




Abstract
Background: The monoclonal antibody cetuximab (IMC-225, Erbitux) inhibits epidermal growth factor receptor (EGFR) signaling and has been approved for metastatic colon cancer therapy. However, to achieve effective titers, passive antibody therapies must be repeatedly administered over long periods. To overcome this limitation, we aimed to generate a vaccine inducing continuously available “cetuximab-like” antibodies in vivo using the mimotope approach. Methods: We used the phage display technique to identify four peptides structurally mimicking the cetuximab epitope. We coupled two of these peptides to an immunogenic carrier protein, and we vaccinated four groups (n = 8) of BALB/c mice intraperitoneally with 10 μg of the mimotope conjugates, a control peptide conjugate, or the carrier protein alone. We assessed antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity mediated by the induced antibodies against EGFR-overexpressing human A431 carcinoma cells. We then tested receptor internalization capacity of the induced antibodies with fluorescently labeled EGFR, and we assayed their growth inhibitory potential toward A431 cells with a [ 3 H]thymidine proliferation assay. Results: Mimotope-induced antibodies recognized EGFR, and both types of antibody-mediated cytotoxic effects were elicited by these antibodies. In both cellular cytotoxicity assays, the mimotope-induced antibodies exhibited specific lysis of more than 50%. The induced antibodies caused internalization of the receptor from the cell surface into endocytic vesicles and inhibited growth of EGFR-expressing cells to a similar extent as cetuximab [67% (95% confidence interval {CI} = 55% to 79%) and 69% (95% CI = 55% to 84%), respectively]. Conclusions: Epitope-specific immunization is feasible for active anti-EGFR immunotherapy. The in vitro biologic features of mimotope-induced antibodies are similar to those of the monoclonal antibody cetuximab.
Members of the epidermal growth factor (EGF) family of membrane receptors are among the best-characterized targets on cancer cells. This family, which contains four members—EGF receptor (EGFR; also known as HER-1 or ErbB1), HER-2 (also known as ErbB2 or neu), HER-3 (also termed ErbB3), and HER-4 (also known as ErbB4)—is essential in regulating cellular growth, differentiation, and proliferation ( 1 ) . Increased EGFR signaling, in particular, has been associated with oncogenic transformation; it results in autonomous cell growth, tumor invasion, angiogenesis, and metastasis ( 2 , 3 ) . EGFR expression and overexpression has been reported in multiple tumor types ( 4 ) , and one-third of epithelial cancers express high levels of the receptor. Often, EGFR overexpression has been associated with poor prognosis ( 5 ) .
Because EGFR has been implicated in cancer initiation and development, therapeutics that target EGFR have been developed. One EGFR inhibitor is cetuximab (IMC-C225, Erbitux) ( 6 ) , a monoclonal antibody that was approved by the U.S. Food and Drug Administration for treatment of metastatic colon cancer ( 7 ) . Cetuximab is a chimeric human–murine antibody that targets the extracellular domain of EGFR ( 8 ) . It binds to EGFR with higher affinity than its naturally occurring ligands, EGF and transforming growth factor α (TGF-α), thus competitively blocking their ability to bind to and activate EGFR ( 8 , 9 ) .
One limitation of this therapeutic approach of passive immunotherapy is the need to repeatedly administer the antibody to achieve titers that effectively inhibit EGFR and elicit antitumor activity. Active immunization that initiates the ongoing production of antibodies of the desired type would be an attractive alternative because it would circumvent both the need for multiple infusions and the danger of inducing an immune response against the nonhuman parts of the artificial antibodies. Our group has previously used mimotopes (epitope mimics) for epitope-specific induction of antibodies in type I allergy ( 10 , 11 ) and malignant disease ( 12 , 13 ) . Mimotopes are small peptides that mimic a given epitope structurally, but not necessarily by amino acid sequence ( 14 , 15 ) . These mimics are isolated from phage display libraries ( 16 ) , often comprising a repertoire of more than 10 9 possible ligands. A special advantage of the phage display technique is that the epitope to be mimicked does not need to be defined; the only prerequisite is that an antibody recognizes it ( 17 ) —in this study, an antibody with proven beneficial antitumor properties.
We recently reported the generation of a mimotope vaccine for induction of “trastuzumab-like” antibodies against HER-2 ( 13 ) , and we were able to show that epitope-specific active immunotherapy is feasible against the EGFR family of growth receptors. In the EGFR family, epitope-specificity of vaccines is crucial, as growth-stimulatory antibody populations must be prevented ( 18 ) . Here our aim was to identify peptide mimics of the EGFR epitope recognized by cetuximab and to develop a mimotope vaccine. Also, we sought to characterize the resulting immune response in vitro.
M ATERIALS AND M ETHODS
Cell Lines and Total Cell Lysates
The EGFR-overexpressing human epidermoid carcinoma cell line A-431 (CRL-1555; American Type Culture Collection [ATCC], Manassas, VA) was grown in Dulbecco's modified Eagle's medium (Gibco BRL, Inchinnan, UK) supplemented with 10% fetal calf serum, 1% glutamine, 1% penicillin–streptomycin, and 50 μg/mL gentamicin sulfate. The human mammary carcinoma cell line SK-BR-3 (ATCC HTB-30), which expresses very low levels of EGFR, was grown in McCoy's medium (Gibco BRL) that was supplemented as described above.
Total cell lysates were made as previously described ( 19 ) using a modified lysis buffer containing 20 m M Tris, 150 m M NaCl, 1 m M EDTA, 1 m M EGTA, 1% Triton X-100, and a protease inhibitor cocktail (Complete; Roche, Basel, Switzerland) at pH 7.5. The protein concentration was determined photometrically using bicinchoninic acid (BCA Protein Assay Kit; Pierce, Rockford, IL). Extracts were aliquoted and stored at −80 °C.
