





Abstract
Rates of molecular evolution vary widely between lineages, but quantification of how rates change has proven difficult. Recently proposed estimation procedures have mainly adopted highly parametric approaches that model rate evolution explicitly. In this study, a semiparametric smoothing method is developed using penalized likelihood. A saturated model in which every lineage has a separate rate is combined with a roughness penalty that discourages rates from varying too much across a phylogeny. A data-driven cross-validation criterion is then used to determine an optimal level of smoothing. This criterion is based on an estimate of the average prediction error associated with pruning lineages from the tree. The methods are applied to three data sets of six genes across a sample of land plants. Optimally smoothed estimates of absolute rates entailed 2- to 10-fold variation across lineages.
Introduction
Estimates of rates of evolution of genes and other elements of the genome have revealed much about molecular evolution across a diversity of taxa (e.g., Li 1997 ). Comparisons of rates have contributed to the development of ideas about modes of selection on different genomic elements, such as introns versus exons, and have permitted correlates of rate differences, such as generation time and metabolic rate, to be identified (Gillespie 1991 ; Martin and Palumbi 1993 ). Comparisons of relative rates between lineages, in particular, have provided abundant evidence for departures from constant rates of substitution (Li and Wu 1985 ; Britten 1986 ; Li 1997 ; Muse 2000 ), a finding that has not, however, dampened enthusiasm for estimating divergence times from molecular data (e.g., Wray, Levinton, and Shapiro 1996 ; Kumar and Hedges 1998 ; Korber et al. 2000 ).
Characterization of the timescale over which molecular rates change and of the extent of their autocorrelation in time has lagged behind characterization of relative rates (Gillespie 1991 ). The reasons for this are several. Such studies require good estimates of absolute substitution rates. Estimates of absolute rates are often made by pairwise comparisons that necessarily average the rate differences on intervening branches (Easteal and Herbert 1997 ; Ayala, Rzhetsky, and Ayala 1998 ), thus underestimating the variability in rate. More comprehensive methods, such as estimating the slope of the regression of pairwise distance against calibrated divergence time (Wray, Levinton, and Shapiro 1996 ; Leitner and Albert 1999 ), have difficulty accounting for phylogenetic nonindependence (Pagel 1997 ; Ayala, Rzhetsky, and Ayala 1998 ). Estimation of absolute rates also requires reliable external information about time, usually from fossils. The use of fossil information in molecular rate estimation problems hinges, in turn, on the correct assignment of fossils to particular nodes in a phylogenetic tree (Marshall 1990 ; Smith and Littlewood 1994 ; Springer 1995 ; Lee 1999 ).
Relative rate comparisons, though robust, provide neither an estimate of absolute rate, nor an indication of how absolute rates change through time. Reconstructing how absolute rates change through time requires estimates of rate differences between two or more sequential branches in a tree. Relative rate comparisons only indicate differences in rate between sister branches, which are not sequential, are not descended from each other, and therefore do not indicate the direction of change. This problem cannot be corrected merely by performing multiple relative rate comparisons, whether they are nested within one another or not. Truly independent comparisons share no timepoint in common, making absolute comparisons impossible, and nested comparisons suffer from great sensitivity to what is assumed about the timing of nested nodes (Sanderson and Donoghue 1996 ).
