





Abstract
In phylogenetic inference by maximum-parsimony (MP), minimum-evolution (ME), and maximum-likelihood (ML) methods, it is customary to conduct extensive heuristic searches of MP, ME, and ML trees, examining a large number of different topologies. However, these extensive searches tend to give incorrect tree topologies. Here we show by extensive computer simulation that when the number of nucleotide sequences (m) is large and the number of nucleotides used (n) is relatively small, the simple MP or ML tree search algorithms such as the stepwise addition (SA) plus nearest neighbor interchange (NNI) search and the SA plus subtree pruning regrafting (SPR) search are as efficient as the extensive search algorithms such as the SA plus tree bisection-reconnection (TBR) search in inferring the true tree. In the case of ME methods, the simple neighbor-joining (NJ) algorithm is as efficient as or more efficient than the extensive NJ+TBR search. We show that when ME methods are used, the simple p distance generally gives better results in phylogenetic inference than more complicated distance measures such as the Hasegawa-Kishino-Yano (HKY) distance, even when nucleotide substitution follows the HKY model. When ML methods are used, the simple Jukes-Cantor (JC) model of phylogenetic inference generally shows a better performance than the HKY model even if the likelihood value for the HKY model is much higher than that for the JC model. This indicates that at least in the present case, selecting of a substitution model by using the likelihood ratio test or the AIC index is not appropriate. When n is small relative to m and the extent of sequence divergence is high, the NJ method with p distance often shows a better performance than ML methods with the JC model. However, when the level of sequence divergence is low, this is not the case.
Introduction
Most of the currently used methods of phylogenetic inference from molecular data are based on some form of optimization principle (Nei 1996 ; Swofford et al. 1996 ). Under this principle, the preferred tree is determined by assigning an optimality score to all possible topologies (or all potentially correct topologies) according to a certain procedure and choosing the topology that shows the highest or lowest optimal score. For example, in the case of maximum-parsimony (MP) methods (Eck and Dayhoff 1966 ; Fitch 1971 ), the minimum number of evolutionary changes that explains the entire sequence evolution (tree length [TL]) is computed for each topology, and the topology showing the smallest TL value is chosen as the preferred tree (MP tree). Similarly, in minimum-evolution (ME) methods (Edwards and Cavalli-Sforza 1963 ; Saitou and Imanishi 1989 ; Rzhetsky and Nei 1992 ), the total sum of branch length estimates (S) is used as the optimality score, and the topology showing the smallest S value is chosen as the preferred tree (ME tree). In contrast, maximum-likelihood (ML) methods use the log likelihood value (lnL) for each topology as the optimality score, and the tree showing the highest lnL is chosen as the ML tree (Felsenstein 1981 ).
One problem with the methods based on the optimization principle is that an enormous amount of computational time is required when the number of sequences is large, especially with ML methods. For this reason, a number of heuristic search algorithms that attempt to find the optimal tree are currently used (Swofford and Begle 1993 ). Yet, it is customary for many researchers to make heroic efforts to find the optimal tree, spending a large amount of computer time (Maddison 1991 ; Chase et al. 1993 ; Wainright et al. 1993 ; Rice, Donoghue, and Olmstead 1997 ). However, it should be noted that the tree with the optimal score is not necessarily the true tree. Nei, Kumar, and Takahashi (1998) showed that when the number of nucleotides examined (n) is small and the number of sequences (m) is large, the optimality scores of the MP and ME trees are always smaller than or equal to those of the true tree, whereas the optimality score of the ML tree is always greater than or equal to that of the true tree. This indicates that the optimization principle tends to give incorrect topologies when n is small relative to m.Nei, Kumar, and Takahashi (1998) also showed that for obtaining the true tree (not the optimal tree), simple topology search algorithms are often as efficient as extensive search algorithms. In the case of ME methods, a simple algorithm called neighbor joining (NJ; Saitou and Nei 1987 ) was shown to be as efficient as the standard ME method in almost all cases examined. However, the simple algorithms used for MP and ML methods were ad hoc and did not work well under certain conditions.
The main purpose of this paper is to examine the efficiencies of various search algorithms for MP, ME, and ML trees when the number of sequences used is large and to find simple algorithms for inferring the true tree with a reasonably high level of accuracy. We study this problem by computer simulation and by examining the relationships between the efficiencies of inferring the optimal and the true trees. We also examine the efficiencies of inferring the true tree when wrong nucleotide substitution models are used.
