





Abstract
Surface glycoproteins are principal receptors used by pathogens to invade target cells. It has been suggested that mammalian erythrocyte surface glycoproteins function as decoy receptors attracting pathogens to the anucleated erythrocyte and away from their target tissues. Glycophorin A (GYPA) is solely expressed on the erythrocyte surface where it is the most abundant sialoglycoprotein, although its function is unknown. The pathogen decoy hypothesis may be relevant here, as GYPA has been shown in vitro to bind numerous viruses and bacteria, which do not infect erythrocytes. However, it is also a receptor for erythrocyte invasion by the malarial parasite Plasmodium falciparum. Analyses of gypa sequence variation among six higher primates and within a human population show that there is a large excess of replacement (nonsynonymous) substitutions along each primate lineage (particularly on exons 2–4 encoding the extracellular glycosylated domain of GYPA) and a significant excess of polymorphisms in exon 2 (encoding the terminal portion of the extracellular domain) within humans. These two signatures suggest that there has been exceptionally strong positive selection on this receptor driving GYPA divergence during primate evolution and balancing selection maintaining allelic variation within human populations. The pathogen decoy hypothesis alone is adequate to explain both these signatures of between-species and within-species diversifying selection. This has implications for understanding the functions of erythrocyte surface components and their roles in health and disease.
Introduction
Host-pathogen interactions are major determinants in evolution, with cell surface receptors likely to be under constant selection to protect tissues from infection. Erythrocytes express an unexplained density and diversity of glycoprotein structures on their surface (Mourant, Kopec, and Domaniewska-Sobczak 1978 , Pp. 1–22) which cannot be explained by their primary function of transporting oxygen. Glycophorin A (GYPA) is the most abundant erythrocyte surface sialoglycoprotein (Cartron and London 1992 ; Huang and Blumenfeld 1995 ) with 0.5–1.0 million copies on each cell. The gypa gene is located on chromosome 4q28–31, in tandem with two paralogous genes (gypb and gype) which arose by duplication during primate evolution (Rearden et al. 1993 ). It is expressed solely on cells of the erythroid lineage (Huang and Blumenfeld 1995 ), and variation in its extracellular portion in humans determines the two major alleles of the MN blood group system, with further genetic changes accounting for rare blood group variants (Huang and Blumenfeld 1995 ). However, its function is still unknown. The dense negative charge that the sugar groups give the erythrocyte surface may play a role in preventing adhesion between cells and vascular surfaces (Huang and Blumenfeld 1995 ) having implications for blood viscosity; alternatively there are suggestions that it may be involved in cell ageing (Aminoff 1988 ). The extremely rare individuals who are GYPA-negative show that the null phenotype is viable (Cartron and London 1992 ), although its rarity would seem to indicate some cost associated with an absence of the receptor.
Many pathogens use sugar groups of surface glycoproteins, in particular sialic acid residues, as receptors for invasion (Karlsson 1995 ). It has therefore been suggested that erythrocyte glycoproteins may function as decoy receptors (Gagneux and Varki 1999 ) attracting pathogens to the erythrocyte, which as a mature cell lacks a nucleus and DNA replication, and away from target tissues (Gagneux and Varki 1999 ). This represents a plausible function for GYPA which carries many complex sugar groups (Tomita and Marchesi 1975 ; Pisano et al. 1993 ) and is supported by studies in which GYPA has been shown to bind numerous viruses (Paul and Lee 1987 ; Nishimura et al. 1988 ; Tavakkol and Burness 1990 ; Wybenga et al. 1996 ) and bacteria (Baseman, Banai, and Kahane 1982 ; Brooks et al. 1989 ; Saada et al. 1991 ). GYPA is, however, also known to be a principal receptor for the malarial parasite Plasmodium falciparum, which infects erythrocytes (Pasvol, Wainscoat, and Weatherall 1982 ; Sim et al. 1994 ). If GYPA generally functions as a decoy receptor, then it is likely to be under selection for targeting the diversity of potential pathogens. It is expected that this would cause diversifying selection on the gypa gene sequence.
