




Abstract
Chemoresistance is a major challenge in cancer treatment. miR-506 is a potent inhibitor of the epithelial-to-mesenchymal transition (EMT), which is also associated with chemoresistance. We characterized the role of miR-506 in chemotherapy response in high-grade serous ovarian cancers.
We used Kaplan-Meier and log-rank methods to analyze the relationship between miR-506 and progression-free and overall survival in The Cancer Genome Atlas (TCGA) (n = 468) and Bagnoli (n = 130) datasets, in vitro experiments to study whether miR-506 is associated with homologous recombination, and response to chemotherapy agents. We used an orthotopic ovarian cancer mouse model (n = 10 per group) to test the effect of miR-506 on cisplatin and PARP inhibitor sensitivity. All statistical tests were two-sided.
MiR-506 was associated with better response to therapy and longer progression-free and overall survival in two independent epithelial ovarian cancer patient cohorts (PFS: high vs low miR-506 expression; Bagnoli: hazard ratio [HR] = 3.06, 95% confidence interval [CI] = 1.90 to 4.70, P < .0001; TCGA: HR = 1.49, 95% CI = 1.00 to 2.25, P = 0.04). MiR-506 sensitized cells to DNA damage through directly targeting the double-strand DNA damage repair gene RAD51. Systemic delivery of miR-506 in 8–12 week old female athymic nude mice statistically significantly augmented the cisplatin and olaparib response (mean tumor weight ± SD, control miRNA plus cisplatin vs miR-506 plus cisplatin: 0.36±0.05g vs 0.07±0.02g, P < .001; control miRNA plus olaparib vs miR-506 plus olaparib: 0.32±0.13g vs 0.05±0.02g, P = .045, respectively), thus recapitulating the clinical observation.
MiR-506 is a robust clinical marker for chemotherapy response and survival in serous ovarian cancers and has important therapeutic value in sensitizing cancer cells to chemotherapy.
Epithelial ovarian cancer remains the most lethal gynecological malignancy (1). The current standard of care consists of radical surgery and platinum-based chemotherapy. The five-year survival rate for patients with advanced ovarian cancer is only 30% to 40%, and acquired resistance to platinum is considered a major factor in disease relapse. Platinum-based drugs form intra- and interstrand adducts with DNA, which causes DNA double-strand breaks and triggers DNA damage and repair pathways. Homologous recombination is a critical pathway for DNA double-strand break repair (2) and is responsible for the resistance of high-grade serous ovarian cancer to frontline platinum-based chemotherapy (3). Cells with compromised homologous recombination machinery are highly sensitive to apoptosis triggered by platinum-induced DNA damage through a mechanism termed synthetic lethality (4). Thus, the ability to block homologous recombination-mediated repair is a focus of intense investigation as an approach to improve treatment outcomes in high-grade serous ovarian cancers.
Recent studies demonstrated that BRCA2 mutations, and to a lesser extent BRCA1 mutations/methylation, are associated with improved survival and response to therapy in serous ovarian cancer (5,6). Whereas BRCA1 plays diverse roles in DNA damage pathways, the primary role of BRCA2 is to mediate homologous recombination by directly loading the RAD51 protein onto damage sites or stalled replication forks (7,8). RAD51 is a critical component of the homologous recombination-mediated double-strand DNA break repair machinery and assembles onto single-stranded DNA as a nucleoprotein filament and catalyzes the exchange of homologous DNA sequences (9). RAD51 suppression can sensitize cancer cells to DNA-damaging drugs (10–14), and RAD51 overexpression contributes to chemotherapy resistance in human soft tissue sarcoma cells (15).
MicroRNAs (miRNAs) are a class of small noncoding RNAs (~22 nt) that regulate gene expression. MiRNAs bind to the 3′-untranslated region (3′-UTR) of target genes, which either leads to mRNA degradation or inhibits protein translation (16). Nearly 2578 miRNAs have been identified in the human genome and are thought to regulate 30% of the transcriptome (17). Increasing evidence has demonstrated that miRNA are highly deregulated in cancer, suggesting they may function as therapeutic tools (17–20).
In a recent high-throughput miRNA signature screen, decreased expression of the chrXq27.3-miRNA cluster that included miR-506 was associated with early relapse in patients with advanced-stage epithelial ovarian cancer (21). Our studies established that miR-506 is a potent inhibitor of the epithelial-to-mesenchymal transition (EMT) (22,23), which is also associated with chemoresistance. In addition, we found that miR-506 could suppress proliferation and induce senescence by directly targeting the CDK4/6-FOXM1 axis in ovarian cancer (24). However, it is unknown whether miR-506 is involved in the chemotherapy response.
Methods
Samples and Clinical Data
Level 3 miRNA isoform expression data based on miR-seq across 468 stage II to IV ovarian cancer cases were downloaded from the open-access The Cancer Genome Atlas (TCGA) data portal on January 14, 2013. Clinical information was obtained from the controlled-access tiers of the TCGA data portal, with National Institutes of Health approval. Detailed patient demographics are described in the Supplementary Methods (available online). The Bagnoli cohort included 130 patients belonging to three data sets that have been previously used to find an miRNA profile possibly associated with disease relapse (21).
Bioinformatic Analysis
The expression levels of 3p/5p mature miRNA were further summarized by the MIRACLE pipeline (22) based on the level 3 data. Briefly, read numbers mapped to the same miRNA isoform (based on MIMAT id) were summed up regardless of their sequence variations. The MIMAT ids were further converted to miRNA mature product names according to miRBase V19 annotation. The numbers of reads that were mapped to the precursors, stemloops, and unannotated/retired miRNAs were summed up as “Precursor/Stemloop/Unannotated” in each tumor sample.
miRNA microarray profiling of 130 stage III or IV epithelial ovarian cancers was performed using Illumina human_v2 MicroRNA chips as described previously (21). Raw data were processed and quantile-normalized through BeadStudio V3.0 software. Nonbiological experimental variations were adjusted using ComBat (25). Batch effects in microarray expression data were adjusted using empirical Bayes methods (25).