Monoclonal Antibodies
Cetuximab (Erbitux), a chimeric immunoglobulin G1 (IgG1) monoclonal antibody, was purchased from Merck (Darmstadt, Germany). A different chimeric IgG1 monoclonal antibody, ch14.18, which is directed against disialogangliside GD2, was used as an isotype control.
Biopanning of M13 Phage Display Library
To isolate M13 phage with the desired properties, i.e., high-affinity binding of peptide inserts to cetuximab, a random peptide phage library was subjected to three successive rounds of biopanning with this antibody. The library CL10, kindly provided by Professor Luca Mazzucchelli, Locarno, Switzerland, expresses cysteine-flanked decapeptides that are circularized by disulfide bridging between the cysteines and fused to pIII of the filamentous phage M13. The library carries a kanamycin resistance ( 20 ) . Biopannings were performed as previously described ( 21 ) , with some modifications. In brief, 96-well enzyme-linked immunosorbent assay (ELISA) plates (Nunc, Roskilde, Denmark) were coated with 40 μg/mL cetuximab in bicarbonate buffer at pH 8.5. Nonspecific binding was blocked with phosphate-buffered saline (PBS) containing 1% dry milk, and wells were incubated with an aliquot of the phage library (∼5 × 10 10 phage particles) in PBS containing 1% dry milk and 0.1% Tween 20 at room temperature for 2 hours. Unbound phage particles were removed by extensive washing with PBS containing 0.5% Tween 20. Bound phage particles were eluted with 150 μL of 0.1 M glycine, pH 2.2, and the solution was immediately neutralized with 45 μL of 1 M Tris, pH 8.0. Phage were amplified in Escherichia coli K91, precipitated from the bacterial culture supernatant with polyethylene glycol, and either used immediately for another round of biopanning or stored at −20 °C. Phage titers were determined by plating serial dilutions of the eluate or the amplificate on Luria broth agar plates containing 75 μg/mL kanamycin and counting colonies after overnight incubation at 37 °C.
Colony-Screening Assay
After each round of biopanning, specific phage clones were selected by colony screening ( 22 ) . Colonies grown on Luria Broth–kanamycin agar plates as described above were transferred to nitrocellulose membranes in duplicate, and these were immunoscreened with either cetuximab or isotype control antibody ch14.18 (2.5 μg/mL in PBS–casein). Bound antibody was detected with 125 I-labeled anti–human IgG (IBL GmbH, Hamburg, Germany). Membranes were washed, dried, and exposed to Biomax-MS films (Kodak, Rochester, NY) at −70 °C. Positive clones were amplified as described above.
DNA Sequencing
Single-strand phage DNA was prepared using a Qiaprep Spin M13 kit (Qiagen, Hilden, Germany). The amount of DNA was determined by examining ethidium bromide–stained 0.7% agarose gel under ultraviolet illumination. DNA sequencing was done by I.B.L. (Vienna, Austria).
Specificity ELISA
Ninety-six-well ELISA plates (Nunc) were coated with 10 μg/mL cetuximab or ch14.18 in PBS by incubating overnight at 4 °C. Plates were then washed with PBS containing 0.05% Tween 20, and nonspecific binding was blocked by incubation with PBS containing 1% bovine serum albumin (BSA). Phage clones that were positive in the colony screening assay or wild-type phage were added at a concentration of 10 6 colony forming units (CFU)/mL in PBS containing 0.1% BSA. Bound phage particles were detected with a peroxidase-conjugated mouse anti–phage M13 monoclonal antibody (Amersham Pharmacia Biotech, Little Chalfont, UK). The reaction was developed with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (Sigma, St. Louis, MO) as substrate. Optical density was measured in an ELISA reader (Dynatech, Denkendorf, Germany) at 405 nm (reference wavelength, 490 nm). All determinations were done in triplicate.
Mimicry ELISA
Ninety-six-well ELISA plates (Nunc) were coated with cetuximab (10 μg/mL) in PBS and blocked as described above. Phage particles (10 7 CFU/mL) and cell extracts (containing 500 μg/mL or 200 μg/mL of protein) were added simultaneously, and plates were incubated for 1 hour at 37 °C and then for 1 hour at 4 °C. After washing as above, bound phage particles were detected as described above. Percent inhibition of phage binding to cetuximab by cell extracts was calculated relative to phage binding to cetuximab in the absence of cell extract (set to 100%). Again, all tests were performed in triplicate.
Synthesis of Vaccine Constructs
The 1,12-cyclic peptides C-QYNLSSRALK-C (constituting a high-affinity mimotope), C-VWQRWQKSYV-C (a lower-affinity mimotope), and C-DGGWLSKGSW-C (control) were chemically synthesized (piChem, Graz, Austria). Conformational accuracy was verified with cetuximab in a dot blot assay. In brief, the peptides were solubilized in PBS containing 20% dimethylformamide at 1 mg/mL and dotted onto nitrocellulose membranes. Blots were blocked with PBS containing 1% BSA and later incubated with 10 μg/mL cetuximab or ch14.18 in PBS containing 0.1 % BSA for 2 hours at room temperature. Bound antibody was detected by incubating with 125 I-labeled anti–human IgG (50 nCi/ml) (IBL GmbH). The blots were washed with PBS containing 0.5 % Tween 20, dried, and exposed to Biomax-MS film (Kodak) at −70 °C.
After recognition by cetuximab was confirmed, each peptide was coupled by way of its C terminus to a succinimidyl-4-( N -maleinimidomethyl)cyclohexan-1-carboxylate–activated immunogenic carrier, keyhole limpet hemocyanin (KLH), through a linker (GPGPG) by S -acetyl-thio-acetate. The mimotope conjugates were again checked for cetuximab binding capability in a dot blot assay, as described above.