To investigate the selective forces affecting the evolution of the GYPA receptor, a survey of sequence variation in the gypa gene was carried out. Tests for departures from neutrality were performed on pairwise comparisons of synonymous and nonsynonymous differences in the region encoding the mature GYPA product among six primate species and on genetic diversity in the region encoding the extracellular portion of GYPA in a human population.
Materials and Methods
Sequences of gypa and Other Nuclear Genes from Different Primates
The available gypa coding sequences (exons 2–6) of six primates were studied: human, chimpanzee, gorilla, orangutan, gibbon, and macaque (GenBank accession numbers AJ309706, AJ309708, AF015174, AF023467, AF023468, and AF023469). Exon sequences for nine other protein-coding loci in the same six higher primates were obtained from the following GenBank accession numbers—CC chemokine receptor-5 (ccr5): (HSU83326, AF177894, AF177901, AF177900, AF177884, AF177890). α(1,2) fucosyltransferase (H) (fut1): (M35531, AF080603, AF080605, AF111935, AF045545, AF080607). Lysozyme (lyz) (J03801, U76912–5, X60236). Aγ globin and Gγ globin (Aγ, Gγ): (U01317, M92294–6, J05174, X53419). Protamine 2 (prm2): (NM_002762, X72968, X71336–9). Rhesus blood group associated glycoprotein (rhag): (NM_000324, AF177621–5). Tyrosinase (tyr): (M27160, AF183588–92, AF183599–603, AF183604–8, AF183629–33, AF183644–48). Zinc finger protein, Y linked (zfy): (NM_003411, AB041908, AB041910, AB041912, AB041914, AB041918).
Allele Sequence of gypa Exons 2–4 Sampled from a Human Population
Purified genomic DNA from 33 unrelated individuals (Yoruba ethnicity, southwestern Nigeria) and a single chimpanzee (sample kindly provided by N. Mundy) were used as templates for amplification of a 3.6-kb region of gypa, from exon 2 to the boundary of exon 5 (see fig. 1 ). gypa-specific amplification primers GYPAIfwd5′-GCTTAGCTCAGGGACTGGAGG-3′ and GYPAIIIrev5′-CACCTTGCCTTTTAATAGAAAGC-3′ were designed to ensure no amplification of a paralogous sequence in gypb or gype, which share strong homology with gypa. PCR products were ligated into pGem®-T Easy Vector (Promega) and transformed by heat shock into JM 109 E. Coli High Efficiency Competent Cells. Isolated plasmids were prepared using a Plasmid Miniprep column (QIAGEN). A single clone representing one allele was isolated and sequenced from each individual to avoid problems associated with determination of haplotypes in heterozygotes. Each template was sequenced using internal sequencing primers (available on request) with 3′ BIG DYE™ dye terminator cycle sequencing premix kit (Applied Biosystems). Sequencing was carried out on a Perkin-Elmer ABI Prism™ 377 DNA Sequencer (Applied Biosystems) and sequences were checked and assembled using Sequence Navigator Version 1.0.1 (Applied Biosystems). Polymorphic sites were identified, and all singletons in the data set were confirmed by reamplification from genomic DNA and sequencing to exclude any errors introduced by PCR or cloning (deposited human sequences: GenBank accession numbers: AJ309828–45, AJ311318–32; chimpanzee AJ309708).