Cell Lines, Cell Culture, Reagents, and miRNA Transfection
Human ovarian cancer cell lines (HeyA8, OVCA433, SKOV3) and HeLa were obtained from the American Type Culture Collection (ATCC, Manassas, VA).The authentication of all cell lines was done by the Characterized Cell Line Core Facility at the University of Texas MD Anderson Cancer Center (MDACC; Houston, TX) by the STR Short Tandem Repeat (STR) Method. Futher detail is given in the Supplementary Methods (available online).
Real-time Polymerase Chain Reaction Analysis and Microarray Gene Expression Analysis
Total RNA was isolated with an mirVana miRNA isolation kit (Ambion, Grand Island, NY). Reverse transcription was performed using SuperScript II Reverse Transcriptase (Invitrogen) according to the manufacturer’s protocol. TaqMan real-time polymerase chain reaction (RT-PCR) assays for miR-506 were obtained from Applied Biosciences, Inc. (Grand Island, NY). Cyclophilin and β-actin were used as normalization controls. Data were analyzed by using the -2ΔΔct method. Futher detail is given in the Supplementary Methods (available online).
Colony-Formation Assay
Twenty-four hours after transfection with 20nm miR-506, miR-Ctrl, or anti-miR-LNA, cells were harvested. Transfected cells were seeded in a six-well plate (500 cells/well) and treated with cisplatin for 48 hours or olaparib for seven days and then allowed to recover for 10 to 14 days, during which time the surviving cells spawned a colony of proliferating cells. Colony formation was quantified by staining the cells with 0.1% crystal violet and counting surviving colonies containing more than 50 cells.
MTT Assay
Twenty-four hours after transfection with 20nm miR-506, miR-Ctrl, or anti-miR-LNA, cells were seeded onto 96-well plates (1×103 cells/well) and treated with a titration of cisplatin or olaparib. The medium and drug were replenished at day 3 for olaparib treatment. After incubation for five (cisplatin) or seven (olaparib) days, cell viability was estimated using the MTT reagent (Sigma Chemical, St. Louis, MO), and surviving fractions were calculated. Cell survival was calculated by normalizing the absorbance to that of untreated controls.
Western Blot Analysis
Primary β-actin antibody (goat) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal Rad51 antibody was purchased from Lab Vision Corporation (Fremont, CA). In brief, 30 μg of whole-cell lysate from each sample was loaded on a 10% polyacrylamide gel for electrophoresis; the membrane was blocked in 5% nonfat milk in 1× Tris-buffered saline solution (pH 7.4) containing 0.05% Tween-20 and probed with primary antibodies at a concentration of 1:1000 (for β-actin) or 1:200 (for RAD51). The secondary antibodies were used at a concentration of 1:10 000. The proteins were visualized using the SuperSignal West Pico or SuperSignal Femtochemiluminescent substrate from Pierce (Rockford, IL).
Luciferase Reporter and Single-Cell Gel Electrophoresis (Comet) Assays
The 3′-UTR of RAD51 containing the predicted binding site of miR-506 was amplified separately from normal fetal genomic DNA by PCR. Primers used and other details are given in the Supplementary Methods (available online).
Comet assay was performed per manufacturer’s instructions (Trevigen, Gaithersburg, MD). Proliferating HeyA8 cells were transfected with miR-Ctrl or miR-506 mimic. Twenty-four hours later, transfected cells were treated with 100 μM cisplatin for one hour, were allowed to repair for 18 hours, and then were analyzed by single-cell gel electrophoresis. Further details are in the Supplementary Methods (available online).
Double Strand Break–Induced Homologous Recombination Repair Assay
A stable derivative of HeLa cells was established by transfection with pDR-GFP using Lipofectamine 2000 and selection in 1.5 μg/mL puromycin. Puromycin-resistant cells were cloned by limiting dilution in 96-well plates. Single colonies were plated in 12-well plates and transfected with cBAS to express the I-SceI endonuclease. Further details are given the Supplementary Methods (available online).
Immunofluorescence Microscopy Imaging and Immunohistochemical Staining
Cells were grown on coverslips and treated with 10 μM cisplatin for 24 hours. Phase images were captured by a ZEISS HAL 100 microscope at a magnification of 200×. The fluorescence images were captured using a ZEISSAxioplan 2 microscope. Immunohistochemical staining was performed on tumor tissues from the mouse orthotopic model and tissuemicroarrays that included samples from 92 patients with serous ovarian cancer, both of which were assembled for our previous study (22). Further details are given the Supplementary Methods (available online).
Animal Orthotopic In Vivo Model
We injected 2.5×105 HeyA8-ip1 cells resuspended in Hanks balanced salt solution (GIBCO, Carlsbad, CA) into the peritoneal cavity of eight to 12-week-old female athymic nude mice (Frederick Cancer Research and Development Center, Frederick, MD). Mice were cared for according to guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the US Public Health Service policy on Human Care and Use of Laboratory Animals. All mouse studies were approved and supervised by the MD Anderson Cancer Center Institutional Animal Care and Use Committee. One week after tumor cell injection, mice were randomly separated into treatment groups (n = 10 mice per group) and treated via intraperitoneal administration with miRNA incorporated in DOPC nanoliposomes—miR-Ctrl/DOPC or miR-506/DOPC (200 µg miRNA/kg/mouse)—with or without cisplatin (160 μg/mouse) or olaparib (50mg/kg, for detailed methods of liposomal preparation, see reference [22]). All groups received twice-weekly miRNA treatments for three to six weeks. Cisplatin was administered once weekly for four to six weeks. Olaparib was solubilized as previously described (26) and was given every day for about three weeks. When treatment was completed, mice were killed and their tumors harvested. Tumor weights, numbers, and locations were recorded.
Statistical Analysis
Survival was analyzed by the Kaplan-Meier and Cox proportional-hazards model using R 2.10.0 software. The Cox proportional-hazards model was used to estimate the hazard ratios (HRs) and the 95% confidence intervals (CIs) for the association of miR-506 expression with overall survival (OS) and progression-free survival (PFS). Age and the tumor residual, ethnic background, and TNM stage were considered potential confounding covariates and were thus included in the multivariable regression. The adherence to the proportional hazards assumption was examined by the log-minus-log survival plots and the Schoenfeld test. Differences between groups were defined as statistically significant at P values of less than .05. Data are means ± standard deviation of at least three independent experiments. The two-sided Student’s t test was used for comparisons of two independent groups. All statistical analyses were done using SPSS 17.0 (SPSS Inc, Chicago, IL) and R 2.10.0.