Immunization of BALB/c Mice
Four groups (n = 8 per group) of BALB/c mice (Charles River Laboratories, Sulzfeld, Germany) were immunized by intraperitoneal injection with 10 µg of the mimotope conjugates, C-QYNLSSRALK-C–KLH or C-VWQRWQKSYV-C–KLH, control peptide–KLH, or KLH alone, on days 1, 15, 44, and 57. Aluminum hydroxide (2% suspension in water) was used as an adjuvant in all groups. Blood was taken from the tail vein on day 0 (preimmunization serum) and on days 22 (first immune serum), 50 (second immune serum), and 64 (third immune serum). Serum was pooled from each group of mice by mixing equal volumes of third immune sera from each mouse. Mice were treated according to European Union Rules of Animal Care, with permission #66.009/35-BrGT/P2004 from the Austrian Federal Ministry of Education, Science, and Culture.
Western Blotting and Titer Determination
Total A431 cell lysates, prepared as described above, were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and proteins were electrophoretically transferred to nitrocellulose membranes. Blots were blocked with PBS containing 1% BSA and then incubated with cetuximab or pooled serum for 2 hours at room temperature. Bound cetuximab was detected by a peroxidase-conjugated goat anti–human IgG antibody (Jackson ImmunoResearch, West Grove, PA), and bound mouse antibodies were detected by a peroxidase-conjugated sheep anti–mouse Ig (Amersham), using the ECL chemoluminescent detection protocol (Amersham).
Titers of sera from the immunized BALB/C mice against KLH, the respective mimotope peptide, and against EGFR were determined by incubation of serial dilutions of sera (1 : 500, 1 : 1000, 1 : 2000, 1 : 5000, 1 : 10 000, 1 : 50 000, 1 : 100 000, 1 : 1 000 000, 1 : 10 000 000) with dotted KLH, either mimotope peptide, or blotted A431 cell lysate. Titers are given as the highest serum dilution at which antigen reactivity still was detectable in these blot experiments.
Fluorescence Microscopy
A431 cells, which overexpress EGFR, were plated at 10 5 cells/mL on four-well Lab-Tek tissue culture chamber slides (Miles Laboratories Inc., Naperville, IL). SK-BR-3 cells, which express very low levels of EGFR, were used as negative controls. Cells were grown overnight until half-confluent. Chamber slides were then cooled to 4 °C and washed with ice-cold PBS, cells were fixed with 4% paraformaldehyde in PBS for 30 minutes, chamber slides were incubated with 50 m M NH 4 Cl in PBS to quench fixation, and blocked with 1% BSA in PBS. Then cells were incubated with cetuximab (positive control) or with pooled third immune sera, and bound antibodies were detected by fluorescein isothiocyanate (FITC)–conjugated goat anti–mouse IgG (Caltag Laboratories, Burlingame, CA). Pooled sera from the KLH-immunized mice were used as a control. Nuclei were stained with 0.1μg/mL Hoechst dye (Sigma) in PBS for 10 minutes. Cells were mounted in Mowiol mounting medium and viewed with a Zeiss Axioplan 2 (Carl Zeiss, Jena, Germany).
Antibody-Mediated Cytotoxicity Assays
Transfection and Internalization Assay
SK-BR-3 cells were plated into four-well Lab-Tek tissue culture chamber slides (Miles Laboratories Inc.) at a concentration of 5 × 10 5 cells/mL. Cells were grown overnight and then transfected with a EGFR/green fluorescent protein (GFP) construct ( 23 ) that was kindly provided by Dr. Philippe Bastiaens (EMBL, Heidelberg, Germany) using SuperFect Transfection Reagent (Qiagen), according to the manufacturer's instructions. The construct encodes a fully functional EGFR that is tagged with GFP and therefore can be visualized with a fluorescence microscope (excitation wavelength of GFP: 395 nm; emission wavelength: 508 nm). After a 2-day expression period, cells were incubated for 15 minutes with 10 μg/mL purified IgG from pooled third immune sera obtained from mimotope-immunized or control mice, with 10 μg/mL cetuximab, or with medium alone to allow for receptor trafficking. Cells were then fixed in 4% paraformaldehyde in PBS and mounted in Mowiol mounting medium as described above. Slides were viewed at the indicated wavelengths with a Zeiss Axioplan 2 fluorescence microscope.
[ 3 H]Thymidine Proliferation Assay
A431 cells were seeded to a 96-well tissue culture plate (Nunc) at 1000 cells/well in 100 μL of Dulbecco's modified Eagle's medium. Cetuximab or ch14.18 was added to the respective wells at concentrations of 2 μg/mL, or mouse sera from mimotope-immunized or control mice at a dilution of 1 : 100 or 1 : 500. Plates were incubated at 37 °C and 5% CO 2 for 4 days. [ 3 H]Thymidine was then added to a concentration of 0.5 μCi/well, and the plates were incubated for 4 more hours. Cells were harvested with an LKB-Wallac 1295-001 Cell Harvester (Wallac Oy, Turku, Finland) and collection filters dried and embedded in scintillation fluid. Assays were read in a 1205 Betaplate Liquid Scintillation Counter (Wallac Oy).
Statistical Analysis
Statistical analysis was performed using the Student's t test. P ≤.01 was considered highly statistically significant, P ≤.05 was considered statistically significant, and P ≤.1 was considered suggestive of statistical significance.
R ESULTS
Biopanning and Colony Screenings
We screened a constrained 10-mer random peptide phage display library by biopanning with cetuximab to identify peptides mimicking the epitope recognized by the antibody. The total phage titers increased with each successive round of panning, indicating the presence of phage particles carrying epitope mimics. The phage titer increased from 4 × 10 3 CFU/mL (first round) to 10 4 CFU/mL (second round), and finally to 8 × 10 7 CFU/mL (third round).
Colony screenings identified 11 cetuximab-specific phage clones. These were amplified and sequenced. Four different insert sequences were found, with C-QFDLSTRRLK-C being found most often (seven times). A highly homologous sequence, C-QYNLSSRALK-C, was detected twice, and two more sequences, C-VWQRWQKSYV-C and C-MWDRFSRWYK-C, were found once each. Sequence analysis of the peptides revealed no homology to EGFR or to any other member of the EGF receptor family in database alignments (EMBL).