Methods of Computer Simulation
Model Trees
To study the efficiencies of various tree-building algorithms, we need model trees with which we can simulate the evolution of nucleotide sequences. Once the sequence data at the final stage of evolution are obtained, we can construct an MP, ME, or ML tree following a given tree-building algorithm and then compare the topology of the tree with that of the model tree (Nei 1991 ). We used six different model trees of 48 sequences, which are presented in figure 1 . Model trees A, B, and C represent the cases of constant-rate (CR) evolution, and model trees D, E, and F represent the cases in which the evolutionary rate varies from branch to branch (varying rate [VR]). The model trees with CR evolution were randomly generated by the branching process method described by Kuhner and Felsenstein(1994) . The model trees with VR evolution were produced by modifying the branch lengths of the CR trees obtained by Kuhner and Felsenstein's (1994) method. This modification was done by replacing each branch length by a random variable that followed a gamma distribution with the mean equal to the original branch length and the gamma shape parameter (a) equal to 10 (see Tateno, Nei, and Tajima [1982] for a similar method). The six model trees used here were generated independently. We also considered two levels of sequence divergence: high divergence (model trees A, C, D, and F) and low divergence (model trees B and E). High divergence refers to the case in which the expected number of nucleotide substitutions per site from the root to the tip of the tree is 0.5, and thus the expected number of substitutions between two most distantly related sequences (dmax) is 1.0. Low divergence refers to the case in which the expected number of substitutions from the root to the tip is 0.05 and dmax is 0.1.
Our simulations were not done for all combinations of sequence levels and tree topologies because of the prohibitive amount of computer time required. However, our preliminary study indicated that different model trees with a constant rate or a varying rate give essentially the same results for a given level of sequence divergence, apparently because all of the model trees were generated at random.
Models of Nucleotide Substitution
We used two different models of nucleotide substitution to generate sequence data that are commonly used for phylogenetic reconstruction: Jukes and Cantor's (1969) (JC) model and Hasegawa, Kishino, and Yano's (1985) model with a gamma distribution of substitution rate among different nucleotide sites (HKY+Γ). In the latter model, we used two different sets of substitution parameters. In the first set, we assumed that the transition/transversion rate ratio (k) was equal to 4, and the equilibrium frequencies of nucleotides A, T, C, and G were as follows: gA = 0.15, gT = 0.15, gC = 0.35, and gG = 0.35, respectively. The gamma parameter (a) used in this case was 1.0. In the second set, we assumed k = 10, gA = 0.10, gT = 0.10, gC = 0.40, gG = 0.40, and a = 0.5. Note that in the present model, k = 2R, where R is the standard transition/transversion ratio (Kumar, Tamura, and Nei 1993 ; Nei and Kumar 2000 ). The numbers of nucleotides used (n) were 300 for high sequence divergence and 1,000 for low sequence divergence. For each model tree, we generated 100 random sets of sequence data and constructed MP and ME trees for each data set. However, for ML methods, we used only the first 50 data sets, because the computational time was prohibitively long.
Phylogenetic Reconstruction
In this paper, we used five different algorithms for constructing MP trees that are available in the computer program PAUP* (Swofford 1998 ). The simplest algorithm used was stepwise addition with the closest option (SA). This algorithm is a rough search of MP trees, and it often fails to find the true optimal (MP) tree. Therefore, the tree obtained by this algorithm is usually subjected to various branch-swapping algorithms. The branch-swapping algorithms generally used are nearest-neighbor interchange (NNI), subtree pruning and regrafting (SPR), and tree bisection-reconnection (TBR) (see Swofford and Begle [1993] for the actual procedures). Among these algorithms, the simplest one is NNI, and the most extensive one is TBR. In this paper, these algorithms are denoted by SA+NNI, SA+SPR, and SA+TBR. To search for the most likely MP tree, however, researchers often repeat the TBR search many times starting with different SA trees obtained by rearranging the input order of sequences. We therefore used an algorithm in which the TBR search was repeated 100 times. This algorithm is denoted by 100SA+TBR.