Results
MiR-506 Expression, Response to Platinum Treatment, and Survival in Serous Ovarian Cancers
The association of miR-506 expression with response to first-line platinum-based therapy was examined in a miRNA expression dataset consisting of 130 epithelial ovarian cancer (EOC) cases (21). Elevated miR-506 expression was statistically significantly associated with platinum sensitivity (Figure 1A). The ability of miR-506 to discriminate therapy response was highlighted by Kaplan-Meier survival analysis of progression-free and overall survival (PFS: high vs low miR506 expression: HR = 3.06, 95% confidence interval (CI) = 1.90 to 4.70, P < .0001; OS: HR = 2.72, 95% CI = 1.55 to 4.76, P = .0005) (Figure 1, B and C). Two of the three datasets used (training and test sets) among the 130 cases had been originally selected for time to relapse (21). Therefore, to avoid selection bias, the survival analysis was performed after extracting the Bagnoli validation set including 45 unselected consecutive serous ovarian cancer patients. The impact of miR-506 expression on patient survival was confirmed; indeed, in the 45 consecutive EOC patients, high miR-506 expression was statistically significantly associated with longer PFS and OS (PFS: high vs low miR506 expression: HR = 4.00, 95% CI = 1.80 to 8.47, P = .0003; OS: HR = 7.67, 95% CI = 2.6 to 22.8, P = .0002) (SupplementaryFigure 1, A and B, available online). Consistent with the data obtained in the Bagnoli validation set, the relationship of miR-506 level and patient prognosis was also demonstrated in the TCGA dataset (27), which included 468 high-grade serous EOC samples. The level of miR-506 was again statistically significantly related to PFS and OS (PFS: high vs low miR506 expression: HR = 1.49, 95% confidence CI = 1.00 to 2.25, P = .04; OS: HR = 1.65, 95% CI = 1.05 to 2.60, P = .02, respectively) (Figure 1, D and E). The association remained statistically significant after adjusting for stage, residual tumor, ethnic background, and age at diagnosis, suggesting miR-506 is an independent prognostic factor in ovarian cancer (PFS: low vs high miR506 expression: HR = .62, 95% CI = 0.40 to 0.94, P = .02) (SupplementaryFigure 2, available online).
![MiR-506 and patient response to therapy and prognosis. A) Expression of miR-506 in 130 epithelial ovarian cancer (EOC) patients from Bagnoli’s datasets (21) categorized for response to platinum treatment as: refractory (relapse during treatment), resistant (progression-free survival [PFS] < 6 months), partially sensitive (PFS from 6 to 12 months), sensitive (PFS > 12 months). P value from one-way analysis of variance with Tukey’s multiple comparison as post-test; only comparisons indicated by asterisks are statistically significant. *P = .011, ***P < .0001. B) Kaplan-Meier survival curves for PFS for 130 EOC patients with miR-506 low expression (green line) and high expression (red line). C) Overall survival. D) Kaplan-Meier PFS. E) OS curves of 468 The Cancer Genome Atlas ovarian cancer patients for miR-506 low expression (green line) and high expression (red line). Ovarian cancer case patients were stratified into miR-506 low and high expression according to mean of miR-506 expression. OS = overall survival; PFS = progression-free survival.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnci/107/7/10.1093_jnci_djv108/4/m_jnci.j_djv108_f0001.jpeg?Expires=1716455877&Signature=hoz9m2wMzaRlLyv7diN1Wd1c5dMkapwH9Yo~7FfyG~IB~De54qKhq2bKLd2rW-EIRwShD5vh6Zm54x40IW-OCAlzpcsYUo~fcqHKOOeHSgA53s0mj83teEyOl4IwtjrB7ojVu~ntQAtdpEGD5gP8CWjvqtmcsHdUeslOrsfsZPNp269tAB-wFlbfnJWIvg3-dtajwc99fhKqx5PwhusmgrTH1AqcPIixzvXvfX1MOwwrWLMIW8bJmRuA1QVYpWDyp8k1ywDy963I3hZz7TnykgpLLGRVVaRarzo1PFl0jcKFTnTYkmjuyQcWyWz8gFya3NrRS9xnkyxGLY-OrHpwqw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MiR-506 and patient response to therapy and prognosis. A) Expression of miR-506 in 130 epithelial ovarian cancer (EOC) patients from Bagnoli’s datasets (21) categorized for response to platinum treatment as: refractory (relapse during treatment), resistant (progression-free survival [PFS] < 6 months), partially sensitive (PFS from 6 to 12 months), sensitive (PFS > 12 months). P value from one-way analysis of variance with Tukey’s multiple comparison as post-test; only comparisons indicated by asterisks are statistically significant. *P = .011, ***P < .0001. B) Kaplan-Meier survival curves for PFS for 130 EOC patients with miR-506 low expression (green line) and high expression (red line). C) Overall survival. D) Kaplan-Meier PFS. E) OS curves of 468 The Cancer Genome Atlas ovarian cancer patients for miR-506 low expression (green line) and high expression (red line). Ovarian cancer case patients were stratified into miR-506 low and high expression according to mean of miR-506 expression. OS = overall survival; PFS = progression-free survival.