Mimotope Characterization
A specificity ELISA was performed to independently confirm the colony-screening results. All four mimotope candidates were specifically recognized by the selecting antibody cetuximab but not by the isotype-matched control antibody ch14.18 ( Fig. 1, A ). Moreover, neither cetuximab nor the control antibody reacted with wild-type M13 phage. As in the colony-screening result analysis, the phage-displayed peptides C-QFDLSTRRLK-C and C-QYNLSSRALK-C exhibited the highest reactivity to cetuximab.
![Characterization of cetuximab mimotopes. A ) Specific recognition of selected phage clones by cetuximab was examined by enzyme-linked immunosorbent assay (ELISA). Solid bars , binding of cetuximab to M13 phage clones displaying specific circular peptide inserts or to wild-type M13 phage. Open bars , binding of isotype-matched control antibody ch14.18 to the same phage clones. B ) ELISA analysis of competitive effects of cell extracts on binding of cetuximab to peptides. Solid bars , no cell extract; oblique hatched bars , extract from human epidermoid carcinoma A431 (epidermal growth factor receptor [EGFR] overexpressing) cells at 500 μg/mL; open bars , extract from A431 tumor cells at 200 μg/mL; horizontally hatched bars , extract from the low EGFR-expressing cell line SK-BR-3 at 500 μg/mL. Data in both panels show means of triplicates from a single representative experiment; error bars represent 95% confidence intervals.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnci/97/22/10.1093_jnci_dji373/2/m_jncidji373f01_lw.jpeg?Expires=1716983130&Signature=zauwaKwl-Bva~OiNiWizuqbBQ9ISbsHNnhMcr6J4vEnHpfEsjn7QmP4VL8QgXa1i4JwsCczMgTvCieU8yQsVHjOcmngeA8zZAAdlhtHfyJgTjouTdGDI~6CXm0EaNxcY79Onax3j67qpbFj4Y9xGxIa02sZUdrDLOqWfzYT1qAD~uNoPiDGFI75R-jWhuYt7qGSB6UB5g~015qdlm1OQk7QHhyp1GOBHAR6-bpavN4CkLFKF~kmpuan3cvqmWEBn9ki0TVgkCnuvE3~-cmn5qgww8Z-1qKRQ6KzXbj7PvwChtRssbQliOtSYwrkOKqdbtVOdtUnZTHZBO-q6ZT87sw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of cetuximab mimotopes. A ) Specific recognition of selected phage clones by cetuximab was examined by enzyme-linked immunosorbent assay (ELISA). Solid bars , binding of cetuximab to M13 phage clones displaying specific circular peptide inserts or to wild-type M13 phage. Open bars , binding of isotype-matched control antibody ch14.18 to the same phage clones. B ) ELISA analysis of competitive effects of cell extracts on binding of cetuximab to peptides. Solid bars , no cell extract; oblique hatched bars , extract from human epidermoid carcinoma A431 (epidermal growth factor receptor [EGFR] overexpressing) cells at 500 μg/mL; open bars , extract from A431 tumor cells at 200 μg/mL; horizontally hatched bars , extract from the low EGFR-expressing cell line SK-BR-3 at 500 μg/mL. Data in both panels show means of triplicates from a single representative experiment; error bars represent 95% confidence intervals.
To demonstrate mimicry between peptides and the original antigen, phage particles and a cell extract from the human EGFR-overexpressing cancer cell line A431 were allowed to compete for cetuximab, and bound phage particles were detected by ELISA. A cell extract from SK-BR-3 cells was used as control. Only the A431 cell extract could specifically displace bound phage from the antibody; moreover, this displacement was dose dependent ( Fig. 1, B ). That is, the phage mimics were displaced by 61% (C-QFDLSTRRLK-C, 95% confidence interval [CI] = 58% to 63%), 71% (C-QYNLSSRALK-C, 95% CI = 69% to 72%), 70% (C-VWQRWQKSYV-C, 95% CI = 69.7% to 70.0%), and 66% (C-MWDRFSRWYK-C, 95% CI = 65% to 66%), respectively, when A431 extract was used at 500 mg/mL. At a lower concentration (200 μg/mL), the mimics were displaced by 21% (95% CI = 4% to 36%), 33% (95% CI = 22% to 43%), 30% (95% CI = 6% to 55%), and 31% (95% CI = 23% to 39%), respectively. Some inhibition was also observed with the control extract (500 μg/mL) that was likely due to its low but detectable EGFR content.
Vaccine Constructs and Immune Responses Induced by Mimotope Vaccination
From the specificity ELISA and mimicry test results, the highly reactive peptide C-QYNLSSRALK-C and the somewhat less reactive peptide C-VWQRWQKSYV-C were chosen for immunogenicity evaluation and manufactured synthetically. The synthetic peptides were, again, recognized by cetuximab, but not by the isotype control antibody ( Fig. 2 , left). The pattern of reactivity was similar to the results of the specificity ELISA ( Fig. 1, A ). Also, mimotope conformation was preserved after conjugation to the immunogenic carrier, KLH. Neither control peptide coupled to KLH, nor KLH itself was recognized by cetuximab ( Fig. 2 , right).
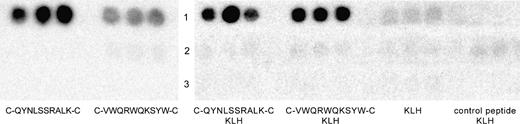
Dot blot analysis of antibody recognition of synthetic cyclic mimotopes. ( Left ) Each peptide of the indicated sequence was dotted in triplicate and blots were probed with cetuximab ( lane 1 ), isotype control antibody ch14.18 ( lane 2 ), or no primary antibody (buffer control, lane 3 ). ( Right ) Both mimotope peptides and a control peptide were conjugated to the immunogenic carrier molecule keyhole limpet hemocyanin (KLH) and dotted in triplicate. Conjugated peptides and carrier KLH alone were again probed with cetuximab ( lane 1 ), isotype control antibody ch14.18 ( lane 2 ), or no primary antibody ( lane 3 ).
