





17β-Hydroxysteroid dehydrogenase-3 (17βHSD3) deficiency is an autosomal recessive form of male pseudohermaphroditism caused by mutations in the HSD17B3 gene. In a nationwide study on male pseudohermaphroditism among all pediatric endocrinologists and clinical geneticists in The Netherlands, 18 17βHSD3-deficient index cases were identified, 12 of whom initially had received the tentative diagnosis androgen insensitivity syndrome (AIS). The phenotypes and genotypes of these patients were studied. Endocrine diagnostic methods were evaluated in comparison to mutation analysis of the HSD17B3 gene. RT-PCR studies were performed on testicular ribonucleic acid of patients homozygous for two different splice site mutations. The minimal incidence of 17βHSD3 deficiency in The Netherlands and the corresponding carrier frequency were calculated. Haplotype analysis of the chromosomal region of the HSD17B3 gene in Europeans, North Americans, Latin Americans, Australians, and Arabs was used to establish whether recurrent identical mutations were ancient or had repeatedly occurred de novo.
In genotypically identical cases, phenotypic variation for external sexual development was observed. Gonadotropin-stimulated serum testosterone/androstenedione ratios in 17βHSD3-deficient patients were discriminative in all cases and did not overlap with ratios in normal controls or with ratios in AIS patients. In all investigated patients both HSD17B3 alleles were mutated. The intronic mutations 325+ 4;A→T and 655–1;G→A disrupted normal splicing, but a small amount of wild-type messenger ribonucleic acid was still made in patients homozygous for 655–1;G→A. The minimal incidence of 17βHSD3 deficiency in The Netherlands was shown to be 1:147,000, with a heterozygote frequency of 1:135. At least 4 mutations, 325 + 4;A→T, N74T, 655–1;G→A, and R80Q, found worldwide, appeared to be ancient and originating from genetic founders. Their dispersion could be reconstructed through historical analysis. The HSD17B3 gene mutations 326–1;G→C and P282L were de novo mutations.
17βHSD3 deficiency can be reliably diagnosed by endocrine evaluation and mutation analysis. Phenotypic variation can occur between families with the same homozygous mutations. The incidence of 17βHSD3 deficiency is 0.65 times the incidence of AIS, which is thought to be the most frequent known cause of male pseudohermaphroditism without dysgenic gonads. A global inventory of affected cases demonstrated the ancient origin of at least four mutations. The mutational history of this genetic locus offers views into human diversity and disease, provided by national and international collaboration.
17β-HYDROXYSTEROID dehydrogenase-3 (17βHSD3) deficiency is an autosomal recessive form of male pseudohermaphroditism due to impaired testicular conversion of androstenedione to testosterone (1). 46,XY homozygotes or compound heterozygotes for mutations in the HSD17B3 gene have testes and normally developed Wolffian duct derivatives. However, they show undervirilization of the external genitalia, which are often female with or without clitoromegaly and/or labial fusion and a blind-ending vagina. Therefore, patients are most often raised as females (2). Less often, ambiguous external genitalia (3–5), male genitalia with micropenis (6), or hypospadias (4, 7) are reported. Virilization occurs at puberty and is probably due to extratesticular conversion of androstenedione to testosterone (4). Thus, the diagnosis 17βHSD3 deficiency should be made before the onset of puberty and followed by gonadectomy in cases with complete female genitalia. In the less frequently seen, partially virilized, 17βHSD3-deficient patients, the diagnosis should be made directly after birth, because androgen treatment may result in a nearly normal male phenotype in adulthood (8, 9), and male sex assignment can be considered. 46,XX 17βHSD3-deficient cases are normal, asymptomatic females (10, 11).
17βHSD3 deficiency is clinically indistinguishable from androgen insensitivity syndrome (AIS) in prepubertal patients, but the diagnosis can be made from elevated serum androstenedione and decreased serum testosterone/androstenedione ratios after hCG stimulation (12, 13). Unfortunately, the diagnostic power of endocrine diagnostics is not optimal because of the lack of normal ranges in strictly age-matched controls. An improved diagnostic procedure became available after cloning of the HSD17B3 gene and detection of 16 different mutations in 21 index patients (5, 10, 11, 14–16). Eleven of these mutations, resulting in amino acid substitutions, were proven to be pathogenic. For the identified splice site mutations, this proof is still lacking.
Except for a high prevalence in an isolated Arabic population (17, 18), 17βHSD3 deficiency is thought to be a rare disease (4, 19). In a nationwide study on male pseudohermaphroditism in The Netherlands (population, 15.5 × 106 in 1998) we found 18 index cases with 17βHSD3 deficiency. Of those, 12 initially received the tentative diagnosis AIS. Here, we evaluate the phenotype/genotype relationship for several mutations. The diagnostic value of testosterone/androstenedione ratios is compared to that of mutation analysis. Molecular genetic proof for the pathogeny of frequently identified splice site mutations is provided. The incidence and carrier frequency of 17βHSD3 deficiency in the Dutch population are investigated. The finding of identical mutations in unrelated families from diverse ethnic background is further investigated. Evidence is provided that some mutations in this gene may be quite ancient.
Subjects and Methods
Design of the study
A nationwide survey on male pseudohermaphroditism among all 9 major pediatric and 7 clinical genetic centers in The Netherlands resulted in the identification of 18 index patients and 2 siblings with a tentative diagnosis of 17βHSD3 deficiency. Some had been diagnosed previously; others were identified during this 5-yr study. In addition, three affected siblings from Turkey were analyzed.
A diagnosis of 17βHSD3 deficiency was established by review of medical history, including prenatal exposures, a 46,XY karyotype, physical examination of the patients, cysto-uroscopy, histological examination of the gonads and Wolffian duct derivatives if possible, additional hCG tests if possible and mutation analysis of the HSD17B3 gene. Furthermore, four generation pedigrees were constructed. Here we report this total of 19 index patients and 4 affected siblings. The study was approved by the medical ethical committee of the University Hospital Rotterdam. Written informed consent was obtained from either the patients or their parents.
DNA samples from patients with 17βHSD3 deficiency from all over the world with similar HSD17B3 gene mutations as those found in these 19 index patients were used in a study on the origin of these mutations.
Endocrine evaluation
Testosterone/androstenedione (T/A) ratios of patients with 17βHSD3 deficiency were compared to those in age-matched normal males and AIS patients.
Androstenedione and testosterone serum levels in 17βHSD3-deficient cases were measured by RIA in different laboratories in The Netherlands; the interlaboratory variation coefficient was maximally 15% for androstenedione and 6% for testosterone (Dutch council for clinical chemistry). The following served as normal controls: 9 normal boys, 1–3 months old; 25 normal prepubertal boys, aged 4 months to 12 yr (20); and 20 normal adult males. For comparison, T/A ratios were determined in AIS patients with proven androgen receptor mutations:1- to 3-month-old infants before (n = 2) and after hCG stimulation (n = 6), in prepubertal cases 4 months to 12 yr after hCG stimulation (n = 3), and in (post)pubertal cases (basal T/A ratios, n = 17; hCG-stimulated T/A ratios, n = 5).