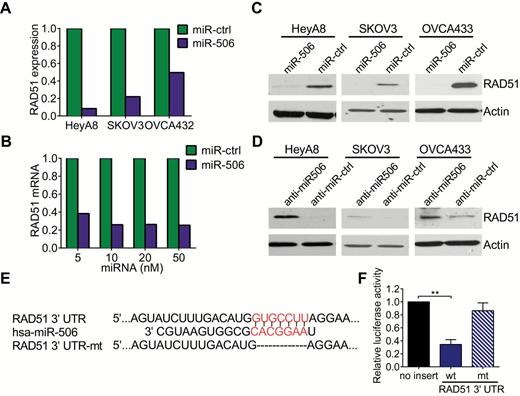
Targeting of RAD51 by miR-506. A) Microarray analysis of RAD51 mRNA level after overexpression of miR-506 in three ovarian cancer cell lines. The expression levels of RAD51 in miR-506 treatments are normalized to RAD51 levels in miR-Ctrl control treatment cells. B) Quantitative polymerase chain reaction results showed that RAD51 mRNA level decreased 48 hours after transfection of miR-506 mimic at different concentrations in HeyA8 cells. (The level of RAD51 mRNA after transfection of miR-ctrl was normalized to 1.) C and D) Three ovarian cancer cell lines were transfected with miR-506 mimics or anti-miR-506 LNA for 48 hours; Western blot showed the changes of RAD51 level. E) TargetScan predicted that the RAD51 3′-UTR has one miR-506 binding site, which is highly conserved among different species. F) Luciferase reporter assay showed that miR-506 directly targets the RAD51 3′-UTR. HeLa cells were cotransfected with RAD51 3′-UTR-luciferase reporter, wild-type or mutant, and miR-Ctrl or miR-506 mimic for 48 hours before analysis. Firefly luciferase activity of the reporter was normalized to the internal Renilla luciferase activity. The means ± SD of three independent experiments are shown (*** P < .001, two-sided Student’s t test). 3’UTR = 3′-untranslated region. mt = mutant; wt = wild-type.
Effect of MiR-506 Expression on RAD51 Levels in Human High-Grade Serous Ovarian Carcinoma
To identify the genes potentially regulated by miR-506, we performed microarray analysis on HeyA8, SKOV3, and OVCA432 ovarian cancer cells that had been transfected with either miR-506 mimic (miR-506) or a scrambled negative miRNA control (miR-Ctrl). The microarray data revealed that RAD51 mRNA was decreased by two- to 11-fold after miR-506 overexpression in all three cell lines (Figure 2A). This result was confirmed with quantitative PCR and Western blotting, which showed that miR-506 overexpression substantially decreased RAD51 mRNA and protein levels (Figure 2, B and C). Conversely, HeyA8, SKOV3, and OVCA433 cells transfected with miR-506 antisense oligonucleotides (locked nucleic acids [LNA]) expressed higher levels of RAD51 protein than those transfected with anti-miR-Ctrl (Figure 2D). These results suggest that miR-506 negatively regulates RAD51 gene expression. Further, the association between miR-506 and RAD51 expression was analyzed in three different cohorts of clinical samples. In the TCGA cohort, levels of miR-506 from small RNA seq expression had a moderate but significant inverse correlation with RAD51 transcript from Agilent array (445 cases with both data, coefficient -0.1, P = .04, Pearson’s product-moment correlation) (SupplementaryFigure 3A, available online). In the relatively smaller Italian cohort (21 cases), there was a strong inverse correlation (a Pearson’s: -0.53 and P = .00947) (SupplementaryFigure 3B, available online). With the Tianjin cohort, we analyzed the expression of miR-506 and RAD51 using in situ hybridization and immunohistochemistry respectively on a microarray of 92 high-grade serous ovarian cancer samples (22). As shown in SupplementaryFigure 3, C and D (available online), miR-506 expression was inversely associated with RAD51 protein expression in the 92 analyzed cases (P = .037).
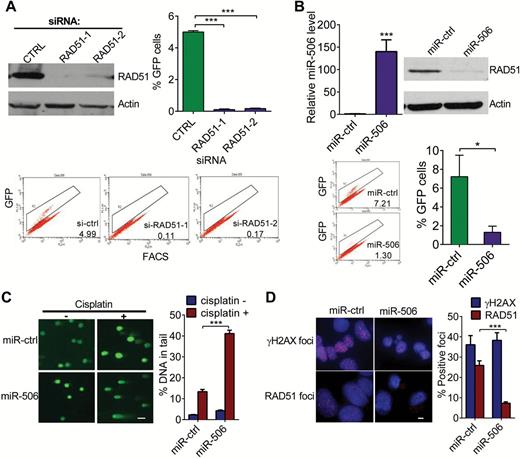
Effect of miR-506 overexpression on homologous recombination-mediated repair of double-strand breaks, DNA double-strand break induction by cisplatin, and DNA repair. A and B) HeLa cells stably carrying the recombination substrate (DR-GFP) were transiently cotransfected with I-SceI expression plasmid and si-RAD51/si-Ctrl or miR-506/miR-Ctrl. GFP-positive cells were quantified 48 hours later by FACS. Means ± SD of three independent experiments are shown. A) Knockdown of RAD51 by si-RAD51-1 or si-RAD51-2 compared with si-Ctrl transfection; *** P < .001. RAD51 expression by Western blot and representative FACS profiles are shown. B) Overexpression of miR-506 and homologous recombination repair compared with miR-Ctrl, * P = 0.013; miR-506 expression by real-time polymerase chain reaction and representative FACS profiles are shown; *** P < .001. C) Overexpression of miR-506 in HeyA8 cells and number of unrepaired double-strand breaks detected by comet assay. Representative images are shown in the left panel and the mean ± SD for each condition on the right. Scale bar = 50 μM. Residual DNA damage after cisplatin treatment in miR-506–transfected cells compared with controls; *** P < 0.001. D) Double-strand break formation and repair after exposure to cisplatin are shown. HeyA8 cells were transfected with miR-Ctrl or miR-506 mimic. Twenty-four hours later, transfected cells were treated with 10 μM cisplatin for 24 hours, and γH2AX foci and RAD51 foci were examined. γH2AX foci indicate DNA double-strand breaks. The number of RAD51 foci in cells transfected with miR-506 mimic compared with controls. Scale bar = 5 μM, *** P < .001. Data represent the mean ± SD from three independent experiments.