For ME methods, we used the NJ method as the fast algorithm, because this method is known to be very fast and as efficient as or more efficient than the exhaustive ME method in obtaining the true tree when the number of sequences used is small (Saitou and Imanishi 1989 ; Kumar 1996 ; Gascuel 1997 ; Nei, Kumar, and Takahashi 1998 ). As the extensive search algorithm, we used the NJ+TBR algorithm, in which the NJ tree is subjected to the TBR search. We used this algorithm because the TBR search does not require much computational time for ME trees. For computing pairwise distances, we used p and JC distances when the JC model was used for generating sequence data and p, JC, and HKY+Γ distances when the HKY+Γ model was used for sequence generation. The HKY+Γ distance was estimated by the maximum-likelihood algorithm available in PAUP*.
For ML methods, we used primarily the SA (“as is” option), SA+NNI, and SA+SPR search algorithms. The SA+TBR search was used only for a few cases because this algorithm required an enormous amount of computer time. When the JC model was used for generating sequence data, we used the same model for constructing trees. When sequence data were generated by the HKY+Γ model, we used both the JC and the HKY+Γ models for tree reconstruction. For the HKY+Γ model, we first constructed an ML tree using the SA algorithm with the JC model and obtained ML estimates of parameters k and a and nucleotide frequencies. Using these parameter estimates, we then constructed ML trees with the HKY+Γ model using the SA, SA+NNI, and SA+SPR algorithms. We used this simplified method to save computer time.
Accuracies of Estimated Tree Topologies
Since we used randomly generated model trees of 48 sequences and some of the interior branches of a realized tree (Nei and Kumar 2000) often had 0 nucleotide substitutions, the probability of obtaining the true topology was negligibly small. We therefore measured the accuracy of the estimated topology by the topological distance (dT) between the estimated tree and the true tree (Robinson and Foulds 1981 ; Penny and Hendy 1985 ; Rzhetsky and Nei 1992 ). In practice, since our simulations were replicated 100 or 50 times, we used the average dT value. This dT value is roughly twice the number of branch interchanges that are required to obtain the true topology from the estimated tree when both trees are bifurcating. For MP methods, several equally parsimonious trees were often obtained for the same data set. In this case, we computed the average dT value for all parsimonious trees for each replication and then obtained the average of this average dT value for all replications. An alternative method is to construct the strict-consensus tree for all MP trees for a given data set and to compute dT between this consensus tree and the true tree using Rzhetsky and Nei's (1992) formula. We can then take the average of this dT for all replications. In practice, however, these two methods give very similar average dT values, so we present only the former average here.
The main purpose of this paper is to find relatively simple algorithms that are as efficient as extensive search algorithms in inferring the true tree. However, to examine the efficiencies of various optimization algorithms, we also computed the average optimality scores of MP, ME, and ML trees obtained by the simple and extensive search algorithms.
Results
Table 1 shows the average topological distances from the true tree (dT) and the average TLs of MP trees obtained by five different algorithms. When model tree A (CR model) is used and the sequence data are generated by the JC model, the dT value for the simplest algorithm, SA, is 10.9, and it declines to 9.5 when the next-simplest algorithm, SA+NNI, is used. However, when the more extensive search algorithms SA+SPR, SA+TBR, and 100SA+TBR are used, dT remains virtually the same. This means that in the present case, the SA+NNI search is quite efficient in inferring the true tree, and there is no need to conduct more extensive searches. However, the SA+NNI search is better than the SA search. The most extensive search, 100SA+TBR, gave a value of dT = 9.6, which is slightly higher than that for less extensive searches (except SA). This happened because the MP trees identified by this extensive search algorithm were not necessarily closer to the true tree.
Note that in the present case, the minimum and maximum possible values of dT are 0 and 90, respectively, because there are 45 interior branches for an unrooted tree of 48 sequences (Nei and Kumar 2000 ). Note also that dT = 10.9 means an error in branching pattern (sequence partition) for 5.5 interior branches, whereas dT = 9.5 means an error for about 5 interior branches. Therefore, the difference in dT between 10.9 and 9.5 is rather small biologically, although the difference is statistically significant.
Table 1 also shows the average TLs for MP trees. This value is highest for SA and becomes smaller for SA+NNI and SA+SPR, but the TLs for SA+SPR, SA+TBR, and 100SA+TBR are essentially the same. In the present case, therefore, even TL does not decrease when extensive search algorithms are used.
When the high-divergence and VR model (tree D) is used, the dT values are nearly the same as those for model tree A. In this case, however, there are no significant differences in dT between different search algorithms. Actually, the SA+NNI search shows a slightly higher dT value (10.2) than the SA search (9.8), although the difference is not statistically significant. This indicates that in the present case, the SA search is as efficient as the 100SA+TBR search. This is so despite the fact that the average TL is smaller for 100 SA+TBR than for SA.