The immunogenicity of the mimotope conjugates (and controls) was then evaluated in four groups (n = 8 per group) of BALB/c mice. All mice developed high anti-KLH titers, indicating successful immunization in all four groups ( Table 1 ). Mice immunized with the mimotope conjugates showed a humoral response toward the respective peptide and, more important, developed antibodies that recognize EGFR in A431 cell lysates ( Table 1 ; Fig. 3 ). Sera from control mice showed no reactivity to the mimotope peptides or EGFR ( Table 1 ; Fig. 3 ).
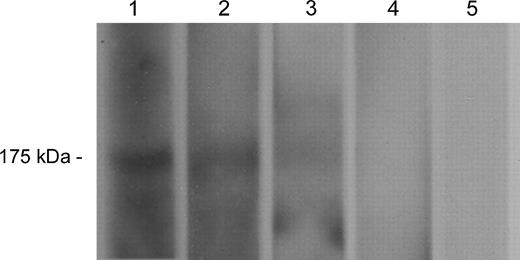
Western blotting with cetuximab or mimotope-induced antibodies for epidermal growth factor receptor (EGFR) recognition in an A431 cell lysate. EGFR detection at 175 kDa by cetuximab ( lane 1 , positive control) and by sera from mice immunized with the C-QYNLSSRALK-C–KLH conjugate ( lane 2 ), with the C-VWQRWQKSYV-C–KLH conjugate ( lane 3 ), with the carrier KLH alone ( lane 4 ), or with a control peptide–KLH conjugate ( lane 5 ).
Titers of the four mouse groups as determined by DotBlot and western blot analysis (third immune serum, day 64), against carrier, peptide, and original antigen
| Titers | |||||
|---|---|---|---|---|---|
| Antigen | anti-KLH | anti-mimotope | anti-EGFR | ||
| C-QYNLSSRALK-C–KLH | 1 : 100 000 | 1 : 100 000 | 1 : 5000 | ||
| C-VWQRWQKSYV-C–KLH | 1 : 100 000 | 1 : 10 000 | 1 : 2000 | ||
| KLH | 1 : 100 000 | – | – | ||
| control peptide–KLH | 1 : 100 000 | – | – | ||
| Titers | |||||
|---|---|---|---|---|---|
| Antigen | anti-KLH | anti-mimotope | anti-EGFR | ||
| C-QYNLSSRALK-C–KLH | 1 : 100 000 | 1 : 100 000 | 1 : 5000 | ||
| C-VWQRWQKSYV-C–KLH | 1 : 100 000 | 1 : 10 000 | 1 : 2000 | ||
| KLH | 1 : 100 000 | – | – | ||
| control peptide–KLH | 1 : 100 000 | – | – | ||
Titers of the four mouse groups as determined by DotBlot and western blot analysis (third immune serum, day 64), against carrier, peptide, and original antigen
| Titers | |||||
|---|---|---|---|---|---|
| Antigen | anti-KLH | anti-mimotope | anti-EGFR | ||
| C-QYNLSSRALK-C–KLH | 1 : 100 000 | 1 : 100 000 | 1 : 5000 | ||
| C-VWQRWQKSYV-C–KLH | 1 : 100 000 | 1 : 10 000 | 1 : 2000 | ||
| KLH | 1 : 100 000 | – | – | ||
| control peptide–KLH | 1 : 100 000 | – | – | ||
| Titers | |||||
|---|---|---|---|---|---|
| Antigen | anti-KLH | anti-mimotope | anti-EGFR | ||
| C-QYNLSSRALK-C–KLH | 1 : 100 000 | 1 : 100 000 | 1 : 5000 | ||
| C-VWQRWQKSYV-C–KLH | 1 : 100 000 | 1 : 10 000 | 1 : 2000 | ||
| KLH | 1 : 100 000 | – | – | ||
| control peptide–KLH | 1 : 100 000 | – | – | ||
Immunofluorescence
We performed immunofluorescence staining of A431 cells to determine whether the induced antibodies also recognized EGFR on the cell surface. Sera of mice immunized with C-QYNLSSRALK-C–KLH ( Fig. 4, A ) or C-VWQRWQKSYV-C–KLH ( Fig. 4, B ) showed marked membrane staining of A431 cancer cells, as did cetuximab (positive control) ( Fig. 4, C ). Only background staining was observed when serum antibodies from mice immunized with KLH were tested ( Fig. 4, D ). SK-BR-3 control cells did not stain with any antibody (data not shown).
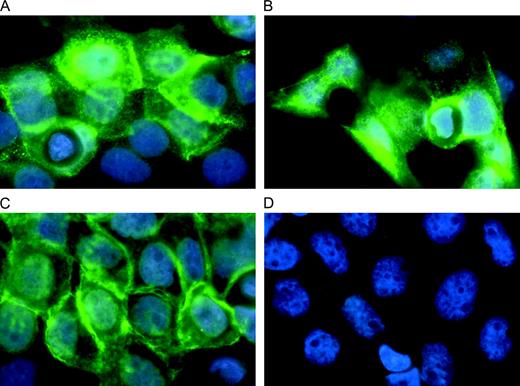
Immunofluorescence staining of epidermal growth factor receptor (EGFR) on A431 cancer cells. Anti-EGFR antibodies were detected by fluorescein isothiocyanate (FITC)–conjugated secondary antibodies. Nuclei were stained with Hoechst dye in phosphate-buffered saline for 10 minutes. Cells were viewed with a Zeiss Axioplan 2 fluorescence microscope. A ) Sera from mice immunized with C-QYNLSSRALK-C–KLH. B ) Sera from mice immunized with C-VWQRWQKSYV-C–KLH. C ) Cetuximab (positive control). D ) Sera from mice immunized with KLH alone (negative control).