In recognition of these obstacles, several general approaches for estimating absolute rate variability have been proposed, mainly in conjunction with estimating divergence times in the presence of rate variation. Because divergence times and absolute rates of evolution are inextricably linked, one cannot be estimated without the other. Some methods involve pruning outlier taxa that appear to depart from a tree-wide rate (Takezaki, Rhetsky, and Nei 1995 ). Some are local molecular clock methods, in which subtrees of the phylogeny are assigned different rates, but the rate is constant within each subtree (Hasegawa, Kishino, and Yano 1989 ; Uyenoyama 1995 ; Cooper and Penny 1997 ; Rambaut and Bromham 1998 ; Bromham and Hendy 2000 ; Yoder and Yang 2000 ). One potential problem with these approaches is that subset selection may be arbitrary, and in large trees, the number of possible ways to assign different rates to subtrees is large (Sanderson 1998 ). More general, but still parametric, methods have been described (Thorne, Kishino, and Painter 1998 ; Huelsenbeck, Larget, and Swofford 2000 ; Kishino, Thorne, and Bruno 2001 ), which assume a specific model for rate variation from branch to branch. Thorne, Kishino, and Painter (1998) assumed a lognormal distribution of rate changes whereas Huelsenbeck et al. assumed a compound Poisson process in which rates change in a stepwise fashion within lineages, and the amount of change is governed by a gamma distribution.
Given that little is known about the timescale for rate variation (Gillespie 1991 ), estimation procedures that are less reliant on parametric assumptions might prove useful. An entirely nonparametric method, nonparametric rate smoothing (NPRS: Sanderson 1997 ), estimated rates and times via a least-squares smoothing criterion that penalized rapid rate changes on a tree (Sanderson 1997 ). However, parametric models, coupled with maximum likelihood estimation methods, have proven to be statistically powerful and highly explanatory descriptions of molecular evolutionary processes. In this paper, I develop a semiparametric approach for estimating rates of molecular evolution. This approach attempts to combine the power of parametric methods and the robustness of nonparametric methods by the use of penalized likelihood (Green and Silverman 1994 ), part of a general class of semiparametric techniques used in smoothing and regression problems.
Penalized likelihood takes a parameter-rich model that would ordinarily overfit the data and constrains fluctuations in its parameters by a roughness penalty. For rate variation, one roughness penalty would penalize how quickly rates varied from branch to nearby branch, as in the NPRS method (Sanderson 1997 ). By adding the parametric component back in, it is possible to examine a broad spectrum of solutions with different levels of rate smoothing, ranging from highly penalized, nearly rate-constant models, to nearly unconstrained rate variability. The key to this approach is to find an objective method for selecting an optimal level of smoothing. After developing such an approach, based on cross-validation, it will be illustrated with three molecular data sets on land plants.
Materials and Methods
Models and Maximum Likelihood Estimation
Consider a rooted phylogenetic tree with M taxa and S + 1 internal nodes, in which the root node is labeled by 0, the remaining internal nodes are labeled by integers from {1, …, S} and the terminal nodes are labeled with integers {S + 1, …, S + M}. Branches are labeled by the node they subtend. Let node k have an age tk (measured backward from the present), and its ancestral node, anc(k), have an age tanc(k). The branch defined by these two nodes has a duration in time given by tanc(k) − tk.
Nucleotide substitution models are commonly cast in a general framework of a four-state Markov process (Rodriguez et al. 1990 ), in which transition probabilities between states are explicitly modeled. This paper extends model realism in a different direction to account for rate variation between lineages and, for simplicity, the standard Markov formulation will be replaced with a simpler substitution process, in which the estimated number of substitutions along a branch is regarded as an observation, xk, drawn from a Poisson process which has a rate rk. The more complex formulation can be reintroduced with considerable computational costs.
Two very different models lie at the opposite extremes of a spectrum of rate variation among lineages. At one extreme is a clock (CL) model, in which the rate parameters are the same for every branch, rk = r. At the other extreme is a saturated (SAT) model, in which each branch is permitted to have a unique rate, rk. The unknown parameters can be written as 𝛉CL = {t0, …, tS; r} for the CL model and 𝛉SAT = {t0, …, tS; r1, …, rS + M} for the SAT model, corresponding to S + 2 or 2S + M + 1 free parameters, respectively.