T/A ratios in the above controls and AIS patients were determined according to the method described by Verjans et al. (21) without chromatography for testosterone and with a coated tube RIA (Diagnostic Products, Los Angeles, CA) for androstenedione.
Genomic DNA isolation and mutation detection
Genomic DNA was extracted from peripheral blood leukocytes or from cultured genital skin fibroblasts following standard procedures (22). In the HSD17B3 gene and androgen receptor gene, exons and flanking intron sequences were screened for mutations using PCR and single strand conformation polymorphism (15, 23). PCR fragments of the introns/exons suspected of harboring mutations were analyzed by automated sequencing.
Ribonucleic acid (RNA) extraction, complementary DNA (cDNA) synthesis, and PCR amplification of cDNA
RNA was extracted as previously described (24) from testes obtained at gonadectomy of patients homozygous for the 325 + 4;A→T mutation (patient 1-I) or the 655–1;G→A mutation (patient 9-III) and from a normal 46,XY male (tissue donor bank). cDNA synthesis was performed with an oligo(deoxythymidine) primer as previously described (24), and further amplification was performed with primers 1AA-11B and 1AA-6BB, 1AA-3BB, or 9AA-11B (for localization of the primers, see Fig. 2).
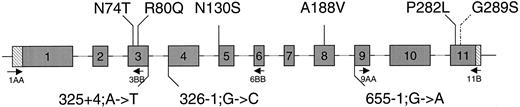
Mutations in the HSD17B3 gene identified in this study. The numbered gray boxes indicate exons; lines indicate intron sequences. 5′ and 3′ untranslated regions are denoted by hatched bars. Mutations leading to substitution of an amino acid are indicated above the gene; mutations in splice sites are indicated below. The neutral polymorphism G289S is denoted with a dashed line. Localization of the primers used for PCR amplification of cDNA is indicated with arrows.
Primer sequences are: 1AA, ACACAGAGAGCCACGGCCAG; 3BB, ATCTCTGTGGCAATGGCCTCTA; 6BB, ACGGAGGTGATGT-TACAATG; 9AA, GCCCTGCAAGAGGAATATAAAGCA; and 11B, GAGGAAAAGGTTGTGCTGGACTCCT.
Fifty microliters of reaction mix for PCR contained 1.5 mmol/L MgCl2. Conditions for the PCR in a Biometra cycle sequencer were as follows: hot start at 94 C for 5 min, then 35 cycles at 94 C for 1 min, at annealing temperature for 1 min, at 72 C for 1 min, and final extension for 10 min at 72 C. Annealing temperatures were as follows: primer pairs 1AA-11B and 1AA-6BB, 55 C; 1AA-3BB, 62 C; and 9AA-11B, 65 C.
The resulting PCR products were subcloned into a plasmid using the TOPO TA cloning kit (Invitrogen, San Diego, CA) and subjected to automated sequencing.
Carrier frequency of the 325 + 4;A→T mutation in the HSD17B3 gene
The carrier frequency of 17βHSD3 deficiency and of the 325 + 4;A→T mutation was calculated under the assumption of a Hardy-Weinberg equilibrium and on basis of the fact that 46,XX homozygous/compound heterozygous cases are asymptomatic (25). Therefore, the carrier frequency is 2pq, with q = √2z/N (z is the number of diseased newborns or the number of 325 + 4;A→T alleles, and N is the total number of newborns during a time period; p = 1 − q). To test the calculated carrier frequency of 325 + 4;A→T, exons 3 of 200 Dutch normal control individuals were screened with PCR-single strand conformation polymorphism (15). As a positive control, a carrier of the 325 + 4;A→T mutation was used.
Haplotyping alleles
Polymorphic extragenic markers on chromosome 9p22.3, AFM023XH8, D9S1786, and D9S1851(Genethon Resource Center, Evry, France) (26) were used to genotype the 17βHSD3-deficient patients described in Table 1 (except for patient 19), and their parents. Consequently, their haplotypes could be derived. Thirty AIS patients that were identified during this same nationwide survey on male pseudohermaphroditism, and 20 of their relatives were used as controls, providing a total of 74 independent alleles. Possible associations of a specific haplotype with a specific mutation were investigated by statistical analysis using the Student’s t test. P values were calculated according to a multiple hypergeometric distribution.
Clinical data for 17β-hydroxysteroid dehydrogenase-deficient patients and mutations in HSD17B3 gene: 23 patients in 19 families
| Family/patient no. | Mutations | Prepubertal external genitalia: Prader stage (27) | Age at referral (yr) | Reason for referral | Endocrine evaluation at age (in years) | T/A basal level | T/A after hCG | Yr of birth | Ethnic origin | Parental consanguinity |
|---|---|---|---|---|---|---|---|---|---|---|
| Homozygotes | ||||||||||
| 1-I | 325+4;A→T | Female | 0.8 | Inguinal mass | 12 | 0.22 | 0.19 | 1982 | Dutch | No |