RAD51 as a Direct Target of miR-506
The miRNA target prediction algorithm TargetScan 6.0 predicted that the 3′-UTR of RAD51 mRNA contains a putative miR-506 binding site (Figure 2E). This potential binding site and its flanking sequences are highly conserved across mammals (SupplementaryFigure 4, available online). To determine whether miR-506 regulates RAD51 through binding to its 3′-UTR, we cloned the RAD51 3′-UTR into the pmirGLO luciferase reporter vector and transfected either this vector (pmirGLO-RAD51-3′-UTR) or the parent luciferase expression vector, along with miR-506 mimic or miR-Ctrl, into HeLa cells. As shown in Figure 2F, cotransfection of pmirGLO-RAD51-3′-UTR and miR-506 mimic resulted in a 61.2% reduction in luciferase activity compared with miR-Ctrl (P < .001), suggesting that miR-506 directly targets RAD51. To further confirm that miR-506 specifically regulates RAD51, we generated a mutant construct, mirGLO-RAD51-3′-UTR-Mu, in which the sequence complementary to the seed sequence of miR-506 on the RAD513′-UTR was deleted. We then transfected cells with the mutant construct and either miR-506 mimic or miR-Ctrl. Deletion of the miR-506 binding site from the RAD513′-UTR abolished the effect of miR-506 on luciferase activity (Figure 2F). Taken together, these results confirmed that miR-506 specifically targets the 3′-UTR of RAD51, thereby inhibiting RAD51 gene expression.
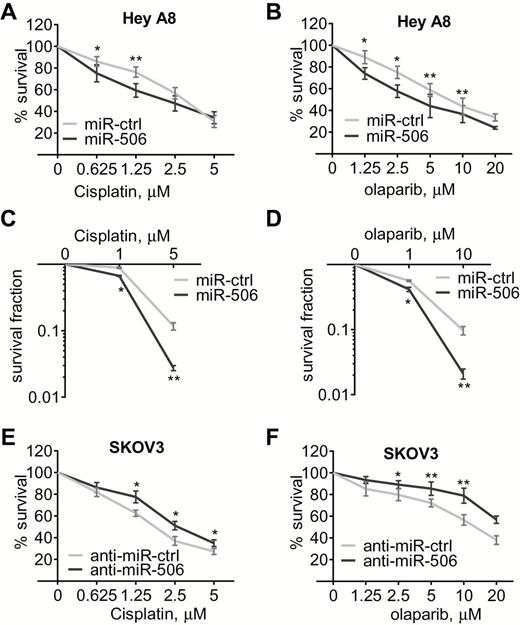
MiR-506–mediated regulation of RAD51 affects sensitivity to cisplatin or a PARP inhibitor in ovarian cancer cells. HeyA8 cells were transfected with either miR-Ctrl or miR-506 mimic. Conversely, SKOV3 cells were transfected with either anti-miR-Ctrl or anti-miR-506. Cell viability was assayed by MTT assay (A, B, E, F) and clonogenic cell-survival assay (C and D). Curves were generated from three independent experiments. miR-506 mimic and sensitivity to cisplatin and PARP inhibitor olaparib (A-D), anti-miR-506 LNA and sensitivity to cisplatin and olaparib (E and F). * P < .05; ** P < .01, two-sided Student’s t test. Data represent the mean ± SD from three independent experiments.
MiR-506 Expression Impacts Homologous Recombination-Mediated Repair
Because RAD51 plays an important role in homologous recombination, we examined the effect of miR-506 on homologous recombination repair activity by performing the homology-directed repair assay. Knockdown of RAD51 via siRNA substantially reduced the homologous recombination efficiency (si-CTRL vs si-RAD51-1 and si-RAD51-2: 4.99±0.09 vs 0.11±0.04 and 0.17±0.03, P < .001, respectively) (Figure 3A). Consistent with the role of RAD51 in homologous recombination, cells overexpressing miR-506 had statistically significantly reduced homologous recombination efficiency (miR-ctrl vs miR-506: 7.21±2.29 vs 1.30±066, P = .013) (Figure 3B). Together these results suggest that miR-506–mediated downregulation of RAD51 impedes the DNA damage response pathway.
Effect of MiR-506–Mediated Suppression of RAD51 on DNA repair
RAD51 is an integral component of the cellular DNA damage response (28,29). To determine whether miR-506–mediated RAD51 downregulation affects DNA repair, we measured the persistence of double-strand breaks after cisplatin treatment as an indicator of unrepaired damaged DNA. Single-cell gel electrophoresis (alkaline comet assay) was carried out to measure DNA damage. HeyA8 cells with ectopic overexpression of miR-506 had lower levels of RAD51 protein and statistically significantly higher residual DNA damage than control cells (% DNA in tail, miR-ctrl plus cisplatin vs miR-506 plus cisplatin: 13.338±1.092 vs 41.134±1.623, P < .001) (Figure 3C).
MiR-506 Leads to Defects in Repair of Combination Lesions Formed by Cisplatin
A key component in DNA repair is the formation of nuclear γH2AX foci at sites of DNA damage, creating a focus for accumulation of proteins involved in DNA repair and chromatin remodeling. Treatment with cisplatin induced the formation of γH2AX to the same extent in miR-506–transfected cells and miR-Ctrl–transfected cells, suggesting that the damage induced is independent of RAD51 function (Figure 3D).
RAD51, which is involved in homologous recombination repair, is relocalized within the nucleus in response to DNA damage. Distinct foci are formed that include proteins assembled at sites of homologous recombination repair; therefore, quantification of RAD51 foci can serve as a marker of homologous recombination. To test further whether miR-506 impairs homologous recombination, we determined the ability of HeyA8 cells to form RAD51 foci in response to cisplatin. Overexpression of miR-506 led to less RAD51 foci formation than in controls 24 hours after cisplatin treatment, confirming the defect in homologous recombination (miR-ctrl vs miR-506: 25.91±2.28 vs 7.27±0.69, P < .001) (Figure 3D).