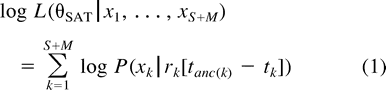
Penalized Likelihood
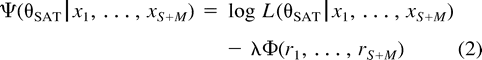
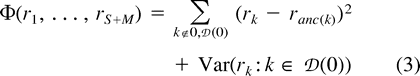
The semiparametric formulation described by equations (2) and (3) is ad hoc, and can really only be justified in terms of its performance in the present problem. For this, it is necessary to develop an objective measure of performance.
Cross-validation and the Choice of Smoothing Parameter
The value of the smoothing parameter, λ, can be seen as indexing an infinite number of semiparametric models. The choice of λ will affect the estimated rates and times, and it is therefore desirable to have an objective, data-driven method for choosing this parameter. A widely used method for model selection in general (Burnham and Anderson 1998 ) and smoothing in particular, is cross-validation (Green and Silverman 1994 ), which sequentially removes small subsets of the data, estimates parameters from the remaining data for a given choice of the smoothing parameter, and then uses the fitted model parameters to predict the data that were removed. Ideally, some choice of smoothing parameter will lead to a best prediction of the removed data, signally the optimal level of smoothing. In practice this is done by constructing a cross-validation criterion (CV) related to prediction error and then selecting the smoothing parameter that minimizes CV.
One method for cross-validation on trees is the pruning of terminal branches from the tree. Removal of a terminal branch, m, from the tree leaves its immediate ancestral node in place, along with all the other branches in the tree. Label the set of observations on numbers of substitutions on the remaining branches {x}(−m). The idea is to use this reduced set of observations to estimate parameters of the model using the penalized likelihood method described above. This generates a set of M estimates, θ̂SAT(−m), corresponding to each pruned taxon.
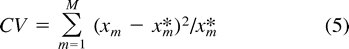
In the data analyzed below, λ was varied on a log scale between 0.1 and 10,000. For comparative purposes, the CV score for two other estimators was also obtained, although these do not depend on λ. These were the NPRS method described in Sanderson (1997) and the maximum likelihood estimation using the CL model, as outlined by Langley and Fitch (1974) . A priori, these might be considered logical extremes. Because the real data sets depart significantly from clocklike substitution rates, a simulated data set was constructed to test the performance of the cross-validation method under those conditions. A tree of 15 taxa was constructed according to a stochastic pure-birth process (Cox and Miller 1977 ), which generates a topology and branch durations. Numbers of substitutions were then assigned to branches by generating random Poisson deviates with means equal to the rate of substitution times the duration of the particular branch. The rate of substitution was set to 50 substitutions per unit time, in which a unit of time consisted of the distance from root to tip of the tree. The simulation protocol has been described elsewhere in more detail (Bininda-Emonds et al. 2001 ) and is implemented in the author's program, r8s (http://ginger.ucdavis.edu/r8s).
Optimization of the Penalized Likelihood
Optimization entails the search for solution(s), θ̂,that maximize the objective function, Ψ, in equation (2) for a given set of observations, {xk}, and choice of λ. The maximization of this objective function is a nonlinear optimization problem which can be solved numerically by standard numerical techniques such as Powell's gradient-free method and quasi-Newton gradient-based methods (Gill, Murray, and Wright 1981 ; Press et al. 1992 ). Termination criteria were based on both the convergence of the objective function and the gradient (to zero). Any cases in which convergence was a problem were reanalyzed with different optimization settings or a different algorithm. The local stability of solutions was checked by perturbing them and restarting the search, and all searches were started from several different initial random guesses at the parameters.
Penalized likelihood optimization is implemented in the author's program, r8s.