| 1-II | 325+4;A→T | Female | 0.3 | Family history | ND | ND | 1989 | |||
| 2 | 325+4;A→T | Female | 0.1 | Inguinal mass | ND | ND | 1993 | Dutch | No | |
| 3 | 325+4;A→T | Female | Birth | Inguinal mass | 0.25 | 0.07 | 0.25 | 1998 | Dutch | Yes |
| 4 | 325+4;A→T | 2 | Birth | Abnormal genitalia | 2 | ND | 0.45 | 1995 | Dutch | No |
| 5 | N74T | 1 | 4 | Abnormal genitalia | 4 | 0.62 | 0.50 | 1986 | Dutch | No |
| 6 | N74T | Not known | 16 | Virilization at puberty | 16 | 0.19 | ND | 1972 | Dutch | Yes |
| 7 | R80Q | 1 | 13 | Virilization at puberty | 13 | 0.84 | 0.94 | 1983 | Dutch | No |
| 8 | 326-1;G→C | Female | 8 | Inguinal mass | ND | ND | 1970 | Dutch | No | |
| 9-I | 655-1;G→A | Not known | 16 | Virilization at puberty | 17 | 0.08 | 0.09 | 1976 | Turkish | Yes |
| 9-II | 655-1;G→A | Not known | 15 | Family history | 16 | 0.1 | 0.09 | 1977 | ||
| 9-III | 655-1;G→A | 1 | 10 | Family history | 10 | 0.05 | 0.15 | 1985 | ||
| 10 | A188V | Female | 2 | Inguinal mass | 10 | 1.08 | 0.56 | 1973 | Turkish | No |
| Compound heterozygotes | ||||||||||
| 11-I | 325+4;A→T/N74T | 1 | 14 | Virilization at puberty | 14 | 0.30 | ND | 1982 | Dutch | No |
| 11-II | 325+4;A→T/N74T | 1 | 10 | Family history | 10 | 0.4 | 0.6 | 1986 | ||
| 12 | 325+4;A→T/N74T | 1 | 0.1 | Inguinal mass | 1.3 | ND | 0.28 | 1985 | Dutch | No |
| 13 | 325+4;A→T/R80Q | 1 | 0.1 | Inguinal mass | 0.1 | 0.16 | ND | 1986 | Dutch | No |
| 14 | 325+4;A→T/R80Q | 1 | Birth | Abnormal genitalia | 0.1 | 0.3 | 0.38 | 1991 | Dutch | No |
| 15 | 325+4;A→T/326-1;G→C | 1 | 2 | Inguinal mass | 2 | ND | 0.35 | 1986 | Dutch | No |
| 16 | 325+4;A→T/326-1;G→C | 1 | 0.1 | Inguinal mass | ND | ND | 1988 | Dutch | No | |
| 17 | 325+4;A→T/P282L | 1 | 3.5 | Abnormal genitalia | 6 | 1.0 | 0.27 | 1989 | Dutch | No |
| Heterozygotes | ||||||||||
| 18 | N130S/–(G289S) | 1 | Birth | Abnormal genitalia | 0.6 | 0.4 | 0.29 | 1982 | West Indian | No |
| 19 | No DNA available | 2 | 2 | Inguinal mass | 15 | ND | 0.34 | 1972 | Dutch | Not known |
| Family/patient no. | Mutations | Prepubertal external genitalia: Prader stage (27) | Age at referral (yr) | Reason for referral | Endocrine evaluation at age (in years) | T/A basal level | T/A after hCG | Yr of birth | Ethnic origin | Parental consanguinity |
|---|---|---|---|---|---|---|---|---|---|---|
| Homozygotes | ||||||||||
| 1-I | 325+4;A→T | Female | 0.8 | Inguinal mass | 12 | 0.22 | 0.19 | 1982 | Dutch | No |
| 1-II | 325+4;A→T | Female | 0.3 | Family history | ND | ND | 1989 | |||
| 2 | 325+4;A→T | Female | 0.1 | Inguinal mass | ND | ND | 1993 | Dutch | No | |
| 3 | 325+4;A→T | Female | Birth | Inguinal mass | 0.25 | 0.07 | 0.25 | 1998 | Dutch | Yes |
| 4 | 325+4;A→T | 2 | Birth | Abnormal genitalia | 2 | ND | 0.45 | 1995 | Dutch | No |
| 5 | N74T | 1 | 4 | Abnormal genitalia | 4 | 0.62 | 0.50 | 1986 | Dutch | No |
| 6 | N74T | Not known | 16 | Virilization at puberty | 16 | 0.19 | ND | 1972 | Dutch | Yes |
| 7 | R80Q | 1 | 13 | Virilization at puberty | 13 | 0.84 | 0.94 | 1983 | Dutch | No |
| 8 | 326-1;G→C | Female | 8 | Inguinal mass | ND | ND | 1970 | Dutch | No | |
| 9-I | 655-1;G→A | Not known | 16 | Virilization at puberty | 17 | 0.08 | 0.09 | 1976 | Turkish | Yes |
| 9-II | 655-1;G→A | Not known | 15 | Family history | 16 | 0.1 | 0.09 | 1977 | ||
| 9-III | 655-1;G→A | 1 | 10 | Family history | 10 | 0.05 | 0.15 | 1985 | ||
| 10 | A188V | Female | 2 | Inguinal mass | 10 | 1.08 | 0.56 | 1973 | Turkish | No |
| Compound heterozygotes | ||||||||||
| 11-I | 325+4;A→T/N74T | 1 | 14 | Virilization at puberty | 14 | 0.30 | ND | 1982 | Dutch | No |
| 11-II | 325+4;A→T/N74T | 1 | 10 | Family history | 10 | 0.4 | 0.6 | 1986 | ||
| 12 | 325+4;A→T/N74T | 1 | 0.1 | Inguinal mass | 1.3 | ND | 0.28 | 1985 | Dutch | No |
| 13 | 325+4;A→T/R80Q | 1 | 0.1 | Inguinal mass | 0.1 | 0.16 | ND | 1986 | Dutch | No |
| 14 | 325+4;A→T/R80Q | 1 | Birth | Abnormal genitalia | 0.1 | 0.3 | 0.38 | 1991 | Dutch | No |
| 15 | 325+4;A→T/326-1;G→C | 1 | 2 | Inguinal mass | 2 | ND | 0.35 | 1986 | Dutch | No |
| 16 | 325+4;A→T/326-1;G→C | 1 | 0.1 | Inguinal mass | ND | ND | 1988 | Dutch | No | |
| 17 | 325+4;A→T/P282L | 1 | 3.5 | Abnormal genitalia | 6 | 1.0 | 0.27 | 1989 | Dutch | No |
| Heterozygotes | ||||||||||
| 18 | N130S/–(G289S) | 1 | Birth | Abnormal genitalia | 0.6 | 0.4 | 0.29 | 1982 | West Indian | No |
| 19 | No DNA available | 2 | 2 | Inguinal mass | 15 | ND | 0.34 | 1972 | Dutch | Not known |
ND, Not determined. Patient 19 is included for disease incidence calculations; no DNA was available.
