in Fig. 5(a′)–(d′) show the formation of new aggregations of cells at distant positions in space. For t ∈ (100, 250), the two tumour populations coexist. For t > 250, population u1 is slowly eliminated and population u2 dominates the dynamics of model (2.22). We also note that despite some chaotic-like dynamics exhibited by the u1 and u2 populations for t < 300, in the long term the system approaches stationary pulses defined by 13 peaks (corresponding to unstable wave number k13).
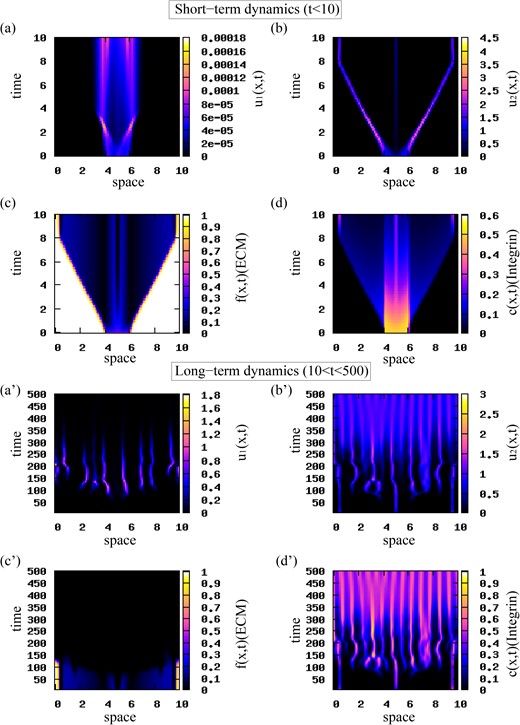
Short-term and long-term patterns exhibited by model (2.22). The initial conditions for the two cancer cell populations are described by a rectangular pulse (see (4.1)) with |${u_{2}^{c}}=1.0$|. We use the interaction kernel given by (3.10), and the model parameters given in Table 2. (a),(a′) Density of u1 population; (b),(b’) density of u2 (highly mutated) population; (c),(c′) ECM density f; (d),(d′) integrin density c.
In Figs 6 and 7 we investigate the effect of various mutation rates on the dynamics of the u1 and u2 populations. In Fig. 6 we choose M = 0.001 (see Table 2), and observe that population u2 vanishes for t > 400, while population u1 persists and forms small high-density aggregations of cells. This unexpected behaviour might be explained by the combined effect of three factors: (i) the mutation rate (M = 0.001) that is much smaller than the growth rates (r1, 2 = 0.1) of the two cancer cell populations, (ii) the lack of diffusion for u2 and (iii) the competition for nutrients between the two cell populations (which is modelled implicitly by the logistic terms G1, 2; see equations (2.14)). This eventually leads to an overall increase in the first population, u1, and a decrease in the second (highly mutated) population, u2. In Fig. 7 we increase the mutation rate to M = 0.05, while chosing the growth rates fixed at r1 = r2 = 0.1. We observe that faster mutation rates lead to a decrease and eventual elimination of the u1 population. The u2 population increases and dominates the long-term dynamics of model (2.22).
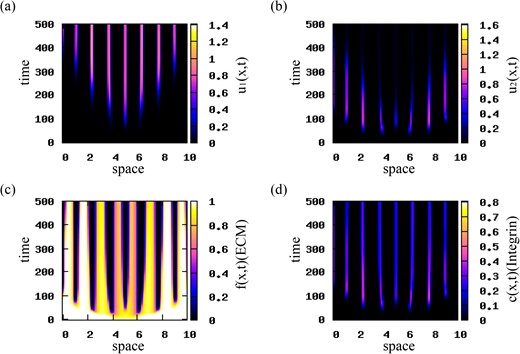
Patterns exhibited by model (2.22). The initial conditions for the cancer cell populations consist of a rectangular pulse (see (4.1)) with |${u_{2}^{c}}=0.001$|. The mutation rate is M = 0.001. The rest of model parameters are given in Table 2. (a) Density of u1 population; (b) density of u2 population; (c) ECM density f; (d) integrin density c.