Molecular Data
The methods are illustrated by reference to three published molecular sequence data sets spanning land plants. The timescale for these taxa extends back 450 MYA to the origin of land plants or to the origin of vascular plants at about 420 MYA. None of the data sets are clocklike, based on likelihood ratio tests (data not shown). The first data set consists of 3,795 nt concatenated from two chloroplast photosystem (PS) genes, psaA and psbB,in 19 species of land plants (Sanderson et al. 2000 : tree from their Fig. 3 ; sequence data available at http://ginger.ucdavis.edu/wwwdata). The second data set consists of 1,428 nt of the plastid rbcL gene sampled in 37 land plants (Sanderson and Doyle 2001; data at http://ginger.ucdavis.edu/wwwdata). Data for the three protein coding genes were partitioned into approximate substitution classes to help distinguish nonsynonymous from synonymous changes: one class for first and second positions and the other for third positions. The third data set consists of 4,744 nt of concatenated sequence from small subunit (SSU) ribosomal DNA sequences from nuclear, chloroplast, and mitochondrial genomes of 28 land plants (Nickrent et al. 2000 : sequence data at http://www.science.siu.edu/landplants/Alignments/Alignments.html; the tree used is the single most parsimonious tree obtained from that data set, excluding rbcL,which was included in the original data set on the website, and excluding two algal outgroups).
Zero-length branches were collapsed to hard polytomies. Estimated numbers of substitutions along each branch of these trees (the observations, xk) were obtained by maximum likelihood using PAUP* 4.0 (Swofford 1999 ) with a Jukes-Cantor model of substitution. This is an extremely simple substitution model but should provide the closest fit to the Poisson assumption described earlier. Subsequently, I discuss extensions to more complex substitution models.
Results
Algorithmic Issues
Successful estimation of parameters—meaning that the optimization procedure converged to a stable solution—depended on the data, the smoothing parameter, and the numerical algorithm used. Data sets with as many as 100 taxa were tested, and for reasonable levels of smoothing, could converge in under 60 s using quasi-Newton methods on a 500 MHz Pentium III running Linux 6.0. All algorithms had trouble with extremely low smoothing parameters, when the optimization problem is ill-conditioned, meaning that a small change in the data translates into a large change in the estimated parameters (Gill, Murray, and Wright 1981 ). Overall, quasi-Newton methods that relied on an explicit calculation of the gradient of the objective function succeeded much better over a wider range of smoothing values. Derivative-free methods ran into trouble both at low smoothing and at very high levels of smoothing. On the other hand, standard implementations of quasi-Newton methods in Press et al. (1992) failed when any terminal branches had zero observed substitutions, apparently because the directional derivative for the rate along those terminal branches cannot be zero even at the obtained solution.
Cross-validation Results
In all the molecular data sets, plots of the CV score versus the level of smoothing, λ, indicate a minimum for intermediate levels of smoothing (fig. 1 ). These optimal points indicate the level of smoothing corresponding to the least prediction error, and therefore these can be viewed as the best semiparametric models for the data. A range of patterns is observed, however, among the various data sets. In the PS and SSU data sets, very high values of λ introduce more prediction error than very low values. One does better with an undersmoothed than with an oversmoothed model. For the rbcL third position data, the reverse is true, and for first and second position data the CV curve is nearly symmetrical. In the data from the simulation that assumed a clock, no minimum is present. Instead, there is a monotonic improvement as smoothing increases in the direction of clocklike evolution.
Comparisons with the CV scores of other methods indicate that the penalized likelihood approach always performs better than either a clock-based method such as Langley-Fitch (CL) or a wholly nonparametric method, such as NPRS (Sanderson 1997 ) whenever the data depart from constant rates. NPRS never achieves the low level of prediction error obtained by optimal smoothing in penalized likelihood. NPRS tends to overfit the data, in that the estimates of local substitution rates obtained from it tend to have higher variance than the variance found at optimal levels of smoothing. Even when the data are clocklike, penalized likelihood would select a value for smoothing that would be essentially clocklike itself.
NPRS outperformed CL in the real data, except in the rbcL first and second position partition. There the optimal smoothing level is nearly clocklike, and lower values of smoothing do progressively worse than assuming a clock, a result anticipated when data are nonclocklike, but there is simply not much of it (Sanderson 1997 ; see Discussion).