Clinical data for 17β-hydroxysteroid dehydrogenase-deficient patients and mutations in HSD17B3 gene: 23 patients in 19 families
| Family/patient no. | Mutations | Prepubertal external genitalia: Prader stage (27) | Age at referral (yr) | Reason for referral | Endocrine evaluation at age (in years) | T/A basal level | T/A after hCG | Yr of birth | Ethnic origin | Parental consanguinity |
|---|---|---|---|---|---|---|---|---|---|---|
| Homozygotes | ||||||||||
| 1-I | 325+4;A→T | Female | 0.8 | Inguinal mass | 12 | 0.22 | 0.19 | 1982 | Dutch | No |
| 1-II | 325+4;A→T | Female | 0.3 | Family history | ND | ND | 1989 | |||
| 2 | 325+4;A→T | Female | 0.1 | Inguinal mass | ND | ND | 1993 | Dutch | No | |
| 3 | 325+4;A→T | Female | Birth | Inguinal mass | 0.25 | 0.07 | 0.25 | 1998 | Dutch | Yes |
| 4 | 325+4;A→T | 2 | Birth | Abnormal genitalia | 2 | ND | 0.45 | 1995 | Dutch | No |
| 5 | N74T | 1 | 4 | Abnormal genitalia | 4 | 0.62 | 0.50 | 1986 | Dutch | No |
| 6 | N74T | Not known | 16 | Virilization at puberty | 16 | 0.19 | ND | 1972 | Dutch | Yes |
| 7 | R80Q | 1 | 13 | Virilization at puberty | 13 | 0.84 | 0.94 | 1983 | Dutch | No |
| 8 | 326-1;G→C | Female | 8 | Inguinal mass | ND | ND | 1970 | Dutch | No | |
| 9-I | 655-1;G→A | Not known | 16 | Virilization at puberty | 17 | 0.08 | 0.09 | 1976 | Turkish | Yes |
| 9-II | 655-1;G→A | Not known | 15 | Family history | 16 | 0.1 | 0.09 | 1977 | ||
| 9-III | 655-1;G→A | 1 | 10 | Family history | 10 | 0.05 | 0.15 | 1985 | ||
| 10 | A188V | Female | 2 | Inguinal mass | 10 | 1.08 | 0.56 | 1973 | Turkish | No |
| Compound heterozygotes | ||||||||||
| 11-I | 325+4;A→T/N74T | 1 | 14 | Virilization at puberty | 14 | 0.30 | ND | 1982 | Dutch | No |
| 11-II | 325+4;A→T/N74T | 1 | 10 | Family history | 10 | 0.4 | 0.6 | 1986 | ||
| 12 | 325+4;A→T/N74T | 1 | 0.1 | Inguinal mass | 1.3 | ND | 0.28 | 1985 | Dutch | No |
| 13 | 325+4;A→T/R80Q | 1 | 0.1 | Inguinal mass | 0.1 | 0.16 | ND | 1986 | Dutch | No |
| 14 | 325+4;A→T/R80Q | 1 | Birth | Abnormal genitalia | 0.1 | 0.3 | 0.38 | 1991 | Dutch | No |
| 15 | 325+4;A→T/326-1;G→C | 1 | 2 | Inguinal mass | 2 | ND | 0.35 | 1986 | Dutch | No |
| 16 | 325+4;A→T/326-1;G→C | 1 | 0.1 | Inguinal mass | ND | ND | 1988 | Dutch | No | |
| 17 | 325+4;A→T/P282L | 1 | 3.5 | Abnormal genitalia | 6 | 1.0 | 0.27 | 1989 | Dutch | No |
| Heterozygotes | ||||||||||
| 18 | N130S/–(G289S) | 1 | Birth | Abnormal genitalia | 0.6 | 0.4 | 0.29 | 1982 | West Indian | No |
| 19 | No DNA available | 2 | 2 | Inguinal mass | 15 | ND | 0.34 | 1972 | Dutch | Not known |
| Family/patient no. | Mutations | Prepubertal external genitalia: Prader stage (27) | Age at referral (yr) | Reason for referral | Endocrine evaluation at age (in years) | T/A basal level | T/A after hCG | Yr of birth | Ethnic origin | Parental consanguinity |
|---|---|---|---|---|---|---|---|---|---|---|
| Homozygotes | ||||||||||
| 1-I | 325+4;A→T | Female | 0.8 | Inguinal mass | 12 | 0.22 | 0.19 | 1982 | Dutch | No |
| 1-II | 325+4;A→T | Female | 0.3 | Family history | ND | ND | 1989 | |||
| 2 | 325+4;A→T | Female | 0.1 | Inguinal mass | ND | ND | 1993 | Dutch | No | |
| 3 | 325+4;A→T | Female | Birth | Inguinal mass | 0.25 | 0.07 | 0.25 | 1998 | Dutch | Yes |
| 4 | 325+4;A→T | 2 | Birth | Abnormal genitalia | 2 | ND | 0.45 | 1995 | Dutch | No |
| 5 | N74T | 1 | 4 | Abnormal genitalia | 4 | 0.62 | 0.50 | 1986 | Dutch | No |
| 6 | N74T | Not known | 16 | Virilization at puberty | 16 | 0.19 | ND | 1972 | Dutch | Yes |
| 7 | R80Q | 1 | 13 | Virilization at puberty | 13 | 0.84 | 0.94 | 1983 | Dutch | No |
| 8 | 326-1;G→C | Female | 8 | Inguinal mass | ND | ND | 1970 | Dutch | No | |
| 9-I | 655-1;G→A | Not known | 16 | Virilization at puberty | 17 | 0.08 | 0.09 | 1976 | Turkish | Yes |
| 9-II | 655-1;G→A | Not known | 15 | Family history | 16 | 0.1 | 0.09 | 1977 | ||
| 9-III | 655-1;G→A | 1 | 10 | Family history | 10 | 0.05 | 0.15 | 1985 | ||
| 10 | A188V | Female | 2 | Inguinal mass | 10 | 1.08 | 0.56 | 1973 | Turkish | No |
| Compound heterozygotes | ||||||||||
| 11-I | 325+4;A→T/N74T | 1 | 14 | Virilization at puberty | 14 | 0.30 | ND | 1982 | Dutch | No |
| 11-II | 325+4;A→T/N74T | 1 | 10 | Family history | 10 | 0.4 | 0.6 | 1986 | ||
| 12 | 325+4;A→T/N74T | 1 | 0.1 | Inguinal mass | 1.3 | ND | 0.28 | 1985 | Dutch | No |
| 13 | 325+4;A→T/R80Q | 1 | 0.1 | Inguinal mass | 0.1 | 0.16 | ND | 1986 | Dutch | No |
| 14 | 325+4;A→T/R80Q | 1 | Birth | Abnormal genitalia | 0.1 | 0.3 | 0.38 | 1991 | Dutch | No |
| 15 | 325+4;A→T/326-1;G→C | 1 | 2 | Inguinal mass | 2 | ND | 0.35 | 1986 | Dutch | No |
| 16 | 325+4;A→T/326-1;G→C | 1 | 0.1 | Inguinal mass | ND | ND | 1988 | Dutch | No | |
| 17 | 325+4;A→T/P282L | 1 | 3.5 | Abnormal genitalia | 6 | 1.0 | 0.27 | 1989 | Dutch | No |
| Heterozygotes | ||||||||||
| 18 | N130S/–(G289S) | 1 | Birth | Abnormal genitalia | 0.6 | 0.4 | 0.29 | 1982 | West Indian | No |
| 19 | No DNA available | 2 | 2 | Inguinal mass | 15 | ND | 0.34 | 1972 | Dutch | Not known |
ND, Not determined. Patient 19 is included for disease incidence calculations; no DNA was available.
Study of genetic origin of recurrent mutations worldwide
The haplotypes of 18 index cases were compared with the genotypes (FM023XH8, D9S1786, D9S1851) of 12 unrelated patients from all over the world. These patients carried the same mutations as the 18 index cases. Some had previously been described as denoted in Table 3 (15); others have not been reported before.