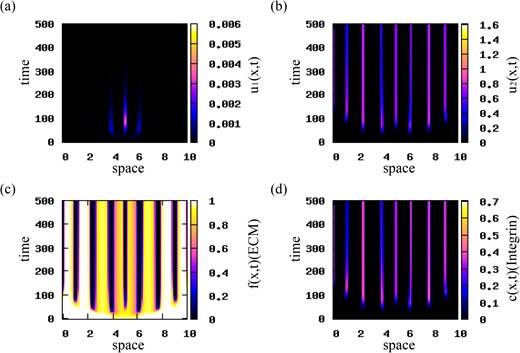
Patterns exhibited by model (2.22). The initial conditions for the cancer cell populations consist of a rectangular pulse (see (4.1)) with |${u_{2}^{c}}=0.001$|. The mutation rate in M = 0.05. The rest of model parameters are given in Table 2. (a) Density of u1 population; (b) density of u2 population; (c) ECM density f; (d) integrin density c.
Travelling wave patterns. Choosing again rectangular pulse initial conditions, we reduce the magnitudes of cell–cell attractive and repulsive interactions to qa = 0.00025 and qr = 0.0005. The numerical simulations in Fig. 8 show travelling waves that propagate in opposite directions at a constant speed. Similar behaviour is obtained for qa > qr. The evolution of the travelling waves is shown in Fig. 9 for |$t=25j,\; j=1,\dots ,9$|. We observe that the waves connect the unstable steady states |$\left (0, 1, 0, p_{2}/q\right )$|, with p2/q = 0.5, and |$\left (0, 0, f^{\ast }, 0\right )$|, with f* = 1.
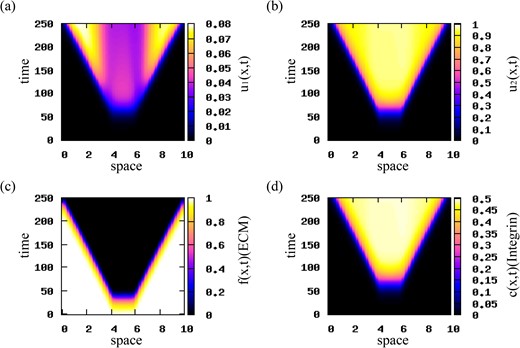
Patterns exhibited by model (2.22) for initial conditions for the two cancer cell populations consisting of a rectangular pulse with |${u_{2}^{c}}=0.001$|. Here, qr = 0.0005, qa = 0.00025 and the rest of the model parameters are given in Table 2. (a) Cell density for u1 population; (b) cell density for u2 population; (c) ECM density f; (d) integrin density c. Travelling waves are obtained for the translated Gaussian kernel given by relation (3.10).
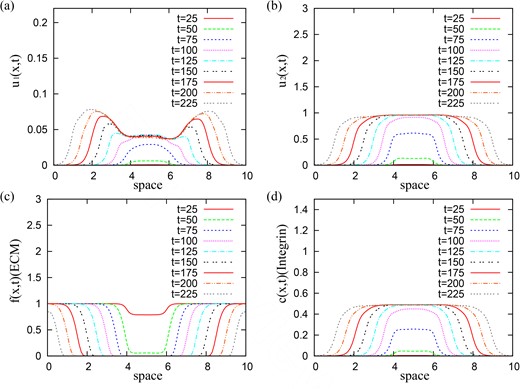
The evolution of the travelling waves under small perturbations for qr = 0.0005, qa = 0.00025 and the rest of the model parameters given in Table 2, of (a) cancer cell density u1 of the first (‘normal’) population; (b) cancer cell density u2 of the second (‘mutant’) population; (c) ECM density f; (d) integrin density c.
Finally, we check the effect that an increase in the mutation rate M has on the travelling wave behaviour exhibited by model (2.22). Figure 10 shows that for high mutation rates (M = 0.05) the first population still exhibits a travelling wave before vanishing.
To understand the long-term behaviour of model (2.22) with odd kernels, we also ran numerical simulations using the interaction kernel given by (3.15). The spatial patterns obtained in this case were similar to the patterns obtained for the translated Gaussian kernel (see relation (3.10)).