Effect of MiR-506 on Sensitivity to Cisplatin or a PARP Inhibitor in Ovarian Cancer In Vitro and In Vivo
Homologous recombination-deficient cells are sensitive to DNA-damaging drugs and PARP inhibitors (30). Because miR-506 overexpression decreased RAD51 levels and homologous recombination efficiency, we tested whether miR-506 overexpression sensitized ovarian cancer cells to cisplatin or to a commercially available PARP inhibitor (olaparib). According to the real-time PCR results of relative miR-506 expression levels (SupplementaryFigure 5, available online) in several ovarian cancer cell lines, we overexpressed miR-506 in HeyA8 cells, which have relatively low levels of miR-506. As expected, miR-506–transfected HeyA8 cells were more sensitive to cisplatin or olaparib than controls (survival percent for 1.25 μM cisplatin treatment, miR-ctrl vs miR-506: 76.169±4.732 vs 59.329±6.217, P = .009; survival percent for 10 μM olaparib treatment, miR-ctrl vs miR-506: 43.627±7.749 vs 36.623±7.850, P = .006) (Figure 4, A and B), with relevant morphological changes (SupplementaryFigure 6, available online). This miR-506–induced sensitization to both cisplatin and olaparib was confirmed in a clonogenic survival assay (Figure 4, C and D). Conversely, anti-miR-506 LNA transfection enhanced RAD51 expression and induced resistance to cisplatin and olaparib in SKOV3 cells (Figure 4, E and F), which have relatively high levels of miR-506 (SupplementaryFigure 5, available online).
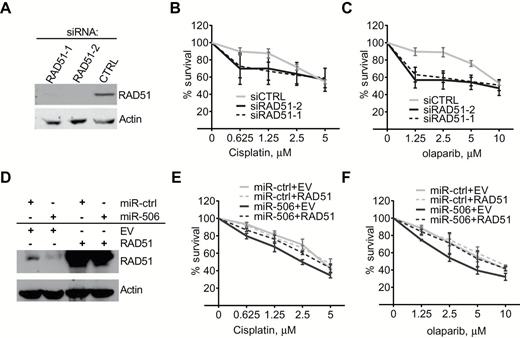
RAD51 and miR-506–induced increases in cisplatin/PARP inhibitor sensitivity in ovarian cancer cells. A-C) HeyA8 cells were transfected with 40nm si-Ctrl or si-RAD51. After 24 hours, cells were reseeded for cisplatin (B) or olaparib (C) sensitivity assay or harvested for Western blot analysis (A). Curves were generated from three independent experiments. D-F) HeyA8 cells were cotransfected with RAD51 without the 3′-UTR or empty vector (EV) together with 20nm miR-Ctrl or miR-506. After 24 hours, cells were reseeded for cisplatin (E) or olaparib (F) sensitivity assay or harvested for Western blot analysis (D). Curves were generated from three independent experiments (data represent the mean ± SD).
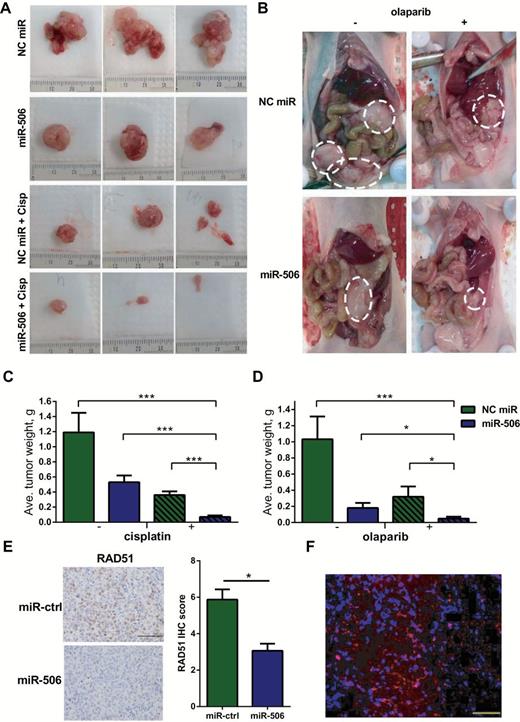
MiR-506 and cisplatin/olaparib sensitivity in an orthotopic mouse model of ovarian cancer. A and B) Images of the representative gross tumors from different treatment groups in the cisplatin/olaparib sensitivity model are shown. The scale was millimeters. C) Mean tumor weights in mice treated with miR-506 plus cisplatin compared with those treated with control miRNA, miR-506, or control miRNA plus cisplatin (*** P < .001, respectively). D) Mean tumor weights in mice treated with miR-506 plus olaparib compared with those treated with control miRNA, miR-506 or control miRNA plus olaparib (*** P < .001, *P = .03 for miR-506 vs miR-506 plus olaparib, *P = .045 for control miRNA plus olaparib vs miR-506 plus olaparib). E) Samples of HeyA8-ip1 tumors from control and miR-506–treated mice were subjected to haematoxylin and eosin and immunohistochemical staining for RAD51; scale bar =100 μm. Expression of RAD51 proteins was calculated as immunohistochemical staining scores; error bars represent standard deviations; *P = .031.F) Tumors were harvested in HeyA8-ip tumor-bearing mice treated with Cy3-miRNA encapsulated in DOPC liposomes and delivery of fluorescently tagged miRNA was assessed; scale bar = 50nm, red: Cy3-miRNA; blue: nucleus. Cisp = cisplatin; NC = negative control.
We further examined whether RAD51 reduction is critical for miR-506–induced cellular sensitivity to cisplatin and olaparib in ovarian cancer cells. RAD51 knockdown via siRNA statistically significantly sensitized cells to cisplatin and olaparib, similar to miR-506 transfection (survival percent for 1.25 μM cisplatin treatment, si-CTRL vs si-RAD51-1 and si-RAD51-2: 87.520±5.306 vs 66.849±10.425 and 69.960±18.570, P < .01, respectively; survival percent for 2.5 μM olaparib treatment, si-CTRL vs si-RAD51-1 and si-RAD51-2: 88.947±5.306 vs 55.051±4.906 and 54.196±8.356, P < .01, P = .011, respectively) (Figure 5, A-C). Moreover, the effect of miR-506 on cisplatin and olaparib sensitivity was fully rescued by overexpressing RAD51 without its 3’-UTR (Figure 5, D-F), suggesting that miR-506–mediated sensitivity to cisplatin and olaparib is primarily a result of RAD51 expression suppression.