Polymorphic marker haplotypes and genotypes of worldwide patients with 17βHSD3 deficiency
| Patient origin | Genotype/haplotype | Mutations |
|---|---|---|
| The Netherlands | 3/4/3,4,5 | 325+4;A→T |
| Munich (15) | 33/44/44 | 325+4;A→T/325+4;A→T |
| Australia | 33/44/44 | 325+4;A→T/325+4;A→T |
| Pittsburg (15) | 23/45/24 | 325+4;A→T/Q176P |
| San Francisco 3 (15) | 3/4/4 | 325+4;A→T |
| The Netherlands | 1/7/5 | P282L |
| San Francisco 3 (15) | 3/4/4 | P282L |
| The Netherlands | 2/7/5,6 or 3/4/5 | 326-1;G→C |
| Brazil 1 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Brazil 2 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| The Netherlands | 3/4/4 | R80Q |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| Gaza (15) | 33/44/66 | R80Q/R80Q |
| Sao Paulo 3 (15) | 33/44/66 | R80Q/R80Q |
| Portugal | 33/44/36 | R80Q/E215D |
| Turkey | 2/6/6 | 655-1;G→A |
| Syria (15) | 22/66/44 | 655-1;G→A/655-1;G→A |
| Greece (15) | 22/66/66 | 655-1;G→A/655-1;G→A |
| Patient origin | Genotype/haplotype | Mutations |
|---|---|---|
| The Netherlands | 3/4/3,4,5 | 325+4;A→T |
| Munich (15) | 33/44/44 | 325+4;A→T/325+4;A→T |
| Australia | 33/44/44 | 325+4;A→T/325+4;A→T |
| Pittsburg (15) | 23/45/24 | 325+4;A→T/Q176P |
| San Francisco 3 (15) | 3/4/4 | 325+4;A→T |
| The Netherlands | 1/7/5 | P282L |
| San Francisco 3 (15) | 3/4/4 | P282L |
| The Netherlands | 2/7/5,6 or 3/4/5 | 326-1;G→C |
| Brazil 1 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Brazil 2 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| The Netherlands | 3/4/4 | R80Q |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| Gaza (15) | 33/44/66 | R80Q/R80Q |
| Sao Paulo 3 (15) | 33/44/66 | R80Q/R80Q |
| Portugal | 33/44/36 | R80Q/E215D |
| Turkey | 2/6/6 | 655-1;G→A |
| Syria (15) | 22/66/44 | 655-1;G→A/655-1;G→A |
| Greece (15) | 22/66/66 | 655-1;G→A/655-1;G→A |
Polymorphic marker haplotypes and genotypes of worldwide patients with 17βHSD3 deficiency
| Patient origin | Genotype/haplotype | Mutations |
|---|---|---|
| The Netherlands | 3/4/3,4,5 | 325+4;A→T |
| Munich (15) | 33/44/44 | 325+4;A→T/325+4;A→T |
| Australia | 33/44/44 | 325+4;A→T/325+4;A→T |
| Pittsburg (15) | 23/45/24 | 325+4;A→T/Q176P |
| San Francisco 3 (15) | 3/4/4 | 325+4;A→T |
| The Netherlands | 1/7/5 | P282L |
| San Francisco 3 (15) | 3/4/4 | P282L |
| The Netherlands | 2/7/5,6 or 3/4/5 | 326-1;G→C |
| Brazil 1 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Brazil 2 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| The Netherlands | 3/4/4 | R80Q |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| Gaza (15) | 33/44/66 | R80Q/R80Q |
| Sao Paulo 3 (15) | 33/44/66 | R80Q/R80Q |
| Portugal | 33/44/36 | R80Q/E215D |
| Turkey | 2/6/6 | 655-1;G→A |
| Syria (15) | 22/66/44 | 655-1;G→A/655-1;G→A |
| Greece (15) | 22/66/66 | 655-1;G→A/655-1;G→A |
| Patient origin | Genotype/haplotype | Mutations |
|---|---|---|
| The Netherlands | 3/4/3,4,5 | 325+4;A→T |
| Munich (15) | 33/44/44 | 325+4;A→T/325+4;A→T |
| Australia | 33/44/44 | 325+4;A→T/325+4;A→T |
| Pittsburg (15) | 23/45/24 | 325+4;A→T/Q176P |
| San Francisco 3 (15) | 3/4/4 | 325+4;A→T |
| The Netherlands | 1/7/5 | P282L |
| San Francisco 3 (15) | 3/4/4 | P282L |
| The Netherlands | 2/7/5,6 or 3/4/5 | 326-1;G→C |
| Brazil 1 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Brazil 2 | 33/22/11 | 326-1;G→C/326-1;G→C |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| The Netherlands | 3/4/4 | R80Q |
| Sao Paulo 1 (15) | 33/24/46 | R80Q/326-1;G→C |
| Gaza (15) | 33/44/66 | R80Q/R80Q |
| Sao Paulo 3 (15) | 33/44/66 | R80Q/R80Q |
| Portugal | 33/44/36 | R80Q/E215D |
| Turkey | 2/6/6 | 655-1;G→A |
| Syria (15) | 22/66/44 | 655-1;G→A/655-1;G→A |
| Greece (15) | 22/66/66 | 655-1;G→A/655-1;G→A |
Results
Patients
All 17βHSD3-deficient patients had a 46,XY karyotype and were initially raised as girls. Table 1 summarizes data on genotype, phenotype, age of- and reason for referral, endocrine evaluation, year of birth, ethnic background and parental consanguinity. Female-like external genitalia were present in all but three patients (patients 6, 9-I, and 9-II). Most cases were referred because of inguinal masses or abnormal external genitalia in infancy or childhood. In a few cases virilization at puberty prompted referral. Virilization consisted of rugged, pigmented skin of labia majora, enlarged clitoris (>3 cm), and male pattern body hair in patients 6, 9-I, 9-II, and 11-I and lowering of the voice in patients 6 and 11-I. Patient 7 was gonadectomized early in puberty, at age 13 yr, Tanner M2, and had an enlarged clitoris at time of gonadectomy.
Interfamilial phenotypic variability was found in homozygotes for the 325 + 4;A→T mutation. Two sisters (no. 1-I and 1-II) and two unrelated patients (no. 2 and 3) had complete female genitalia at birth. Another unrelated patient (no. 4) had virilized genitalia at birth that allowed a gender reversal from female to male when the diagnosis of 17βHSD3 deficiency was made at age 2 yr.
Patients 1-I, 1-II, 2, 5, 6, 7, 8, 10, 12, 13, 14, 16, and 17 had initially received the tentative diagnosis AIS. No interfamilial relationships were found. Clinical data and in vivo and in vitro testosterone synthesis studies of patient 18 were described previously (28).