Estimated Rates and Times as a Function of Smoothing Level
Specific results are illustrated with examples primarily from the two larger molecular data sets, SSU and PS. Detailed analyses of these data will be presented elsewhere. In all the data sets, higher levels of smoothing led to estimated absolute substitution rates that varied less from branch to branch, as indicated by the coefficient of variation of rates across branches (fig. 2 ). The rate variation for individual branches is shown for two contrasting examples: the branch subtending the Gnetales and the branch subtending angiosperms (fig. 3 ). See figure 4 for the phylogenetic placement of these clades. Rates of substitution for the branch subtending Gnetales varied by a factor of 2–3 over the ranges of smoothing values described here, depending on the gene, whereas the range of variation for the branch subtending angiosperms was considerably less than that.
Differences in levels of smoothed rate estimates can be visualized across an entire tree in rate-calibrated phylogenies (fig. 4 ), which are tree diagrams in which branch lengths are drawn proportional to the absolute rates of substitution. These clearly show how smoothing can decrease the estimated variation in rate across the tree. For example, for first and second positions in PS, the rates of branches become steadily more similar to each other in moving from less smoothing to more smoothing. The rate along the branches leading to Huperzia and to Angiopteris, two low-rate lineages, increases, whereas the rate along the branch leading to Welwitschia and Ephedra, two high-rate lineages, decreases (albeit only slightly for this gene partition).
Estimated divergence times also depend on the choice of smoothing parameter. This is illustrated by the estimated age of the two nodes corresponding to the age of Gnetales and angiosperms (fig. 5 ). The estimated age of Gnetales is very sensitive to the smoothing level, showing a nearly monotonic increase in age as the model is made more clocklike, especially for the SSU data. The angiosperm age is a bit less sensitive to smoothing, but again the SSU data show relatively more sensitivity than the PS data.
Optimal Estimated Rates and Times
Given the results of the cross-validation analysis, estimates of ages and rates can also be obtained for each data set at the optimal level of smoothing. These are indicated for the exemplar branches and nodes in figures 3 and 5 . The variation in rates across each tree is substantial for all data sets (table 1 ): about twofold variation in rbcL first and second position data, threefold variation in the PS first and second position data, fivefold variation in the PS third position data, eightfold variation in the SSU data, and 10-fold variation in rbcL third position data.
Discussion
Optimal Levels of Smoothing
In four of the five data partitions among the three data sets, cross-validation indicated an optimal level of smoothed rate variation that led to performance significantly better than that permitted by the assumption of a clock. In the fifth, rbcL first and second position, the improvement was only marginal but the method did no worse. These patterns can be understood intuitively. Consider the case in which rates vary substantially across a tree. For large λ, the predicted values will essentially be the prediction under a molecular clock, but the observed number of substitutions along many branches will deviate from that expectation because rates are variable, leading to poor prediction of the observations in pruned branches. When λ is small, on the other hand, the model is overfit, and small changes in the data (i.e., subsamples of the data constructed during pruning) will lead to large changes in the parameter estimates. This leads, in turn, to a poor ability to predict pruned branches. For these reasons, one would expect some intermediate value of λ to give the best (smallest) CV score.
If the data are truly clocklike, on the other hand, CV should monotonically decrease with increasing λ, as it does for the simulated data in figure 1 , mainly because the noise in a constant-rate Poisson process causes unstable estimates of rates and times at low values of smoothing. However, if data are sufficiently noisy, as in the rbcL first and second positions, this pattern may well occur even for nonclocklike data because the simultaneous inference of both divergence times and absolute rates simply cannot be any more effective than a clock-based method. Previous simulation studies of the NPRS method (Sanderson 1997 ) indicated that it performed best when the average numbers of substitutions on branches were relatively high. With less data, the assumption of a clock could often provide results as good as those with more sophisticated methods.