4.1. Summary of model variables and parameters
Here we present two tables with the model variables and parameters. In Table 1 we list the model variables with their units. In Table 2 we list the parameters of our model and their corresponding units and non-dimensional values used in the simulations.
Parameter estimation.
For the diffusion coefficient, Chaplain & Lolas (2006) has shown a range between |$\tilde {D}= 10^{-5}$| and |$\tilde {D}=10^{-3}$|. In this study, we choose |$\tilde {D}=10^{-4}$|.
The sensing radius was based on the range of values given in (Armstrong et al., 2006; Gerisch & Chaplain, 2008). In this study, we choose |$\tilde {R}=0.99$|.
Attraction and repulsion ranges were chosen to be smaller or equal to sensing radius, with the repulsion range to be smaller than the attraction range (Green et al., 2010).
Various experimental studies (Cunningham & You, 2015; Morani et al., 2014) have shown that doubling times for tumour cells range from 1–10 days. This corresponds to growth rates between |$(\ln (2)/10, \ln (2)/1)=(0.07, 0.7)$|. In this study, we assume that |$\tilde {r_{1}}=\tilde {r_{2}}=0.1$|.
Experimental studies (Cillo et al., 1987; Hill et al., 1984; Mareel et al., 1991) have shown that the mutation rate ranges between M = 10−3/day and M = 0.1/day. Thus the non-dimensional value of the mutation rate is in the range between |$\tilde {M}=0.001$| and |$\tilde {M}=0.1$| (for highly aggressive tumours). In this study, we choose |$\tilde {M}=0.001$|.
The parameters |$a_{i}, d, b_{i}, s_{i}^{\ast }, s^{\ast }, c_{i}^{\ast }, i=1, 2$|, were based on the range of the adhesion strength parameters used in Armstrong et al. (2006).
Experimental studies (Kidera et al., 2010) have shown greater production of integrins for highly mutated cancer cells. Thus, we choose |$\tilde {p_{1}}<\tilde {p_{2}}$|.
Experimental studies (Delcommenne & Streuli, 1995; Davis et al., 2001; Liu et al., 2011; Lobert et al., 2010) have shown that the half-lifes of the integrins range from 0.04–4 days. This corresponds to decay rate between |$\left (\ln \left (2\right )/4, \ln \left (2\right )/0.04\right )=\left (0.17, 17.3\right )$|. In this study, we assume that |$\tilde {q}=0.2$|.
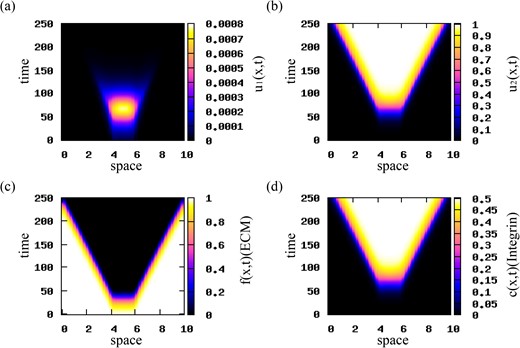
Patterns exhibited by model (2.22) for higher mutation rates (M = 0.05) and initial conditions for the two cancer cell populations consisting of a rectangular pulse with |${u_{2}^{c}}=0.001$|. Here, qr = 0.0005, qa = 0.00025 and the rest of the model parameters are given in Table 2. (a) Cell density for u1 population; (b) cell density for u2 population; (c) ECM density f; (d) integrin density c. Travelling waves are obtained for the translated Gaussian kernel given by relation (3.10)
5. Speed of travelling waves
Let s1, s2, s3, s4 > 0 be the steepness (i.e. exponent of an exponential decay/increase profile) of u1, u2, f, c. The minimum of |$w\left (s_{j}\right )$| for |$s_{j}>0, \; j=1,\dots ,4$|, gives an upper bound on the rightward travelling wave speed of the nonlinear system. Thus, we make the general exponential ansatz
A list of model variables with their units. Since we are in 1D, length and volume coincide and we express the variables in terms of domain length.