For low divergence (dmax = 0.1), the dT values are substantially smaller than those for high divergence, as expected. In this case, the dT for SA is significantly greater than that for SA+NNI or other search algorithms. However, for inferring the true tree, SA+NNI is sufficient for both CR and VR cases, and there is no need to conduct the SA+TBR or 100SA+TBR search.
Table 1 also shows the dT values for the CR and VR cases when sequence data were generated by the HKY+Γ model with a high divergence level (dmax = 1.0). The dT values are considerably greater than those for sequence data generated by the JC model. This indicates that the MP method is less efficient when the pattern of nucleotide substitution is complex than when it is simple. In this case, SA+SPR is slightly better than SA+NNI, but the dT values for SA+TBR and 100SA+TBR are nearly the same as the dT for SA+SPR.
Table 2 shows the values of dT and S for ME trees when the sequence data were generated by the JC model. In this case, two distance measures, p distance and JC distance, were used. For both distance measures, NJ, a simplified ME method, always shows a smaller dT than the NJ+TBR search. Note also that the p distance gives a smaller dT than the JC distance in all cases, although the sequence data were generated by the JC model. This is true for both CR and VR models, irrespective of the extent of sequence divergence.
Table 2 also shows the simulation results (dT and negative log ML value [−lnL]) for ML methods. In this case, SA is less efficient than SA+NNI and SA+SPR in inferring the true tree. The latter two algorithms show nearly the same dT value. Therefore, the SA+NNI search appears to be sufficient for inferring the true tree by ML methods. For the first 10 data sets (model tree D), we conducted the SA+TBR search, but the dT value was essentially the same as that for SA+NNI (data not shown). (In the present case, the SA+TBR search for an ML tree required a computational time of about 60 h for one replication with our 8500/132 PowerPC computer.) The −lnL value is also greater for SA than for SA+NNI and SA+SPR, but the latter two algorithms show essentially the same −lnL value.
Table 3 shows the results for ME and ML methods for the case in which the sequence data were generated by the HKY+Γ model. Here, we considered only the case of high divergence (dmax = 1.0) with the first and second parameter sets of the HKY+Γ model mentioned earlier (see also table 3 ). Because the HKY+Γ model is more complicated than the JC model, the dT values for this model are higher than those for the JC model (table 2 ). For ME methods, the NJ algorithm again shows a smaller dT than NJ+TBR in all cases, irrespective of the distance measure. One of the surprising results is that the distance measure (HKY+Γ) based on the same model as the one used for generating sequence data shows a much poorer performance than p or JC distance. This again indicates that a simple distance measure often shows a better performance than complex distance measures in inferring the true tree.
For ML methods, we present the results for only two search algorithms, SA and SA+NNI, because an enormous amount of computer time was required for SA+SPR, and the simulation results in table 2 show that SA+SPR gives essentially the same results as those obtained by SA+NNI. (Actually, we constructed ML trees for SA, SA+NNI, and SA+SPR for the first 10 data sets for model tree F with the first parameter set of the HKY+Γ model, but the latter two algorithms gave essentially the same dT value.) Here, however, we used the JC model as well as the HKY+Γ model to construct ML trees. The results show that the SA+NNI search gives a significantly smaller dT than does SA in all cases. Therefore, at least the SA+NNI search should be performed for inferring the true tree.
One of the most paradoxical results in this study is that the use of the correct model, HKY+Γ, does not necessarily give a better topology (a smaller dT value) than the use of an incorrect model, JC. Actually, the JC model generally gives a better performance than the HKY+Γ model, particularly when the SA algorithm is used. Interestingly, however, the average lnL value is much higher for HKY than for JC, with the difference being more than 1,000 for the second set of HKY+Γ parameters. Obviously, if we conduct the likelihood ratio test for individual data sets, the difference will become highly significant. In other words, if we choose a substitution model by applying the likelihood ratio test to this type of data set (Swofford et al. 1996 ), we will certainly choose the HKY+Γ model, yet this model does not give a better performance for topology reconstruction.