Functional Assays
To assess whether the biologic activities of the antibodies induced by the mimotopes are similar to those of cetuximab, we performed ADCC and CDC assays and internalization and proliferation inhibition studies. In both cellular cytotoxicity assays, the mimotope-induced antibodies exhibited specific lysis of more than 50% of A431 cells ( Fig. 5 ). Anti–C-QYNLSSRALK-C antibodies elicited specific lysis of 54% (95% CI = 32% to 76%) of target cells in the ADCC assay and 51% (95% CI = 39% to 63%) specific lysis in the CDC assay. Anti–C-VWQRWQKSYV-C antibodies caused lysis of 50% (95% CI = 39% to 61%) of target cells as measured by the ADCC assay and of 65% (95% CI = 54% to 76%) in the CDC assay. No ADCC (1%, 95% CI = 0% to 19%), although some background CDC (13%, 95% CI = 4% to 22%), was seen with antibodies from the KLH-immunized control group. The differences were highly statistically significant ( P ≤.01) both for ADCC and CDC results. When anti-mimotope antibodies were tested on SK-BR-3 control cells, no specific cytotoxicity was observed (data not shown). Because all effectors (cells and complement) in these assays were murine, cetuximab could not be used as a positive control.
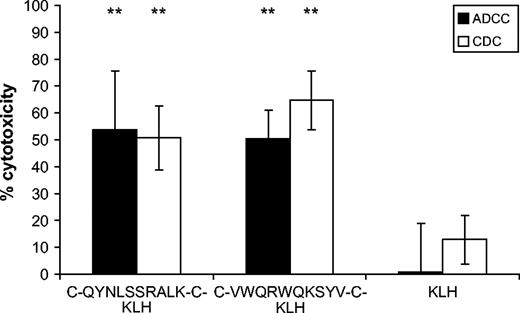
Antibody-mediated cytotoxicity against A431 cells elicited by antibodies induced after vaccination with mimotopes or keyhole limpet hemocyanin, expressed as percent cytotoxicity. Solid bars , antibody-mediated cellular cytotoxicity (ADCC). Open bars , complement-mediated cytotoxicity (CDC). Columns represent means of triplicate analysis; error bars represent 95% confidence intervals. Specific lysis was compared with lysis incurred by the anti-KLH control antibodies. Statistical analysis was performed using the Student's t test (**, P ≤.01; exact P values: comparison of ADCC mediated by anti–C-QYNLSSRALK-C–KLH antibodies to anti-KLH antibodies, P = .01; comparison of ADCC mediated by anti–C-VWQRWQKSYV-C–KLH antibodies to anti-KLH antibodies, P = .006; comparison of CDC mediated by anti–C-QYNLSSRALK-C–KLH antibodies to anti-KLH antibodies, P = .005; comparison of CDC mediated by anti–C-VWQRWQKSYV-C–KLH antibodies to anti-KLH antibodies, P = .001).
To assess antibody-mediated internalization of the receptor, we transfected SK-BR-3 cells with a GFP-labeled EGFR construct so that it could be visualized with a fluorescence microscope. (The low constitutive level of EGFR in SK-BR-3 cells reduces the level of unlabelled receptors that could interfere in the assay.) After a 15-minute incubation of transfected cells with either cetuximab ( Fig. 6, A ) or mimotope-induced antibodies ( Fig. 6, B and C ), we observed a reduction of receptor surface density with later appearance of the receptor in vesicles. Mimotope-induced antibodies in particular led to pronounced internalization and aggregation of EGFR in perinuclear vesicles, which are indicative of receptor degradation. Sera from mice immunized with the carrier KLH alone did not cause receptor internalization, but EGFR remained at the cell surface ( Fig. 6, D ), like in cells treated with medium alone ( Fig. 6, E ).
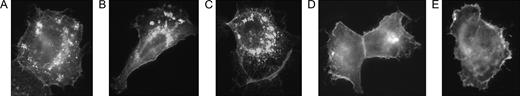
Antibody-mediated internalization of epidermal growth factor receptor (EGFR). Cells were transfected with a fluorescence-labeled EGFR and incubated with antibodies for 15 minutes; receptor trafficking was viewed with a Zeiss Axioplan 2 fluorescence microscope. A ) Cetuximab. B ) Purified antibodies from mice immunized with C-QYNLSSRALK-C–KLH. C ) Purified antibodies from mice immunized with C-VWQRWQKSYV-C–KLH. D ) Purified anti-keyhole limpet hemocyanin antibodies. E ) Medium alone.
We performed a proliferation inhibition assay to analyze the direct growth-inhibitory capacities of the induced antibodies ( Fig. 7 ). Cetuximab inhibited growth of A431 cancer cells by 69% (95% CI = 55% to 84%). Anti–C-QYNLSSRALK-C antibodies at a dilution of 1 : 100 also inhibited A431 cancer cell growth (67%, 95% CI = 55% to 79%) to an extent comparable to cetuximab, whereas anti–C-VWQRWQKSYV-C antibodies exhibited weaker inhibition (47%, 95% CI = 38% to 57%). Both cetuximab and anti–C-QYNLSSRALK-C antibodies inhibited growth to a statistically significantly ( P = .05 and P = .03, respectively) greater extent than the isotype control antibody (16%, 95% CI = 0% to 43%). The weaker inhibition of anti–C-VWQRWQKSYV-C antibodies still was suggestive of statistical significance ( P = .1) but was not statistically significant when compared with the inhibition caused by the isotype control antibody. The observed growth inhibitions were dose dependent; sera diluted to 1 : 500 caused less inhibition, 46% (95% CI = 17% to 75%) for anti–C-QYNLSSRALK-C antibodies, and 31% (95% CI = 7% to 55%) for anti–C-VWQRWQKSYV-C antibodies, than sera diluted to 1 : 100. Antibodies from the KLH-immunized group exhibited similar nonspecific growth inhibitions of 21% (95% CI = 3% to 38%) at 1 : 100 and 18% (95% CI = 0% to 42%) at 1 : 500 as the isotype control for cetuximab.