| Variable | Description | Dimensional Units |
|---|---|---|
| u1 | ‘Normal’ cancer cell density | cell/length |
| u2 | ‘Mutant’ cancer cell density | cell/length |
| f | ECM density | mg/length |
| c | Integrin density | integrins/cell |
| Variable | Description | Dimensional Units |
|---|---|---|
| u1 | ‘Normal’ cancer cell density | cell/length |
| u2 | ‘Mutant’ cancer cell density | cell/length |
| f | ECM density | mg/length |
| c | Integrin density | integrins/cell |
A list of model variables with their units. Since we are in 1D, length and volume coincide and we express the variables in terms of domain length.
| Variable | Description | Dimensional Units |
|---|---|---|
| u1 | ‘Normal’ cancer cell density | cell/length |
| u2 | ‘Mutant’ cancer cell density | cell/length |
| f | ECM density | mg/length |
| c | Integrin density | integrins/cell |
| Variable | Description | Dimensional Units |
|---|---|---|
| u1 | ‘Normal’ cancer cell density | cell/length |
| u2 | ‘Mutant’ cancer cell density | cell/length |
| f | ECM density | mg/length |
| c | Integrin density | integrins/cell |
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| D | Diffusion coefficient | length2/time | 10−4 | Chaplain & Lolas (2006) |
| R | Sensing radius | length | 0.99 | Armstrong et al. (2006) |
| qa | Magnitude of attraction | length2/cell | 0.09 | Estimated |
| qr | Magnitude of repulsion | length2/cell | 0.01 | Estimated |
| sa | Attraction range | length | 0.99 | Estimated |
| sr | Repulsion range | length | 0.25 | Estimated |
| ma | Width of attraction kernel | length | 0.99/8 | Estimated |
| mr | Width of repulsion kernel | length | 0.25/8 | Estimated |
| r1 | Growth rate of u1 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| r2 | Growth rate of u2 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| M | Mutation rate | 1/time | 0.001 | Cillo et al. (1987); Hill et al. (1984); Mareel et al. (1991) |
| a1 | Coeff. related to the number of integrins necessary for max self-adhesion between u1 | cell/integrins | 0.3 | Estimated |
| a2 | Coeff. related to the number of integrins necessary for max self-adhesion between u2 | cell/integrins | 0.01 | Estimated |
| d | Coeff. related to the number of integrins necessary for max cross-adhesion | cell/integrins | 0.5 | Estimated |
| b1 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u1 | cell/integrins | 1.8 | Estimated |
| b2 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u2 | cell/integrins | 5 | Estimated |
| |$s^{\ast }_{1}$| | Magnitude of self-adhesion forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.9 | Estimated |
| |$s^{\ast }_{2}$| | Magnitude of self-adhesion forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.2 | Estimated |
| s* | Magnitude of cross-adhesion forces | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Estimated |
| (continued). |
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| D | Diffusion coefficient | length2/time | 10−4 | Chaplain & Lolas (2006) |
| R | Sensing radius | length | 0.99 | Armstrong et al. (2006) |
| qa | Magnitude of attraction | length2/cell | 0.09 | Estimated |
| qr | Magnitude of repulsion | length2/cell | 0.01 | Estimated |
| sa | Attraction range | length | 0.99 | Estimated |
| sr | Repulsion range | length | 0.25 | Estimated |
| ma | Width of attraction kernel | length | 0.99/8 | Estimated |
| mr | Width of repulsion kernel | length | 0.25/8 | Estimated |
| r1 | Growth rate of u1 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| r2 | Growth rate of u2 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| M | Mutation rate | 1/time | 0.001 | Cillo et al. (1987); Hill et al. (1984); Mareel et al. (1991) |
| a1 | Coeff. related to the number of integrins necessary for max self-adhesion between u1 | cell/integrins | 0.3 | Estimated |
| a2 | Coeff. related to the number of integrins necessary for max self-adhesion between u2 | cell/integrins | 0.01 | Estimated |
| d | Coeff. related to the number of integrins necessary for max cross-adhesion | cell/integrins | 0.5 | Estimated |