Discussion
Fast Heuristic Algorithms
The primary purpose of this paper was to examine the efficiencies of several fast heuristic algorithms of phylogenetic inference under the MP, ME, and ML criteria when a large number of sequences are used. For the ME criterion, we reached the conclusion that the simple NJ algorithm is almost always better than other sophisticated algorithms in inferring the true tree. This is consistent with the results obtained by Kumar (1996) , Gascuel (1997) , and Nei, Kumar, and Takahashi (1998) .
Currently, the most popular methods for obtaining MP and ML trees use extensive search algorithms such as SA+TBR or 100SA+TBR. This is based on the general belief that the MP or ML tree is likely to be the true tree or close to it. However, when the number of sequences (m) is large and the number of nucleotides examined (n) is relatively small, this is incorrect, and the MP and ML trees tend to give incorrect topologies, as was shown by Nei, Kumar, and Takahashi (1998) . In this study, we have shown that relatively simple search algorithms such as SA+NNI or SA+SPR are sufficient for inferring the true tree.
Of course, when m is large and n is relatively small, it would be very difficult to find the true tree by any method. In this case, the important thing is to identify the major branching patterns of the tree and leave the other branching patterns unresolved. This can be done easily if we use the bootstrap test (Felsenstein 1985 ). For this purpose, a simple search algorithm has an added bonus, because it does not take much computer time.
Nevertheless, it should be noted that the results obtained by this type of simulation study depend on the model trees used. Although we used several model trees that were generated at random and our conclusion was largely independent of the model tree used, these model trees still represent only a small proportion of many possible trees. It is therefore desirable to examine more different topologies and to find general properties of different algorithms in the future. Note also that the pattern of nucleotide substitution in real data is much more complicated than the models used in this paper.
Models of Nucleotide Substitution
We have shown that for the ME or NJ method, the simple p distance is more efficient than the JC or HKY+Γ distance in inferring the true tree. The superiority of the p distance over the JC distance in phylogenetic reconstruction has been noted by Saitou and Nei (1987) , Sourdis and Krimbas (1987) , and Tajima and Takezaki (1994) , but our results indicate that it is also superior to the HKY+Γ distance even when the sequence data are generated by the HKY+Γ model. This superiority is apparently caused by the small variance of this distance compared with the variances of other distance measures. In recent years, a number of investigators have used ML estimates of the HKY or HKY+Γ distance to construct an NJ or ME tree (e.g., Berntson, France, and Mullineaux 1999 ; Silberman et al. 1999 ; Zardoya, Economidis, and Doadrio 1999 ). Our study suggests that this practice should be discouraged. It appears that the HKY or HKY+Γ distance is useful only when the evolutionary rate varies extensively with evolutionary lineage and the number of nucleotides examined is very large (>10,000) (Takezaki and Gojobori 1999 ).
Similarly, we have shown that in phylogenetic inference by ML methods, the HKY+Γ model is less efficient than the JC model even if the HKY+Γ model is used for generating sequence data. Somewhat similar results were obtained by Yang (1997) and Bruno and Halpern (1999) when they compared the efficiencies of topology reconstruction by the JC and the JC+Γ models for various model trees of four to eight sequences. Complex relationships between substitution models and the efficiency of topology construction have also been noted by Tateno, Takezaki, and Nei (1994) and Gaut and Lewis (1995) . It appears that these results are partly caused by a larger number of parameters to be estimated in complex models than in simple models and are partly due to the complex nature of phylogenetic inference by ML methods as discussed by Nei (1987, 1996) , Yang, Goldman, and Friday (1995) , and Yang (1997) . Note that the mathematical basis of phylogenetic analysis by ML methods is quite different from the standard parameter estimation in a single probability space (Yang, Goldman, and Friday 1995 ; Nei 1996 ). The pattern of nucleotide substitution in real sequence data is also much more complicated than the HKY+Γ model, and for this reason some authors (e.g., McArthur and Koop 1999 ; Silberman et al. 1999 ; Waits et al. 1999 ) have used even more complicated models. It has also been suggested that selection of a model to be used for constructing ML trees should be determined by conducting the likelihood ratio test (Swofford et al. 1996 ; Huelsenbeck and Crandall 1997 ) or by using the Akaike information criterion (AIC) (Hasegawa and Kishino 1989 ). The present study raises serious questions about these procedures. A more careful study of the theoretical foundation of phylogenetic inference by ML methods is necessary.
In this paper, we were concerned primarily with topology construction, and our conclusion does not apply to the estimation of branch lengths for a given topology. For estimating branch lengths, it would be better to use a model close to the real substitution pattern.