The estimation of λ is itself subject to error, of course. For the present paper, this error has been ignored, but it clearly will contribute to the error variance of the estimates of rates and divergence times, just as model misspecification does in conventional parametric inference. Determination of the error on λ is likely to be computationally expensive, at least if resampling methods are used.
Comparisons to Other Methods
Penalized likelihood outperformed CL and NPRS in every data set that departed from a clock, as long as cross-validation was used to determine the optimal level of smoothing. Even under the simulated clocklike data, cross-validation would lead the investigator to choose a level of smoothing that was clocklike and thereby retain optimal levels of prediction error. Penalized likelihood always outperforms NPRS, which tends to overfit the data, allowing too much rate variation and thereby losing predictive power. This does not, however, imply that NPRS is worse than assuming a clock. Usually NPRS is better, except perhaps in the case of few substitutions along branches.
The CV criterion proposed here provides an empirical method for comparing other recently proposed rate and time estimation procedures (Thorne, Kishino, and Painter 1998 ; Huelsenbeck, Larget, and Swofford 2000 ), which have mainly been evaluated on an absolute scale by simulation studies (Sanderson 1997 ; Rambaut and Bromham 1998 ). Studies of relative performance of methods on the same data should illuminate the strengths and weaknesses of these approaches.
Extensions
Penalized likelihood can be applied to better models of the substitution process, such as the full four-state Markov model commonly used in maximum likelihood phylogenetic inference—albeit at considerable computational cost. Unfortunately, the cross-validation procedures are computationally expensive. A tree with M terminal taxa will have M independent estimation steps corresponding to each pruning. The running time of the numerical algorithms is also polynomial in M. Thus, implementation of a full Markov model version of cross-validation will add one degree to the exponent in the running-time scaling factor. For anything but the smallest trees, addition of the full Markov model will require good algorithm engineering.
The simplification used here may not be too bad in general. Suitably corrected branch length estimates may be nearly sufficient, in a statistical estimation sense, to estimate rates and divergence times. The main problem may well be accounting properly for rate variation across sites, which seems to exert a very substantial influence on length estimates (Yang 1996 ). Rate variation can be incorporated directly by using a negative binomial distribution for branch lengths rather than a Poisson as used here. If sites have rates that are chosen randomly from a gamma distribution—the usual approach (Yang 1996 )—then the probability of a substitution at any site is a negative binomial (Uzzell and Corbin 1971 ), and the distribution of branch lengths for a particular branch is then the sum of R negative binomial distributions (where R is the number of sites), which is also a negative binomial.
The increasing availability of multigene data sets for many taxa suggests extensions of the model in a different direction. Different genes may well have different patterns of rate variation, but the divergence times are held in common between them (unless coalescence times of the separate gene trees differ significantly). A comprehensive model should then include a single set of divergence time parameters but a separate set of rate parameters for each gene. This could be easily implemented if all genes were subject to the same smoothing parameter, but if each gene is optimally smoothed to a different extent, then the cross-validation procedure must simultaneously optimize multiple smoothing parameters, a fairly daunting prospect. Obviously, the problem would be much easier if the divergence times themselves could be fixed prior to analysis. In that case, the problem would reduce to one that is much closer to semiparametric regression problems that are well characterized. Given enough data, this may become possible for some clades.
Rates of Evolution of Plant Genes
Recent studies have suggested dramatic differences in rates of molecular evolution among land plant lineages based on differences in branch lengths on estimated trees (Chaw et al. 2000 ; Nickrent et al. 2000 ; Sanderson et al. 2000 ). This study confirms these inferences but places bounds on the rate variation. Certain lineages, especially Gnetales and some ferns, show much higher than average rates of evolution in all data sets and in both codon partitions of the protein-coding genes. Other lineages have much lower rates than average. The variation in rate is highest for rbcL third positions and SSU data, largely owing to extremely long branches in the Gnetales. In the PS genes, codon position partitions do not differ much in the level of variation in rate, despite the third codon partition mainly reflecting synonymous changes. The implications of this and its generality remain to be explored, but it agrees with previous findings in plant genes, which suggest strong lineage effects for both synonymous and nonsynonymous sites (Muse 2000 ).