Endocrine evaluation
T/A ratios in 17βHSD3-deficient patients, controls, and AIS patients are summarized in Fig. 1. No overlap between the gonadotropin-stimulated T/A ratios in 17βHSD3-deficient patients and controls or AIS patients was observed. Depending on the presence or absence of the physiological LH surge at the time of serum sampling, T/A ratios did show some overlap in the age group of 1–3 months. T/A ratios in 17βHSD3-deficient patients initially diagnosed with AIS did not differ from ratios in the other 17βHSD3 patients. The highest T/A ratios in 17βHSD3-deficient patients of 0.84 before hCG and 0.94 after hCG were found in one pubertal patient (no. 7) homozygous for a known partially inactivating mutation (14).
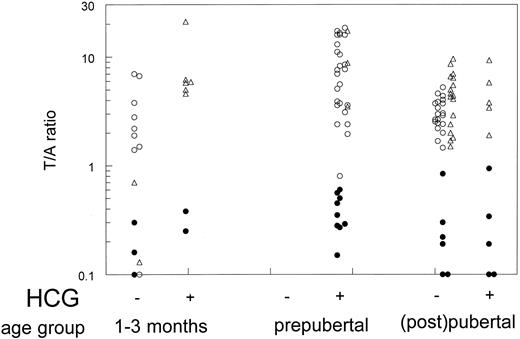
T/A ratios in serum of 17βHSD3-deficient patients (•), age-matched controls (○), or age-matched AIS patients (▵) before (−) or after (+) hCG stimulation. Ratios in individual 17βHSD3-deficient patients from this study are also shown in Table 1.
Mutations
In 18 index patients, 9 different splice site or amino acid substitution mutations were identified (Table 1 and Fig. 2). No DNA could be obtained from patient 19. Recurrent mutations found among unrelated Dutch index patients were 325 + 4;A→T (15 alleles), N74T (6 alleles), R80Q (4 alleles), and 326–1;G→C (4 alleles). One patient (no. 18; Table 1) was heterozygote for a proven pathogenic mutation, N130S (16). The second identified mutation, G289S, is supposed to be a neutral polymorphism (16); we assume that the other allele contained another mutation outside the coding region, because males heterozygous for mutations in the HSD17B gene are normal, as was established in the fathers of these patients.
RNA splicing in homozygotes for 325 + 4;A→T or 655–1;G→A
PCR amplification of cDNA with primer pair 1AA-11B resulted in a 1016-bp product in the control (Fig. 3, A and B). In patient 1-I, mutation 325 + 4;A→T, no wild-type transcript was detected. Instead, a transcript with deletion of exon 3 (941 or 454 bp when using primer pair 1AA-11B or 1AA-6BB, respectively) and in minor amounts a transcript with deletion of exons 3 and 4 (833 and 346 bp with the respective primer combinations) were present (Fig. 3A). Both HSD17B3 gene transcripts render the message out of frame.
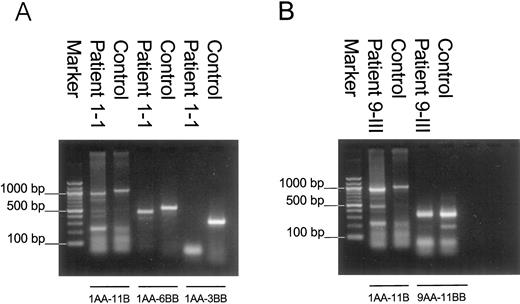
RT-PCR of testicular RNA from patient 1-I homozygous for mutation 325 + 4;A→T (A) and patient 9-III homozygous for mutation 655–1;G→A (B) vs. testicular RNA from a normal healthy male (control).
In the patient with mutation 655–1;G→A, a transcript with an in-frame deletion of exon 9 was present (with primer pair 1AA-11B, 950 bp in weight). In minor amounts a transcript with an in-frame deletion of exons 9 and 10 of 800 bp was present. A wild-type transcript was found only with use of primer pair 9AA-11B (Fig. 3B). Therefore, only minute amounts of wild-type transcript are present compared to the mutant transcripts.
The 175-bp band is found in cDNA of patient 9-III as well as in cDNA of the control and is therefore not specific for the patient (Fig. 3B). Additional bands of 123, 230, 492, and 738 bp were unknown sequences and were present in cDNA of patient 1-I, 9-III as well as in cDNA of the control (Fig. 3, A and B). The transcript with a deletion of exon 3 till 11 of 396 bp (primer pair 1AA-11B), present in cDNA of patient 1-I, patient 9-III, and the control (Fig. 3, A and B), can also be regarded as aspecific for the mutants.
Incidence of 17βHSD deficiency in The Netherlands
Of the total of 23 patients (19 families), 20 were born in The Netherlands between 1969–1999, including patients 10 and 18 (Table 1). The mean annual birth rate during that period was 190,000 (29). Thus, a minimal incidence at birth of 28/20 × 190.000 = 1:266,000 can be calculated. However, the first 17βHSD3-deficient patient was described in 1971 (30). Very likely many earlier cases of 17βHSD3 deficiency will have received other diagnoses. Affected cases that manifest only with virilization at puberty born after 1987 will not be diagnosed until 1999 or later. The number of patients born in the 1980s is probably the most representative group for calculation of incidence data; most cases born during that period will be symptomatic by 1998. For the 1980s (mean birth rate, 176,000) the calculated minimal incidence is 1:147,000.
Carrier frequency
The heterozygote frequency for the Dutch population, as calculated from the incidence of 17βHSD3 deficiency of 1:147,000 in the 1980s, is 1:135. For the 325 + 4;A→T mutation it is 1:210, based on eight 325 + 4;A→T alleles found in index patients born in the 1980s. In a study of 200 random controls (400 chromosomes) no 325 + 4;A→T mutations were found. This finding does not contradict the calculated carrier rate, as there is a (209/210) (200) = 38% chance for this outcome.
Founders of HSD17B3 mutations in the Dutch population
The haplotypes for chromosomes in the Dutch patients carrying either mutation 325 + 4;A→T or N74T vs. controls are shown in Table 2. The 325 + 4;A→T mutations were observed on the same 3/4 haplotype for the flanking markers AFM023XH8/D9S1786 (∼100 kb on the centromeric and telomeric sides, respectively). For the more distant marker (∼1500 kb) D9S1851, recombination (allele 3, 4, or 5) had occurred. This confirmed that there was no close genetic relationship between families, as was established by pedigree analysis.