Efficiencies of Different Tree-Building Methods
Although it was not our primary purpose to compare the efficiencies of different tree-building methods in inferring the true tree, our study provides some information on this problem. First, our study indicates that the efficiencies of the NJ and the ML methods depend on the mathematical model (or distance measure) used and that the correct model does not necessarily give the highest efficiency. Therefore, comparison of different tree-building methods should be done using the model that is most appropriate for each method. Our results showed that in the case of high divergence, the NJ algorithm with p distance generally gives a smaller dT than the MP+NNI and the ML+NNI algorithms, particularly when the HKY+Γ model is used for generating nucleotide sequences. In this case, even NJ with JC distance shows a better performance than MP and ML methods.
When the extent of sequence divergence is low and dmax = 0.1, however, NJ with p distance tends to give a slightly higher dT than the MP+NNI and ML+NNI methods. This is understandable, because a phylogenetic tree for closely related sequences with a relatively small n is expected to have many multifurcating nodes, and for identifying multifurcating nodes, MP and ML methods are superior to NJ, since the latter is intended to construct a bifurcating tree (Nei and Kumar 2000). In practice, of course, an inferred tree is usually subjected to the bootstrap test, and these multifurcating nodes are judged as unresolved in the inference of bifurcating trees. Therefore, it is difficult to compare the efficiencies of different tree-building methods in terms of dT alone when dmax is low and n is small.
As mentioned above, the relative efficiencies of different tree-building methods depend on the model tree, the substitution model, the number of nucleotides, etc., and it is very difficult to make any general conclusions from a limited number of computer simulations. In the past, it was customary to compare different tree-building methods by using the same substitution model for all of the methods and a small number of sequences. Our study shows that this is not the right method of comparison if we want to have conclusions that can be used for practical application. In any theoretical study of phylogenetics, it is important to find the best method of analysis for each tree-building method and then compare different methods. Also, the mathematical models currently used in phylogenetic analysis are crude approximations to reality. Therefore, we should not make unwarranted extrapolations of simulation results to real sequence data. Actually, many recent empirical studies with real data sets (Andersson et al. 1999 ; Honda, Yokota, and Sugiyama 1999 ; Marko and Vermeij 1999 ; Meireles et al. 1999 ; Tan et al. 1999 ) suggest that MP, NJ, and ML methods generally give essentially the same phylogenetic inference when the bootstrap test is applied. Phylogenetic inference is a biological problem rather than a mathematical one, and it is important to use biological information in the final judgment of the reliability of inferred trees.
Naruya Saitou, Reviewing Editor
Keywords: phylogenetic inference maximum parsimony minimum evolution maximum likelihood topological distance tree-building algorithms
Address for correspondence and reprints: Masatoshi Nei, 328 Mueller Laboratory, The Pennsylvania State University, University Park, Pennsylvania 16802. E-mail: nxm2@psu.edu.