| b1 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u1 | cell/integrins | 1.8 | Estimated |
| b2 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u2 | cell/integrins | 5 | Estimated |
| |$s^{\ast }_{1}$| | Magnitude of self-adhesion forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.9 | Estimated |
| |$s^{\ast }_{2}$| | Magnitude of self-adhesion forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.2 | Estimated |
| s* | Magnitude of cross-adhesion forces | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Estimated |
| (continued). |
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| D | Diffusion coefficient | length2/time | 10−4 | Chaplain & Lolas (2006) |
| R | Sensing radius | length | 0.99 | Armstrong et al. (2006) |
| qa | Magnitude of attraction | length2/cell | 0.09 | Estimated |
| qr | Magnitude of repulsion | length2/cell | 0.01 | Estimated |
| sa | Attraction range | length | 0.99 | Estimated |
| sr | Repulsion range | length | 0.25 | Estimated |
| ma | Width of attraction kernel | length | 0.99/8 | Estimated |
| mr | Width of repulsion kernel | length | 0.25/8 | Estimated |
| r1 | Growth rate of u1 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| r2 | Growth rate of u2 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| M | Mutation rate | 1/time | 0.001 | Cillo et al. (1987); Hill et al. (1984); Mareel et al. (1991) |
| a1 | Coeff. related to the number of integrins necessary for max self-adhesion between u1 | cell/integrins | 0.3 | Estimated |
| a2 | Coeff. related to the number of integrins necessary for max self-adhesion between u2 | cell/integrins | 0.01 | Estimated |
| d | Coeff. related to the number of integrins necessary for max cross-adhesion | cell/integrins | 0.5 | Estimated |
| b1 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u1 | cell/integrins | 1.8 | Estimated |
| b2 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u2 | cell/integrins | 5 | Estimated |
| |$s^{\ast }_{1}$| | Magnitude of self-adhesion forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.9 | Estimated |
| |$s^{\ast }_{2}$| | Magnitude of self-adhesion forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.2 | Estimated |
| s* | Magnitude of cross-adhesion forces | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Estimated |
| (continued). |
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| D | Diffusion coefficient | length2/time | 10−4 | Chaplain & Lolas (2006) |
| R | Sensing radius | length | 0.99 | Armstrong et al. (2006) |
| qa | Magnitude of attraction | length2/cell | 0.09 | Estimated |
| qr | Magnitude of repulsion | length2/cell | 0.01 | Estimated |
| sa | Attraction range | length | 0.99 | Estimated |
| sr | Repulsion range | length | 0.25 | Estimated |
| ma | Width of attraction kernel | length | 0.99/8 | Estimated |
| mr | Width of repulsion kernel | length | 0.25/8 | Estimated |
| r1 | Growth rate of u1 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| r2 | Growth rate of u2 | 1/time | 0.1 | Cunningham & You (2015); Morani et al. (2014) |
| M | Mutation rate | 1/time | 0.001 | Cillo et al. (1987); Hill et al. (1984); Mareel et al. (1991) |
| a1 | Coeff. related to the number of integrins necessary for max self-adhesion between u1 | cell/integrins | 0.3 | Estimated |
| a2 | Coeff. related to the number of integrins necessary for max self-adhesion between u2 | cell/integrins | 0.01 | Estimated |
| d | Coeff. related to the number of integrins necessary for max cross-adhesion | cell/integrins | 0.5 | Estimated |
| b1 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u1 | cell/integrins | 1.8 | Estimated |
| b2 | Coeff. related to the number of integrins necessary for max cell-ECM adhesion for u2 | cell/integrins | 5 | Estimated |
| |$s^{\ast }_{1}$| | Magnitude of self-adhesion forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.9 | Estimated |
| |$s^{\ast }_{2}$| | Magnitude of self-adhesion forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 0.2 | Estimated |
| s* | Magnitude of cross-adhesion forces | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Estimated |
| (continued). |
Continued.