Allele frequencies of mutant chromosomes vs. Dutch control chromosomes
| Allele | AFM023XH8 | D9S1786 | D9S1851 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | |
| 1 | 0 | 5 | 0 | 0 | 0 | 4 | 0 | 0 | 3 |
| 2 | 0 | 0 | 33 | 0 | 0 | 8 | 0 | 0 | 1 |
| 3 | 12 | 0 | 40 | 0 | 0 | 1 | 1 | 0 | 8 |
| 4 | 0 | 0 | 1 | 12 | 0 | 17 | 7 | 2 | 34 |
| 5 | 0 | 0 | 9 | 2 | 3 | 13 | |||
| 6 | 0 | 0 | 29 | 0 | 0 | 10 | |||
| 7 | 0 | 0 | 4 | 0 | 0 | 1 | |||
| 8 | 0 | 5 | 2 | ||||||
| Total alleles tested | 12 | 5 | 74 | 12 | 5 | 74 | 10 | 5 | 70 |
| Allele | AFM023XH8 | D9S1786 | D9S1851 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | |
| 1 | 0 | 5 | 0 | 0 | 0 | 4 | 0 | 0 | 3 |
| 2 | 0 | 0 | 33 | 0 | 0 | 8 | 0 | 0 | 1 |
| 3 | 12 | 0 | 40 | 0 | 0 | 1 | 1 | 0 | 8 |
| 4 | 0 | 0 | 1 | 12 | 0 | 17 | 7 | 2 | 34 |
| 5 | 0 | 0 | 9 | 2 | 3 | 13 | |||
| 6 | 0 | 0 | 29 | 0 | 0 | 10 | |||
| 7 | 0 | 0 | 4 | 0 | 0 | 1 | |||
| 8 | 0 | 5 | 2 | ||||||
| Total alleles tested | 12 | 5 | 74 | 12 | 5 | 74 | 10 | 5 | 70 |
Allele frequencies of mutant chromosomes vs. Dutch control chromosomes
| Allele | AFM023XH8 | D9S1786 | D9S1851 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | |
| 1 | 0 | 5 | 0 | 0 | 0 | 4 | 0 | 0 | 3 |
| 2 | 0 | 0 | 33 | 0 | 0 | 8 | 0 | 0 | 1 |
| 3 | 12 | 0 | 40 | 0 | 0 | 1 | 1 | 0 | 8 |
| 4 | 0 | 0 | 1 | 12 | 0 | 17 | 7 | 2 | 34 |
| 5 | 0 | 0 | 9 | 2 | 3 | 13 | |||
| 6 | 0 | 0 | 29 | 0 | 0 | 10 | |||
| 7 | 0 | 0 | 4 | 0 | 0 | 1 | |||
| 8 | 0 | 5 | 2 | ||||||
| Total alleles tested | 12 | 5 | 74 | 12 | 5 | 74 | 10 | 5 | 70 |
| Allele | AFM023XH8 | D9S1786 | D9S1851 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | 325+4;A→T | N74T | Control | |
| 1 | 0 | 5 | 0 | 0 | 0 | 4 | 0 | 0 | 3 |
| 2 | 0 | 0 | 33 | 0 | 0 | 8 | 0 | 0 | 1 |
| 3 | 12 | 0 | 40 | 0 | 0 | 1 | 1 | 0 | 8 |
| 4 | 0 | 0 | 1 | 12 | 0 | 17 | 7 | 2 | 34 |
| 5 | 0 | 0 | 9 | 2 | 3 | 13 | |||
| 6 | 0 | 0 | 29 | 0 | 0 | 10 | |||
| 7 | 0 | 0 | 4 | 0 | 0 | 1 | |||
| 8 | 0 | 5 | 2 | ||||||
| Total alleles tested | 12 | 5 | 74 | 12 | 5 | 74 | 10 | 5 | 70 |
Likewise, mutation N74T was associated with haplotype 1/8/4,5 (AFM023XH8/D9S1786/D9S1851).
The association between mutation 325 + 4;A→T and haplotype 3/4 is significant [P < 0.05 (AFM023XH8) and P < 0.01 (D9S1786)] and also between N74T and haplotype 1/8 [P < 0.00000001 (AFM023XH8) and P < 0.00001 (D9S1786)]. Thus, it is likely that both mutations were introduced by two genetic founders for all Dutch patients. Mutation 326–1;G→C, on the other hand, occurred on different haplotypes (2/7/5, 6 and 3/4/5; Table 3), which suggests a recurrent de novo mutation.
Haplotypes of disease chromosomes found in patients worldwide
The geographic distribution of mutations reported in this study as being found worldwide is shown in Fig. 4; haplotypes and marker genotypes are shown in Table 3. The 325 + 4;A→T mutations in Dutch, Germans, white Australians, and white Americans share the same marker genotype and are likely to be identical by descent. Likewise, the mutation R80Q in Dutch, in Arabs in Gaza, in white Brazilians, and in white Portuguese patients and the mutation 655–1;G→C in Turkish, Syrian, and Greek patients (Table 3 and Fig. 3) are due to common founders. The mutations 326–1;G-C and P282L have different intra and/or interethnic haplotypes; therefore, these mutations must have recurrently occurred de novo.
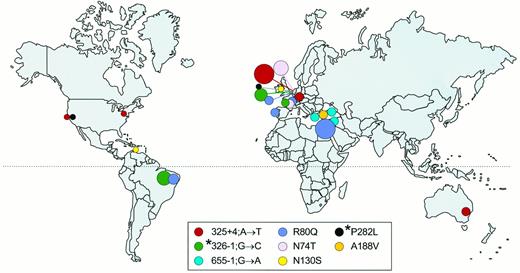
Global distribution of identical HSD17B3 gene mutations as found in this study (5, 15, 16, 39 ). The sizes of the circles correspond to the number of alleles that have been found for a specific mutation in a specific area. However, the number of R80Q alleles in Israel is much larger than indicated (10 ). The different mutations are color coded. *, Mutations with proven de novo recurrence.
Discussion
Pathogeny of splice site mutations
We obtained evidence for the pathogeny of the frequently found 325 + 4;A→T and 655–1;G→A splice site mutations. Both mutations disrupt normal splicing. The transcripts found in the patient homozygous for 325 + 4;A→T are out of frame and therefore nonfunctional. No wild-type transcript was identified in the patient homozygous for mutation 325 + 4;A→T.
As all tested substitution mutations in exon 9 completely abolish enzyme activity (14, 31), the transcripts with a deletion of exon 9 or with a deletion of exons 9 and 10, found in the patient homozygous for 655–1;G→A, are likely to be nonfunctional. A wild-type transcript was found in lesser amounts than the mutant transcripts in a patient homozygous for mutation 655–1;G→A.
Genotype-phenotype relationship
Recurrence of several mutations in multiple patients offered the opportunity for genotype/phenotype comparison. Prepubertal compound heterozygotes had clitoromegaly and labial fusion regardless of whether the mutations had been shown to cause truly female genitalia in homozygote form (325 + 4;A→T or 326–1;G→C; patients 15 and 16) or to render the enzyme completely defective in in vitro studies (P282L, patient 17) (15).