Table 1 Average Topological Distances from the True Tree (dT) (±SE) and Tree Lengths (TLs) of Inferred Trees Under the Maximum-Parsimony Criterion
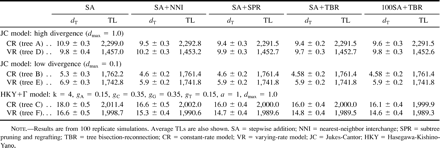
Table 1 Average Topological Distances from the True Tree (dT) (±SE) and Tree Lengths (TLs) of Inferred Trees Under the Maximum-Parsimony Criterion
Table 2 Average Topological Distances from the True Tree (dT) (±SE), Sums of Branch Lengths (S), and Negative Log Likelihood Values (−lnL) of Inferred Trees Under the Minimum-Evolution (ME) and Maximum-Likelihood (ML) Criteria
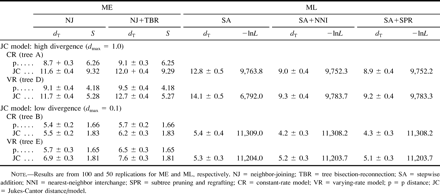
Table 2 Average Topological Distances from the True Tree (dT) (±SE), Sums of Branch Lengths (S), and Negative Log Likelihood Values (−lnL) of Inferred Trees Under the Minimum-Evolution (ME) and Maximum-Likelihood (ML) Criteria
Table 3 Average Topological Distances from the True Tree (dT) (±SE), Sums of Branch Lengths (S), and Negative Log Likelihood Values (−lnL) of Inferred Trees Under the Minimum-Evaluation (ME) and Maximum-Likelihood (ML) Criteria with Different Nucleotide Substitution Models
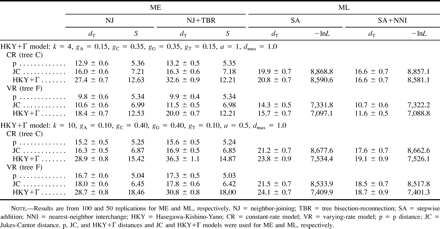
Table 3 Average Topological Distances from the True Tree (dT) (±SE), Sums of Branch Lengths (S), and Negative Log Likelihood Values (−lnL) of Inferred Trees Under the Minimum-Evaluation (ME) and Maximum-Likelihood (ML) Criteria with Different Nucleotide Substitution Models
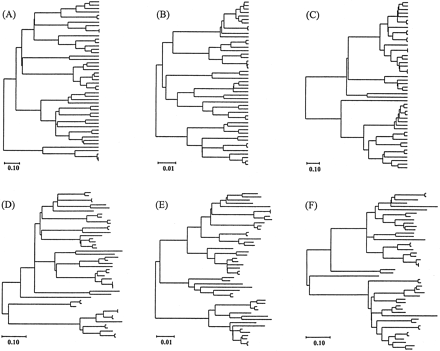
Fig. 1.—Six different model trees used for computer simulation. Trees A–C represent the cases of constant-rate evolution, and trees D–F represent the cases in which the substitution rate varies with evolutionary lineage. These trees were randomly generated by the methods described in the text
We thank James Lyons-Weiler and Mike Miyamoto for their comments on an earlier version of this paper. This study was supported by grants from NIH (GM20293) and NASA (NCC2-1057).
literature cited
Andersson, S. G. E., D. R. Stothard, P. Fuerst, and C. G. Kurland.
Berntson, E. A., S. C. France, and L. S. Mullineaux.
Bruno, W. J., and A. L. Halpern.
Chase, M. W., D. E. Soltis, R. G. Olmstead et al. (42 co-authors).
Eck, R. V., and M. O. Dayhoff.
Edwards, A. W. F., and L. L. Cavalli-Sforza.
Felsenstein, J.
Fitch, W. M.
Gascuel, O.
Gaut, B. S., and P. O. Lewis.
Hasegawa, M., and H. Kishino.
Hasegawa, M., H. Kishino, and T. Yano.
Honda, D., A. Yokota, and J. Sugiyama.
Huelsenbeck, J. P., and K. A. Crandall.
Jukes, T. H., and C. R. Cantor.
Kuhner, M. K., and J. Felsenstein.
Kumar, S.
Kumar, S., K. Tamura, and M. Nei.
McArthur, A. G., and B. F. Koop.
Maddison, D. R.
Marko, P. B., and G. J. Vermeij.
Meireles, C. M., J. Czelusniak, M. P. C. Schneider, J. A. P. C. Muniz, M. C. Brigido, H. S. Ferreira, and M. Goodman.
———.
Nei, M., and S. Kumar.
Nei, M., S. Kumar, and K. Takahashi.
Rice, K. A., M. J. Donoghue, and R. G. Olmstead.
Robinson, D. F., and L. R. Foulds.
Rzhetsky, A., and M. Nei.
Saitou, N., and M. Imanishi.
Saitou, N., and M. Nei.
Silberman, J. D., C. G. Clark, L. S. Diamond, and M. L. Sogin.
Sourdis, J., and C. Krimbas.
Swofford, D. L.
Swofford, D. L., and D. P. Begle.
Swofford, D. L., G. J. Olsen, P. J. Waddell, and D. M. Hillis.
Tajima, F., and N. Takezaki.
Takezaki, N., and T. Gojobori.
Tan, I. H., J. Blomster, G. Hansen, E. Leskinen, C. A. Maggs, D. G. Mann, H. J. Sluiman, and M. J. Stanhope.
Tateno, Y., M. Nei, and F. Tajima.
Tateno, Y., N. Takezaki, and M. Nei.
Wainright, P. O., G. Hinkle, M. L. Sogin, and S. K. Stickel.
Waits, L. P., J. Sullivan, S. J. O'Brien, and R. H. Ward.
Yang, Z.
Yang, Z., N. Goldman, and A. E. Friday.

{kind=link}