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| |$c^{\ast }_{1}$| | Magnitude of cell-ECM forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1.5 | Estimated |
| |$c^{\ast }_{2}$| | Magnitude of cell-ECM forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 5.5 | Estimated |
| α | Rate of ECM degradation by u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Sherratt et al. (2009) |
| β | Rate of ECM degradation by u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 2 | Sherratt et al. (2009) |
| p1 | Production rate of c by u1 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.05 | Estimated |
| p2 | Production rate of c by u2 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.1 | Estimated |
| q | Decay rate of c | 1/time | 0.2 | Liu et al. (2011) |
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| |$c^{\ast }_{1}$| | Magnitude of cell-ECM forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1.5 | Estimated |
| |$c^{\ast }_{2}$| | Magnitude of cell-ECM forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 5.5 | Estimated |
| α | Rate of ECM degradation by u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Sherratt et al. (2009) |
| β | Rate of ECM degradation by u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 2 | Sherratt et al. (2009) |
| p1 | Production rate of c by u1 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.05 | Estimated |
| p2 | Production rate of c by u2 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.1 | Estimated |
| q | Decay rate of c | 1/time | 0.2 | Liu et al. (2011) |
Continued.
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| |$c^{\ast }_{1}$| | Magnitude of cell-ECM forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1.5 | Estimated |
| |$c^{\ast }_{2}$| | Magnitude of cell-ECM forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 5.5 | Estimated |
| α | Rate of ECM degradation by u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Sherratt et al. (2009) |
| β | Rate of ECM degradation by u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 2 | Sherratt et al. (2009) |
| p1 | Production rate of c by u1 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.05 | Estimated |
| p2 | Production rate of c by u2 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.1 | Estimated |
| q | Decay rate of c | 1/time | 0.2 | Liu et al. (2011) |
| Param. | Description | Dimensional Units | Non-dim. value (|$\tilde {p}$|) | Reference |
|---|---|---|---|---|
| |$c^{\ast }_{1}$| | Magnitude of cell-ECM forces of u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1.5 | Estimated |
| |$c^{\ast }_{2}$| | Magnitude of cell-ECM forces of u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 5.5 | Estimated |
| α | Rate of ECM degradation by u1 | |$length/\left (time\cdot \textit {cell}\right )$| | 1 | Sherratt et al. (2009) |
| β | Rate of ECM degradation by u2 | |$length/\left (time\cdot \textit {cell}\right )$| | 2 | Sherratt et al. (2009) |
| p1 | Production rate of c by u1 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.05 | Estimated |
| p2 | Production rate of c by u2 | |$integrins/\left (time\cdot \textit {cell}\right )$| | 0.1 | Estimated |
| q | Decay rate of c | 1/time | 0.2 | Liu et al. (2011) |
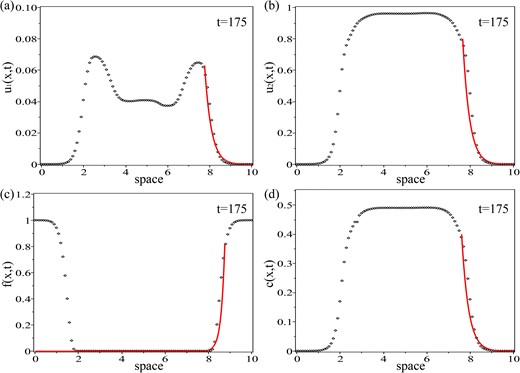
Plot of the numerical simulations of the travelling wave profile, shown in Figs 8 and 9, for (a) population u1 and the exponential ansatz function |$E_{1}\left (x\right )$| given by relation (5.3) with |$A_{1}=0.063\cdot \exp \left (24.5675\right )$|; (b) population u2 and the exponential ansatz function |$E_{2}\left (x\right )$| given by relation (5.4) with |$A_{2}=0.8\cdot \exp \left (24.2505\right )$|; (c) ECM density, f, and the exponential ansatz function |$E_{3}\left (x\right )$| given by relation (5.5) with |$A_{3}=0.82\cdot \exp \left (-52.5\right )$|; (d) integrin density, c, and the exponential ansatz function |$E_{4}\left (x\right )$| given by relation (5.6) with |$A_{4}=0.4\cdot \exp \left (24.092\right )$|. Here, qr = 0.0005, qa = 0.00025 and the rest of the model parameters are given in Table 2.
Since we are looking for the minimum positive speed with respect to s > 0 and s3 > 0, in Fig. 12(a) we plot implicitly equation (5.8) in the |$\left (s, s_{3}, w\right )$|-space. Note that the steepness coefficient s3 (for the ECM degradation profile) does not have any significant effect on the minimum positive wave speed w (of the invading u1 and u2 populations). For this reason, in Fig. 12(b) we plot the relation (5.8) in the |$\left (s, w\right )$|-plane for fixed s3 > 0. We see that indeed we cannot have a travelling wave with positive speed unless the wave has a steepness s > 3.17. Moreover, faster waves have higher steepness.