To further assess the ability of miR-506 to induce cisplatin and olaparib sensitivity, we tested the therapeutic efficacy of a miR-506 and cisplatin/olaparib combination in an established ovarian cancer model. We used an aggressive HeyA8 clone generated from ascites developed in a nu/nu mouse by administering HeyA8 cells intraperitoneally (HeyA8ip1). As previously described, miRNAs were incorporated into neutrally charged DOPC nanoliposomes (22). For the cisplatin treatment model, following intraperitoneal injection of HeyA8ip1 cells, mice were randomly distributed and assigned to the following treatment groups: 1) control miRNA/DOPC, 2) miR-506/DOPC, 3) control miRNA/DOPC + cisplatin, or 4) miR-506/DOPC + cisplatin. As compared with the control miRNA group (mean tumor weight = 1.19±0.26), tumors in the miR-506 group (mean tumor weight = 0.53±0.09) had statistically significantly less tumor burden based on aggregate mass (55.5 percent reduction, P = .013) (Figure 6, A and C). While the addition of cisplatin to control miRNA led to decreased tumor burden (mean tumor weight = 0.36±0.05, 69.7 percent reduction in tumor weight), the combination of miR-506 + cisplatin led to marked reductions in disease progression (mean tumor weight = 0.07±0.02, 94.1% reduction in tumor weight compared with control miRNA group, P < .001) (Figure 6C). Next, we assessed the ability of miR-506 to sensitize cancer cells to PARP inhibition in vivo. The treatment groups were as follows: 1) control miRNA/DOPC, 2) miR-506/DOPC, 3) control miRNA/DOPC + olaparib, or 4) miR-506/DOPC + olaparib. Compared with the control group (mean tumor weight = 1.03±0.28), we observed statistically significant reduction in tumor burden in the group treated with PARP inhibitor or miR-506 alone (mean tumor weight = 0.32±0.13 and 0.18±0.06, P = .035, P = .010 respectively). However, we observed marked reduction of tumor weight in the group treated with combination of miR-506 and olaparib (mean tumor weight = 0.05±0.02, P < .001 compared with control, P = .045 compared with control miRNA plus olaparib) (Figure 6D). These results showed that miR-506 delivered via nanoparticles may serve as a potential cisplatin/olaparib sensitizer for ovarian cancer. Further, RAD51 expression was examined by immunohistochemistry (IHC) in samples of HeyA8-ip1 tumors from control and miR-506–treated mice. Consistent with in vitro results, miR-506–treated tumors (IHC score = 3.061±0.389) showed lower RAD51 expression compared with control, (IHC score = 5.874±0.560, P = .031) (Figure 6E). To confirm delivery of DOPC-miRNA nanoparticles in vivo, we treated HeyA8 tumor–bearing mice with Cy3-miRNA incorporated in DOPC nanoliposomes. We harvested the tumor and assessed for delivery of fluorescently tagged miRNA. There was substantial delivery of Cy3-labeled miRNA, as evident from the immunofluorescence images taken from tumor sections (Figure 6F). Together, these data suggest a greater sensitivity to cisplatin and PARP inhibitor upon RAD51 downregulation mediated by miR-506 in vivo.
Discussion
In this study, we interrogated two clinically annotated genomics datasets (TCGA and Bagnoli) and showed that miR-506 expression was associated with better response to therapy and longer survival in ovarian cancer patients. Results of microarray analysis demonstrated that, except for cell cycle and EMT, the DNA repair pathway was among the top three defective pathways after overexpression of miR-506. In computational analyses, we demonstrated that miR-506 directly targets RAD51 and sensitizes ovarian cancer cell lines to DNA damage and cell death induced by cisplatin or a PARP inhibitor. Furthermore, nanoparticle delivery of miR-506 enhanced the effect of cisplatin and olaparib in orthotopic ovarian cancer models. The discovery of a miR-506-RAD51-DNA repair axis supports the approach that combining miR-506 expression with DNA-damaging agents may substantially benefit ovarian cancer management.
DNA-damaging agents are important chemotherapeutic interventions for cancer therapy. However, chemoresistance is a major cause of death in women with ovarian carcinoma. Homologous recombination is involved in tumor chemoresistance, and RAD51 plays a critical role in this pathway. Indeed, knockdown of RAD51 sensitizes cancer cells to DNA-damaging drugs, including cisplatin (10–14). In this study, we showed that miR-506 regulates DNA repair and increases chemosensitivity in vitro and in vivo by suppressing the expression of RAD51.
A recent miRNA microarray analysis demonstrated that a cluster of eight miRNAs, located on chrXq27.3, was downregulated in patients with early-relapsing ovarian cancer compared with late-relapsing groups, and miR-506 is among these miRNAs (21). Our previous studies indicated that miR-506 downregulation was at least partially from methylation of the promoter region (22). A recent study reported that transcription factors MAFB and STAT4 negatively regulated miR-506, suggesting another regulatory network that could control miR-506 expression (31).
Currently, our understanding of the biological functions of miR-506 is still limited and sometimes inconsistent (32–34). A recent study showed that restoration of miR-506 in transformed human bronchial epithelial cells led to a decrease in cell proliferation (34), while Streicher et al. reported that the miR-506–514 cluster played an oncogenic role in initiating melanocyte transformation and promoting melanoma growth (32). To test the potential role of miR-506 in ovarian cancer, we performed statistical analysis on two clinically annotated genomics datasets (TCGA and Bagnoli) and showed that high level of miR-506 expression was associated with better response to therapy and longer survival in ovarian cancer patients. We further analyzed downregulated genes via microarray after miR-506 overexpression and observed a decrease in RAD51 levels in a panel of ovarian cancer cell lines. Subsequently, the role of miR-506 in mediating ovarian cancer chemoresistance was confirmed by validating RAD51 as a direct target of miR-506 and by demonstrating that miR-506 could induce defects in DNA repair and increase drug sensitivity to cisplatin and PARP inhibitors.
As an important protein in mediating homologous recombination, RAD51 is a target for decreasing DNA repair and increasing sensitivity to DNA-damaging chemotherapies (13). In this study, the observation that the miR-506–induced defects in DNA repair and sensitivity was largely rescued by overexpressing RAD51 suggests that RAD51 is the key target for miR-506–enhanced DNA repair and drug sensitivity. Reduction of RAD51 levels and enhanced sensitivity to cisplatin or the PARP inhibitor olaparib following miR-506 overexpression may have important clinical relevance.