The interplay between estimates of divergence times and rates is exceedingly complex. The sensitivity of an age estimate for a node to the level of smoothing is partly influenced by the sensitivity of estimated rates in the local region around that node. For example, in the SSU data, the rate for the Gnetales branch varies over a factor of two along the smoothing axis and so does the age estimate. However, the estimated rate for the PS third position data along that branch shows the same level of sensitivity, and yet its age estimates are much less sensitive to different levels of smoothing. Clearly, the discovery of striking differences in substitution rates across land plants does not automatically dim the prospects for using molecular data to reconstruct ancient divergence times.
Michael Hendy, Reviewing Editor
Abbreviations: CL, clock; CV, cross-validation (criterion); NPRS, nonparametric rate smoothing; PS, photosystem; SAT, saturated; SSU, small subunit.
Keywords: penalized likelihood molecular clock evolutionary rates
Address for correspondence and reprints: Section of Evolution and Ecology, One Shields Avenue, University of California, Davis, California 95616. mjsanderson@ucdavis.edu .
Table 1 Range of Estimated Absolute Ratesa of Substitution at Optimal Smoothing Levels
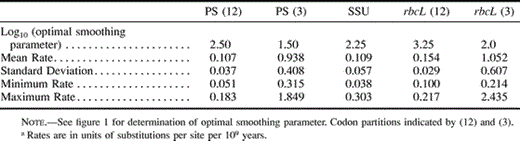
Table 1 Range of Estimated Absolute Ratesa of Substitution at Optimal Smoothing Levels
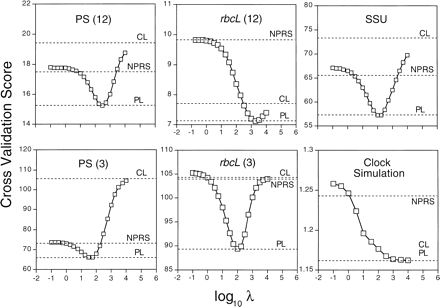
Fig. 1.—Cross-validation analysis for SSU, the two PS codon partitions, the two rbcL partitions, and for data set consisting of a simulated clocklike substitution process. The CV score is given by equation (5) . The horizontal dashed line labeled PL indicates the CV score for penalized likelihood under optimal smoothing. The dashed line labeled CL indicates the score for maximum likelihood estimation assuming a clock. The dashed line labeled NPRS indicates the score for the NPRS method (Sanderson 1997 ). The separate codon partitions for the PS data set are indicated by PS(12) for the first and second positions, PS(3) for the third positions, and similarly for rbcL
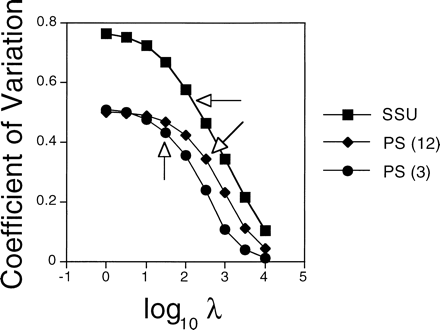
Fig. 2.—Variation in the absolute rates of substitution across lineages as a function of smoothing parameter. The coefficient of variation is the standard deviation divided by the mean. Optimal values based on cross-validation analysis are indicated by arrows (see fig. 1 )
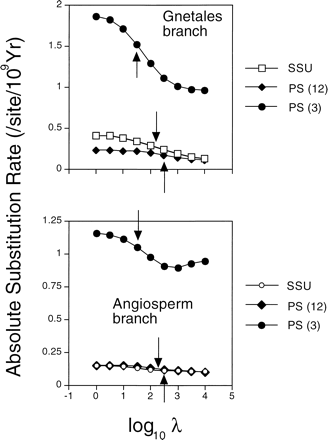