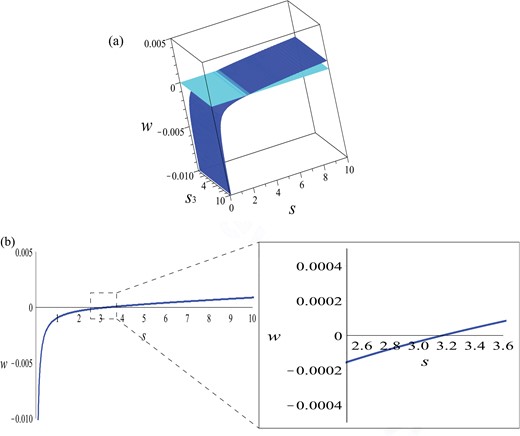
Finally, we are interested in investigating how the speed of the invading waves correlates with their steepness, as we vary different model parameters. In Fig. 13(a) we see that a decrease in the diffusion coefficient D by one order of magnitude leads to a reduction in the velocity w > 0. In Fig. 13(b) we see that an increase in the mutation rate M (from 0.001 to 0.05) leads to a slightly lower velocity w > 0 and a higher steepness. (Note that for s > 100, the invading speed obtained for M = 0.001 and M = 0.05 is the same—not shown here.)
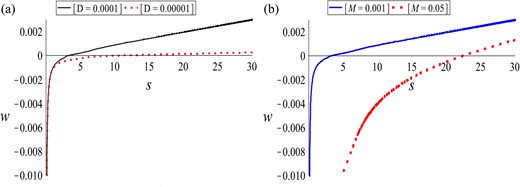
Plot of the relation (5.8) for qr = 0.0005, qa = 0.00025, and the rest of the model parameters as given in Table 2, as we vary: (a) diffusion coefficient D: D = 10−4 (black solid curve) and D = 10−5 (red dashed curve); (b) mutation rate M: M = 0.001 (blue solid curve) and M = 0.05 (red dashed curve).
6. Conclusions and discussion
In this paper we introduced a model of integro-differential equations describing the dynamics of two cancer cell populations: a ‘normal’ cell population that exhibits both random and directed movement, and a ‘mutant’ cell population that exhibits only directed movement. The model incorporated both nonlocal cell–cell interactions and cell–matrix interactions. Moreover, unlike other models in the literature, in our model these interactions were not constant but depended on the cellular level of integrins.
Linear stability analysis of the non-dimensional model showed that aggregations could arise only via real bifurcations (the system could not exhibit Hopf bifurcations). Numerical results showed that these aggregations were described by a large number of stationary pulses (e.g. 13 pulses corresponding to the critical unstable wave number k13). Moreover, numerics also showed the existence of travelling waves. We investigated the speed of these waves, which seemed to be affected by the diffusion coefficient and the mutation rate of cells.
The rate at which cancer cells mutate seemed to play a critical role in our model. In Figs 6 and 7 (as well as Figs 8 and 10) we showed that depending on the magnitude of the mutation rate, either the u1 or the u2 cell populations can be eliminated (or, in some cases they can co-exist—see Figs 4 and 8). The existence of these dominant behaviours exhibited by the u1 or u2 populations are consistent with the principle of competitive exclusion of clonal sub-populations in heterogeneous tumours (Egan et al., 2012; Keats et al., 2012; Leith et al., 1989). In Fisher et al. (2013) the authors interpreted the experimental data, which showed suppression and reappearance of cancer clones in myeloma patients (Keats et al., 2012) and chronic lymphocytic leukaemia patients (Schuh et al., 2012), by suggesting that two subclones can exist in a dynamic equilibrium. While all these experimental studies record the survival/suppression of tumour clones in various cancers, they do not offer a mechanistic explanation for the factors that could lead to these behaviours. In contrast, our numerical results (see Figs 6 and 7, 8 and 10) offer such a mechanistic explanation by identifying the magnitude of the mutation rate as a factor that could explain the experimentally observed suppression and reappearance of cancer clones.