Recent findings (4,35) demonstrate that PARP inhibitors have particularly robust cytotoxic effects on BRCA1- or BRCA2-deficient cells. The prevailing explanation for these findings is synthetic lethality (36). Promising results from clinical trials of PARP inhibitors in BRCA-associated carcinomas (including ovarian carcinoma) have been reported (37–42). Moreover, cancer cells deficient in homologous recombination showed sensitivity to PARP inhibitors (30), which may become a useful therapeutic strategy for tumors displaying properties of “BRCAness,” or with defects in the homologous recombination pathway. Consistent with this, suppression of the RAD51 protein can sensitize tumor cells to treatment with PARP inhibitors (30). Recently, other miRNA modulators have also been identified to regulate homologous recombination and sensitize tumor cells to PARP inhibitors or radiation by suppressing expression of RAD51 (43,44).
Whereas the inverse association of miR-506 and Rad51 was seen in three different cohorts in our analyses, the number of cases in some cohorts is still relatively small. Thus, further validation in additional cohorts is important. Further, because more than one miRNAs are known to modulate Rad51, future analyses of combined effect will also be clinically relevant.
Consistent with the results of in vitro experiments, delivery of miR-506 incorporated in DOPC nanoliposomes effectively enhanced the effect of cisplatin and olaparib in an orthotopic ovarian cancer model and resulted in statistically significantly lower tumor weight than miR-Ctrl. Packaging into lipid-based nanoparticles allows successful delivery of miRNAs to the tumor and its microenvironment therapeutically regulating their targets. The ability to target multiple oncogenes relevant to cancer pathways poses a unique advantage of using miRNA mimics as a therapeutic tool. As such, miR-506 inhibits SNAI2-mediated EMT processes during which cancer cells acquire stem cell-like phenotype and become more resistant to chemotherapy and radiotherapy. In addition, miR-506 targets RAD51-mediated DNA repair that can reverse chemotherapy-induced double-stranded breaks, making the tumor cells more sensitive to DNA damaging therapies (Figure 7). Our data support the idea that nanoparticle delivery of miR-506 combined with DNA-damaging agents may lead to substantial benefit for chemoresistant ovarian cancer management by mimicking a BRCAness phenotype.
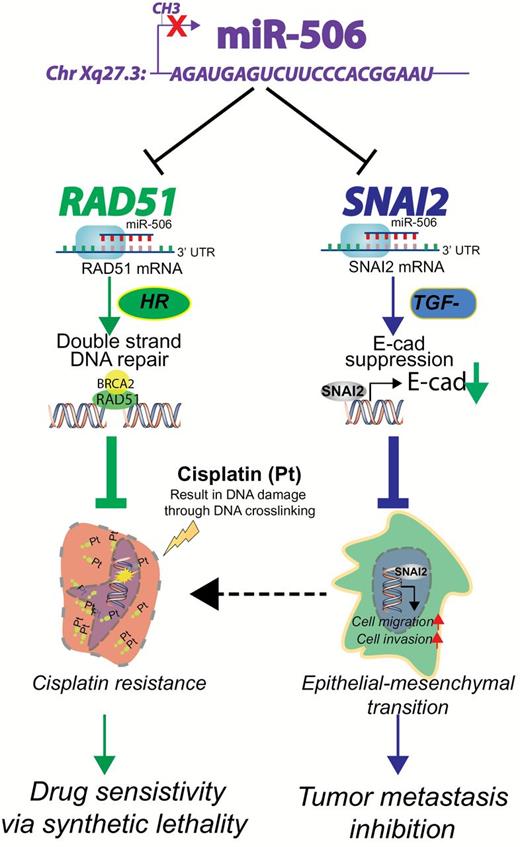
A summary of the therapeutic role of miR-506. MiR-506, located on Xq27.3, is downregulated by methylation (22). MiR-506 directly targets RAD51 (this study) and SNAI2 (22) suppressing double-stranded DNA damage repair via homologous recombination and epithelial-mesenchymal transition via activation of E-cad expression (22), respectively. As a result, miR-506 sensitizes cancer cells to chemotherapy and inhibits epithelial-mesenchymal transition–mediated metastasis. E-cad = E-cadherin; EMT = epithelial-mesenchymal transition; HR = homologous recombination; SNAI2 = snail family zinc finger2, TGF-β = transforming growth factor beta.
Funding
This work was supported by grants from the National Institutes of Health (U24CA143835, P50 CA083639, P50 CA098258, U54 CA151668, CA016672), CPRIT (RP110595), the Ovarian Cancer Research Fund, Inc., and the Blanton-Davis Ovarian Cancer Research Program, and by a grant from the Asian Fund for Cancer Research. The work also was supported by AIRC grant IG10302, from Associazione Italiana per la Ricerca sul Cancro, and by grants from the National Science Foundation of China to GL (#81101673, #81472761) and YS (#81201651), Tianjin Science and Technology Committee Foundation (14JCYBJC25300) to GL, grants from the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) in China and the National Key Scientific and Technological Project (2011ZX0 9307-001-04), and Tianjin Science and Technology Committee Foundation (09ZCZDSF04700) to KC and (15RCGFSY00108) FX. The TCH Tumor Tissue Facility is jointly funded by the National Foundation for Cancer Research. DY was an Odyssey Fellow at the MD Anderson Cancer Center and supported by the Diane Denson Tobola Fellowship in Ovarian Cancer Research, the Harold C. and Mary L. Daily Endowment Fund. DY is supported by the Elsa Pardee Foundation in University of Pittsburgh. CVP was supported by a grant from the National Cancer Institute (T32 training grant CA009666) and the DOM Advanced Scholar Program. YS was supported by the A. Lavoy Moore Endowment Fund.
We would like to thank Kathryn Hale from the Department of Scientific Publications at the MD Anderson Cancer Center for editing this manuscript and Drs. Marina Bagnoli and Valentina Tinaglia from Department of Experimental Oncology and Molecular Medicine at Fondazione IRCCS Istituto Nazionale dei Tumori for helpful discussion. We thank Brittany Parker for assistance with the manuscript.
The authors declare no competing financial interests.
References
Author notes
Current affiliations: School of Medicine, UNC Lineberger Comprehensive Cancer Center, Chapel Hill, NC (CVP); Center of Pharmacogenetics, Department of Pharmaceutical Sciences, University of Pittsburgh, Pittsburgh, PA (DY).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}