![A431 cell proliferation inhibition assessed by [ 3 H]thymidine incorporation. Cells were grown for 4 days with medium alone or with antibodies present as indicated on the x -axis, and then incubated with [ 3 H]thymidine. Uptake of [ 3 H]thymidine, which represents cell growth, was measured in a scintillation counter (counts per minute are given on the y -axis). Columns represent means of triplicate analysis; error bars represent 95% confidence intervals. Differences in A431 cell growth inhibition were analyzed using the Student's t test. P values≤.05 (*) were considered statistically significant ( P = .05 for inhibition caused by cetuximab compared with isotype control, and P = .03 for inhibition caused by anti–C-QYNLSSRALK-C–KLH antibodies compared with isotype control); P ≤.1 (°) was considered suggestive of statistical significance ( P = .1 for anti–C-VWQRWQKSYV-C–KLH antibodies compared with isotype control).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnci/97/22/10.1093_jnci_dji373/2/m_jncidji373f07_lw.jpeg?Expires=1716983130&Signature=dTusbNehmfJUbAiR-u9zI0IhsjmCscFdRF0zPnN6iMIdupCeSP1ESA5x0Oy~TUNIvRINx6t71M2apU-JaibLwjcBLkJGCwHW8pXyJDyJ0FamEP36q~SkN0KOxxbVGwScrvPqKPWlwnglIwJZYrig6~dL6gvlkMbapMaBZhP2lIA7FGJzhXDt-LQHWHe9mR0yR7IgFuuD26tGn1s9ek0N-fCr6BCDhf7xG1scL8SoIlnkahzfx96HbkH0Aoj02FFftH4M9~mwfQjDut387ssBrF8Pzm5aOuzMxe-yFqx8bm4NuF0FKo1sGJDDMYXlb0HFjWmmpBsUK5K5D~JSUpEjQg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A431 cell proliferation inhibition assessed by [ 3 H]thymidine incorporation. Cells were grown for 4 days with medium alone or with antibodies present as indicated on the x -axis, and then incubated with [ 3 H]thymidine. Uptake of [ 3 H]thymidine, which represents cell growth, was measured in a scintillation counter (counts per minute are given on the y -axis). Columns represent means of triplicate analysis; error bars represent 95% confidence intervals. Differences in A431 cell growth inhibition were analyzed using the Student's t test. P values≤.05 (*) were considered statistically significant ( P = .05 for inhibition caused by cetuximab compared with isotype control, and P = .03 for inhibition caused by anti–C-QYNLSSRALK-C–KLH antibodies compared with isotype control); P ≤.1 (°) was considered suggestive of statistical significance ( P = .1 for anti–C-VWQRWQKSYV-C–KLH antibodies compared with isotype control).
D ISCUSSION
The continuous availability of “natural” antibodies induced by active immunization compared with the need of periodic treatment with an exogenous antibody such as cetuximab could open new avenues for the early treatment or even prevention of EGFR-overexpressing tumors. Interest in different vaccination approaches has been strengthened by the finding that immunologic tolerance to the EGFR family is not absolute and can be overcome ( 24 ) . An important consideration when targeting growth receptors is the need to rule out the induction of growth-stimulatory antibodies ( 18 ) . One way to address this problem might be epitope-specific vaccination with peptide mimics (mimotopes) of an epitope that is recognized by a growth-inhibitory antibody.
The goal of this study was to determine whether a mimotope vaccine that could induce cetuximab-like antibodies could be developed using the phage display technique. We isolated four peptide mimics of the epitope recognized by cetuximab from a cyclic decamer peptide library. None of the four peptide mimics showed sequence similarities to EGFR or any of the other EGFR family members in database alignments, suggesting either that the cetuximab epitope is discontinuous, i.e., it is composed of different regions of the polypeptide chain that come together through protein folding, or that carbohydrates participate in the formation of the epitope. It is a strength of the phage technology that even complex epitopes like this, which exist only in the native form of the protein, can be mimicked by amino acid sequences of mimotype peptides.
All four mimotope peptides represent true epitope mimics because they were specifically recognized by cetuximab and showed mimicry with EGFR in an inhibition assay, meeting the previously established guidelines for specificity and mimicry testing ( 25 ) . To investigate the mimotopes' ability to elicit anti-EGFR antibodies, two peptides were selected for immunization studies. One peptide showed high reactivity with cetuximab and specific mimicry with EGFR, and the other had lesser reactivity but still good mimicry results. Because small peptides on their own are not immunogenic and may even induce tolerance ( 26 ) , we coupled the mimotopes to an immunogenic carrier, KLH. We observed that the mimotope conjugates were immunogenic and induced antibodies not only against the peptide and carrier but also against the natural epitope on EGFR.
We then analyzed the biologic properties of the induced antibodies to determine whether they were comparable to those described for cetuximab. The antitumor activity of cetuximab itself is due to the two Fc-mediated mechanisms, antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity ( 27 ) , as well as to the direct effects of this antibody's binding to EGFR. Direct effects include reduction of EGFR on the cell surface due to internalization of the receptor–antibody complex into endocytotic vesicles ( 28 ) and growth inhibition due to blockade of EGFR-signaling ( 29 ) . Like cetuximab, both mimotopes induced antibodies mediating substantial antibody-dependent cellular and complement-dependent cytotoxic effects against EGFR-expressing cells. These antibodies also led to effective internalization of the receptor from the cell surface. The perinuclear distribution of the endocytic vesicles suggests routing of the receptor into a degradation pathway. In direct growth-inhibition assays, antimimotope antibodies caused dose-dependent inhibition of A431 cancer cell proliferation. Interestingly, the antibodies induced by C-QYNLSSRALK-C (the higher affinity mimotope) were more potent growth inhibitors than those induced by C-VWQRWQKSYV-C. These findings parallel the results of the specificity tests, where cetuximab was found to react more strongly with C-QYNLSSRALK-C than with C-VWQRWQKSYV-C. The observed differences in growth-inhibitory potential could indicate that biologic activity of antibodies may be associated not only with their epitope specificity but also with the differences in the antibodies' affinities to their target antigen. High reactivity of a mimotope peptide with the selecting antibody thus appears to be an important criterion in selecting promising peptide candidates for subsequent immunizations. Taken together, these results indicate that antibodies induced by vaccination with an epitope mimic can elicit the same biologic responses as the original antibody. Because the induced antibodies are polyclonal, it may even be speculated that they could mediate a broader spectrum of immunologic effects, as they can interact with more Fc receptors than a monoclonal antibody of a single given isotype.