This issue of cancer heterogeneity has significant implications on cancer drug therapies, since it can lead to drug resistance. For example, clinical studies have shown the emergence of imatinib-resistant mutation clones in patients with chronic myeloid leukaemia, which can co-exist with subclones that carry different imatinib-resistant mutations in treatment-naïve patients (Shah et al., 2002). Overall, these clinical observations suggest that a single drug might not be successful in treating a genetically heterogeneous tumour, since sub-populations of cancer cells with drug-resistant mutations could become dominant, thus leading to therapeutic failure.
We note that the spatial distribution of the two cancer sub-populations, with the original u1 population in the centre of the aggregation and the mutated u2 clones towards the edges of the aggregation (see Fig. 5 for |$t\in \left [0, 10\right ]$| or Fig. 6 for |$t\in \left [100, 300\right ]$|), is consistent with experimental studies on the spatial relationship between clonal sub-populations of hepatocellular carcinoma (HCC) tumour (Ling et al., 2015). In this experimental study, the authors investigated the clonal diversity of a HCC-15 tumour (and the spatial distribution of these clones), and showed the ancestral clones being positioned in the middle of the tumour, with the descendant clones radiating outward.
The numerical results of the model presented in this paper show that cell–cell/cell–matrix adhesion combined with cell proliferation (in the presence of cell competition) and cell mutation, can impact which tumour clones survive (see Figs 4–8 and 10). Therefore, our model puts forward the idea that in order to predict the emergence of a particular cancer clone, we should first quantify cell–cell and cell–matrix adhesive interactions for the various tumour clones, and their proliferative capacities.
In this paper we focused on a 1D model. However, real life cell dynamics occurs in 2D or 3D. In Appendix A we extended model (2.22) to two spatial dimensions. Moreover, to verify if the linear stability results for the 1D model (2.22) were valid also in 2D, we applied linear stability analysis. We showed that for similar kernels, we obtained similar dispersion relations with no imaginary parts (and hence no Hopf bifurcations). Therefore, we expect that the 2D model (A1) would exhibit stationary pulses similar to the ones exhibited by the 1D model (2.22). Future work will consider extending the numerical results in 2D.
Acknowledgements
V. B. acknowledges support from an Engineering and Physical Sciences Research Council (UK) grant number EP/L504932/1. R. E. acknowledges support from an Engineering and Physical Sciences Research Council (UK) grant number EP/K033689/1.
Appendix A. Extension of the model to two dimensions
This expression is similar to the one obtained in the 1D case (see equation (3.17)). Therefore we deduce that there is a range of |$\underline {k}\text{-}$|values for which |$Re\left (\lambda _{3}\left (\underline {k}\right )\right )$| is positive, and thus spatial aggregations could develop also for the 2D model (A1) when applying small spatial perturbations of the steady state |$\left (0, 1, 0, p_{2}/q\right )$|.
Appendix B. A structured-population model for cancer–ECM interactions via integrins
The incorporation of this internal cell structure c into the mesoscopic model (B1) follows the same approach as in Erban & Othmer (2004, 2005); Xue et al. (2011); Lorenz & Surulescu (2014); Engwer et al. (2015); Perthame et al. (2016); Engwer et al. (2017); Hunt & Surulescu (2017).
We note here that while different ‘microscale–macroscale’ models (Stinner et al., 2014, 2015, 2016; Meral et al., 2015a,b) and ‘mesoscale’ models (Lorenz & Surulescu, 2014; Engwer et al., 2015; Perthame et al., 2016; Engwer et al., 2017; Hunt & Surulescu, 2017) for tumour-ECM interactions via integrins have been developed over the last two decades, to our knowledge there are not many studies that compare and contrast the outcomes of different ways the integrins are incorporated into these different models.
A comparison between the effects of integrins on the dynamics of the structured-population model (B1) and the original model (2.22) (both assuming heterogeneous cell populations), including an investigation into the possibility of having travelling wave solutions for the structured model (B1), will be the subject of a future study. (Such an investigation is beyond the scope of this study due to its complexity, which would increase considerably the length of this paper.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}