Fig. 3.—Absolute rates of substitution along two selected individual branches of the phylogenetic tree as a function of the smoothing parameter. Optimal values based on cross-validation analysis are indicated by arrows (see fig. 1 ). The phylogenetic position of these branches is indicated by the named clades shown in figure 4
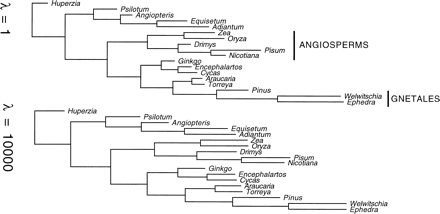
Fig. 4.—Rate-calibrated phylogenies for the first and second codon positions of the PS gene data set for two levels of smoothing. These trees have branch lengths drawn proportional to the absolute rates of substitution. These may appear misleadingly similar to conventional phylograms (Swofford 1999 ), but branch lengths in phylograms are proportional to the amount of sequence divergence rather than the absolute rate of substitution
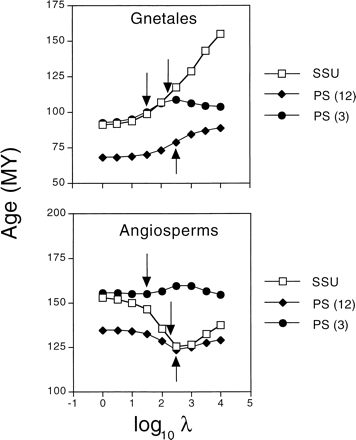
Fig. 5.—Estimated divergence times for two selected nodes as a function of the smoothing parameter. Optimal values based on cross-validation analysis are indicated by arrows (see fig. 1 )
Thanks are due to J. A. Doyle, J. Kim, S. Magallón, and M. F. Wojciechowski for useful suggestions. This research was supported by NSF grant 9726856.
References
Ayala J. A., A. Rzhetsky, F. J. Ayala,
Bininda-Emonds O., S. G. Brady, J. Kim, M. J. Sanderson,
Britten R. J.,
Bromham L. D., M. D. Hendy,
Chaw S.-M., C. L. Parkinson, Y. Cheng, T. M. Vincent, J. D. Palmer,
Cooper A., D. Penny,
Cutler D. J.,
Easteal S., G. Herbert,
Green P. J., B. W. Silverman,
Hasegawa M., H. Kishino, T. Yano,
Huelsenbeck J. P., B. Larget, D. Swofford,
Kishino H., J. L. Thorne, W. J. Bruno,
Korber B., M. Muldoon, J. Theiler, F. Gao, R. Gupta, A. Lapedes, B. H. Hahn, S. Wolinsky, T. Bhattacharya,
Langley C. H., W. Fitch,
Leitner T., J. Albert,
Lee M. S. Y.,
Li W.-H., C.-I. Wu,
Martin A. P., S. R. Palumbi,
Muse S. V.,
Nickrent D. L., C. L. Parkinson, J. D. Palmer, R. J. Duff,
Press W. H., B. P. Flannery, S. A. Teukolsky, W. T. Vetterling,
Rambaut A., L. Bromham,
Rodriguez F., J. L. Oliver, A. Marin, J. R. Medina,
Sanderson M. J.,
———.
Sanderson M. J., M. J. Donoghue,
Sanderson M. J., J. A. Doyle,
Sanderson M. J., M. F. Wojciechowski, J.-M. Hu, T. Sher Khan, S. G. Brady,
Smith A. B., D. T. J. Littlewood,
Springer M.,
Swofford D. S.,
Takezaki N., A. Rzhetsky, M. Nei,
Thorne J. L., H. Kishino, I. S. Painter,
Uyenoyama M. K.,
Uzzell T., K. W. Corbin,
Wray G. A., J. S. Levinton, L. H. Shapiro,
Yang Z.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}