During childhood, homozygotes for mutation 325 + 4;A→T had either truly female genitalia or ambiguous genitalia. Thus, distinct phenotypic variation can occur between homozygotes for the same mutation. Possibilities for the residual, prenatal source of androgen in patient 4 are 1) testosterone formation by another 17βHSD isoenzyme; 2) the variable formation of a small amount of wild-type transcript; or 3) somatic mosaicism for one wild-type allele, sometimes caused by reverse mutations (32). However, somatic mosaicism for the mutation and a normal allele was excluded by allele-specific oligonucleotide hybridization analysis (data not shown). Therefore, the activity of another prenatally expressed isoenzyme or the possible presence of a wild-type transcript are more plausible explanations. As the outcome of aberrant splicing is variable, the absence of a wild-type transcript in one homozygous patient for 325 + 4;A→T does not exclude the possible presence of a wild-type transcript in another patient. This could not be tested in patient 4 because this patient was raised as a boy and consequently was not gonadectomized.
The presence of a wild-type transcript in testicular RNA of the 655–1;G→C homozygous patient (no. 9-III), predicts that phenotypic variation between homozygotes for this mutation could occur depending on the amount of wild-type transcript formed. Indeed, phenotypic variation between families is observed, as the affected children in family 9 were thought to be normal girls during childhood, which is distinctly different from the ambiguous genitalia with which two other unrelated 655–1;G→C homozygous patients had been born (5, 33). Again, androgen formation in peripheral tissues or even in the testes by another 17β-hydroxysteroid dehydrogenase isoenzyme could also be the cause of this phenotypic difference. It seems clear that no specific phenotype is associated with a specific mutation.
Diagnostics
Gonadotropin-stimulated T/A ratios allowed accurate selection of 17βHSD3-deficient cases. However, low T/A ratios are not specific for 17βHSD3 deficiency, but are sometimes also found in patients with other defects in testosterone synthesis or Leydig cell hypoplasia. Therefore, T/A ratios should only be used when a hCG-stimulated response of serum testosterone or/and serum androstenedione is observed. With additional mutation analysis, the diagnosis can hardly be missed. All but 1 of the 18 tested patients were identified as homozygous or compound heterozygous for HSD17B3 mutations. The remaining case, a 46,XY female with testes, had unmistakable endocrine evidence of 17βHSD3 deficiency: an abnormally low T/A ratio and the absence of androstenedione to testosterone conversion in testicular tissue (28).
Based on the ethnic descent of a patient, a prediction on the expected mutations can be made. This greatly facilitates mutation analysis. Furthermore, the West Europeans in this study all had mutations in exon 3, in both splice sites of intron 3, or in exon 11. Mutation analysis can therefore initially be focussed on these particular, relatively small parts of the gene.
In conclusion, endocrine evaluation is an important tool for the selection and diagnosis of patients suspected of 17βHSD3 deficiency. Mutation analysis, facilitated by knowledge of the ethnic distribution of mutations, provides additional proof.
Incidence and carrier frequency
17βHSD3 deficiency is a relatively common cause of male pseudohermaphroditism in The Netherlands, minimally in 1:147,000 newborns. In comparison, the minimal incidence of AIS in the Netherlands is 1:99,000 (unpublished data, based on this same nationwide survey). Previous incidence data for AIS vary between 1:40,800 and 1:128,000 births (34–37) and are based on antiquated diagnostic criteria such as inguinal hernia in girls or X-chromatin-negative bodies in buccal smear of affected girls. Quite likely these series include unidentified 17βHSD3-deficient patients and give a biased, too high incidence rate for AIS.
Like other autosomal recessive diseases, 17βHSD3 deficiency may show increased frequencies among populations with a high intermarriage rate. In Arabs in Gaza, among whom intermarriage is frequent (38), the incidence is 1:200–300, most likely all homozygotes for the R80Q mutation (10). In contrast, the Caucasian Dutch population is heterogeneous, the intermarriage rate is low, and the disease is caused by several different mutations. The carrier frequency for 17βHSD3 deficiency in The Netherlands was calculated to be 1:135.
Founders
Recurrence of mutations N74T and 325 + 4;A→T in the Dutch is very likely due to common founders. Unfortunately, founder analysis of the other described patients with N74T (39) was not possible. 325 + 3;A→T is also carried by other Caucasians living worldwide. All patients with mutation 325 + 4;A→T have the same haplotype for 17HSDB3 gene flanking markers. Thus, the common founders may have lived in Europe, and European immigrants brought the mutation to the U.S. and Australia.
An interesting founder effect may be present in the R80Q mutation, common among Arabs in various parts of Israel, some with Druze ancestors from Lebanon and Syria (40). Their relationship with the same founder of the mutation in Dutch, Portuguese, and white Brazilians prompts the speculation that this mutation became introduced by the Phoenicians who migrated from an area in present day Syria, Lebanon, and Israel around 750 BC toward Portugal and Spain to search for metals and timber (41–43). From there, the mutation was brought to Brazil by the Portuguese colonists and to The Netherlands during the Spanish rule in the 16th-17th century. Alternatively, the mutation may have been introduced in Portugal and Spain by the Moors who had empires on the Iberian Peninsula from AD 711 until 1492 and came from, for example, Lebanon and Syria. The Lebanese and Druze are genetically descendants of the Phoenicians (43), and large numbers of Arabs from Lebanon and Syria, among which were Druze, immigrated in the 19th century in South America (44).
The 655–1;G→A mutation, found in Turks, Greeks, and Syrians might have spread over these populations during the Ottoman empire, which included these three countries between AD 1359 and 1565 (45). The Ottoman empire is known to have contributed to the racial admixture of that area (43, 46).
The recurrent de novo occurrence of other mutations such as 326–1;G→C and P282L supports the conclusion that the genetic basis of 17βHSD deficiency is determined by multiple founders as well as recurrent de novo mutations.
Acknowledgements
We are grateful for the participation of the studied families and for the expert technical assistance of Arienne L.W. Hesseling-Janssen, for the graphic assistance of Tom de Vries Lentsch, and for the bibliographical assistance of Monique L. van Rooijen-Dekkers. We thank Dr. S. M. P.F. de Muinck Keizer-Schrama for providing the endocrine data for individual patients from a previous study (20).
This work was supported by grants from the Netherlands Organization for Scientific Research (GB-MW 900–716-606; to A.L.M.B.) and NIH Grant RO1-DK-52167 (to S.A.).
Farkas A, Rösler A. 1993 Ten years experience with masculinizing genitoplasty in male pseudohermaphroditism due to 17β-hydroxysteroid dehydrogenase deficiency. Eur J Pediatr. 152(Suppl 2):s88–s90.
Prader A.
Statistics Netherlands.
Cavalli-Sforza LL, Menozzi P, Piazza A. 1994 The history and geography of human genes. Princeton: Princeton University Press; 217, 242–245, 260.

{kind=link}
{kind=link}
{kind=link}
{kind=link}