Advantages of synthetic peptide vaccines are that they are cost-effective, they are simple to produce, and quality control during the manufacturing process is straightforward. Also, they are chemically stable and contain no oncogenic, toxic, or infectious material. However, all effects of mimotope-induced anti-EGFR antibodies in vaccinated individuals will undoubtedly have to be closely monitored, even though the induction of antibodies that stimulate tumor growth has been excluded by deliberate selection of the cetuximab epitope. Thus far, no toxicities have been observed in the immunized mice, but further toxicity studies are warranted. Because cetuximab can be safely given in doses that fully inhibit EGFR signaling ( 29 ) , we expect that an immune response narrowly focused to the same epitope should show a similar safety profile. However, it remains to be seen whether similar safety and efficacy would be observed in patients.
We are aware that this study is only the first step and that further work is necessary before being able to predict success in humans. To assess whether the described mimotopes would be able to overcome immunological tolerance against EGFR or affect EGFR-induced tumor growth in vivo, immunizations will have to be conducted in animals transgenic for the human receptor. If all these experiments still show promise for a translation of an anti-EGFR mimotope vaccine to humans, toxicity studies should ideally be performed in primates.
We conclude that active immunization inducing in vivo production of cetuximab-like anti-EGFR antibodies is feasible with mimotope vaccination. With active immunization, the obstacles of passive immunotherapy—such as a comparatively short antibody half-life, high antibody peaks, unequal tissue distribution, and the immunogenicity of artificial antibodies—may be overcome. Also, protein vaccination leads to the induction of immunologic memory, possibly providing ongoing protection for vaccinated individuals. Thus, epitope-specific active vaccination against EGFR may represent a novel therapeutic option in the neoadjuvant setting.
Supported by BioLife Science GmbH, Vienna, Austria, and the Center of Excellence in Clinical and Experimental Oncology (CLEXO), Austrian Federal Ministry of Education, Science and Culture (GZ 200.062/2-VI/1/2002).
References
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network.
Di Fiore PP, Pierce JH, Fleming TP, Hazan R, Ullrich A, King CR, et al. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells.
Mendelsohn J. Targeting the epidermal growth factor receptor for cancer therapy.
Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies.
Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis.
Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model.
U.S. Food and Drug Administration. FDA approves Erbitux for colorectal cancer. Available at: http://www.fda.gov/bbs/topics/NEWS/2004/NEW01024.html .
Sato JD, Kawamoto T, Le AD, Mendelsohn J, Polikoff J, Sato GH. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors.
Gill GN, Kawamoto T, Cochet C, Le A, Sato JD, Masui H, et al. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity.
Ganglberger E, Grunberger K, Sponer B, Radauer C, Breiteneder H, Boltz-Nitulescu G, et al. Allergen mimotopes for 3-dimensional epitope search and induction of antibodies inhibiting human IgE.
Hantusch B, Krieger S, Untersmayr E, Scholl I, Knittelfelder R, Flicker S, et al. Mapping of conformational IgE epitopes on Phl p 5a by using mimotopes from a phage display library.
Riemer AB, Hantusch B, Sponer B, Kraml G, Hafner C, Zielinski CC, et al. High-molecular-weight melanoma-associated antigen mimotope immunizations induce antibodies recognizing melanoma cells.
Riemer AB, Klinger M, Wagner S, Bernhaus A, Mazzucchelli L, Pehamberger H, et al. Generation of peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu.
Felici F, Luzzago A, Folgori A, Cortese R. Mimicking of discontinuous epitopes by phage-displayed peptides, II. Selection of clones recognized by a protective monoclonal antibody against the Bordetella pertussis toxin from phage peptide libraries.
Luzzago A, Felici F, Tramontano A, Pessi A, Cortese R. Mimicking of discontinuous epitopes by phage-displayed peptides, I. Epitope mapping of human H ferritin using a phage library of constrained peptides.
Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface.
Cwirla SE, Peters EA, Barrett RW, Dower WJ. Peptides on phage: a vast library of peptides for identifying ligands.
Yip YL, Smith G, Koch J, Dubel S, Ward RL. Identification of epitope regions recognized by tumor inhibitory and stimulatory anti-ErbB-2 monoclonal antibodies: implications for vaccine design.
Klinger M, Kudlacek O, Seidel MG, Freissmuth M, Sexl V. MAP kinase stimulation by cAMP does not require RAP1 but SRC family kinases.
Mazzucchelli L, Burritt JB, Jesaitis AJ, Nusrat A, Liang TW, Gewirtz AT, et al. Cell-specific peptide binding by human neutrophils.
Parmley SF, Smith GP. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes.
Barbas CF 3rd, Kang AS, Lerner RA, Benkovic SJ. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site.
Wouters FS, Bastiaens PI. Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells.
Bei R, Masuelli L, Moriconi E, Visco V, Moretti A, Kraus MH, et al. Immune responses to all ErbB family receptors detectable in serum of cancer patients.
Wauben MH. Immunological mechanisms involved in experimental peptide immunotherapy of T-cell-mediated diseases.
Naramura M, Gillies SD, Mendelsohn J, Reisfeld RA, Mueller BM. Therapeutic potential of chimeric and murine anti-(epidermal growth factor receptor) antibodies in a metastasis model for human melanoma.
Sunada H, Magun BE, Mendelsohn J, MacLeod CL. Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}