Statistical Analyses of Between-species and Within-species Diversity
The ratio of nonsynonymous substitions per nonsynonymous site to synonymous substitutions per synonymous site (dN/dS) was calculated for all pairs of the six primate species (pairwise analysis) using the method of Nei and Gojobori (Nei and Gojobori 1986 ) by the program MEGA 2.1 (http://www.megasoftware.net/) (Kumar, Tamura, and Nei 1994 ), with standard errors for dN and dS calculated using a bootstrap method in the MEGA program. Maximum likelihood estimates of dN/dS (the ω parameter) for each lineage in the phylogeny (see fig. 2 ) were derived by a method employing different evolutionary models, using the CODEML program of PAML Version 3.0b (http://abacus.gene.ucl.ac.uk/software/paml.html) (Nielsen and Yang 1998 ; Yang 1998 ; Yang et al. 2000 ). Investigation of evolutionary rates between lineages was carried out using alternative likelihood models, one with a single dN/dS ratio (M0, one-ratio) (Goldman and Yang 1994 ) estimated for all branches and another that allowed independent dN/dS ratios for each branch (FR, free-ratio) (Yang 1998 ). Statistical testing of differences between the different models (Nielsen and Yang 1998 ; Yang 1998 ; Yang et al. 2000 ) was carried out using twice the log likelihood difference (2Δl) which conforms to a chi-square distribution (the Likelihood Ratio test, LRT), with the degrees of freedom based on the difference between the number of parameters estimated from the models.
Three tests of neutrality (Hudson, Kreitman, and Aguade 1987 ; Tajima 1989 ; Fu and Li 1993 ) based on analyses of patterns of intraspecific and interspecific nucleotide differences, using the human population data set and a chimpanzee sequence out-group were performed using the program DNAsp Version 3.5 (http://www.bio.ub.es/∼julio/DnaSP.html) (Rozas and Rozas 1999 ).
Prediction of O-linked Glycosylation Sites in Primate GYPA Sequences
Published primate gypa sequences (as above) were translated into amino acid sequences and entered into NetOGlyc Version 2.0 (Hansen et al. 1998 ) (www.cbs.dtu.dk/services/NetOGlyc). This predicts O-glycosylation sites on serine and threonine residues in peptides based on the amino acid sequence context.
Results
Analysis of Sequence Differences in the gypa Gene Between Primate Species
The sequences encoding the mature GYPA product (378 bp, exons 2–6; fig. 1 ) in humans and five other species (chimpanzee, gorilla, orangutan, gibbon, and macaque) were studied. All the 15 pairwise ratios of dN/dS (number of nonsynonymous differences per nonsynonymous site vs. the number of synonymous differences per synonymous site) (Nei and Gojobori 1986 ), among the six species were greater than 1.0 (mean 2.1, minimum 1.4, and maximum 3.3) (table 1 ). This indicates an excess of amino acid replacement changes compared with neutral expectations.
To further examine this excess of nonsynonymous change across primate lineages, a maximum likelihood approach, which takes into account transition or transversion rate bias or nonuniform codon usage (Nielsen and Yang 1998 ; Yang 1998 ; Yang et al. 2000 ), was used to estimate the dN/dS ratio for each separate lineage in the phylogeny. Different likelihood models were tested, including one with a fixed dN/dS ratio (one-ratio) across all branches (Goldman and Yang 1994 ) and a second with independent ratios (free-ratio) for each branch (Yang 1998 ) (fig. 2 ). Both models gave dN/dS ratios well in excess of 1.0 (one-ratio = 2.6, see fig. 2 for the free-ratios which are >1.0 for individual branches). The likelihood of these two models did not differ significantly (log likelihood value for one-ratio model = −1064.0, for free-ratio model = −1060.2; χ2df = 8 = 8.88, P = 0.47), indicating that positive selection has operated broadly to cause divergence of the GYPA protein in different primate lineages. Furthermore, the dN/dS ratio for the part of the sequence encoding the extracellular region is particularly high (exons 2–4: maximum likelihood dN/dS = 4.7, average pairwise dN/dS = 2.4) compared with the transmembrane and intracellular region (exon 5–6: maximum likelihood dN/dS value = 1.4, average pairwise dN/dS = 1.6).
Analysis of Nine Other Nuclear Genes in Primates
A comparable analysis of the nine other nuclear genes, for which coding sequences in the same six primates were available (CCR5,fut1, Aγ, Gγ, lyz,prm2,rhag,tyr, and zfy), indicated the exceptional nature of GYPA diversity in primates. None of these nine genes have an estimated excess of nonsynonymous changes in all lineages. All have average pairwise dN/dS ratios of less than 1.0 (except for rhag for which dN/dS = 1.00) (fig. 3a ). As indicated in figure 3b the average dN pairwise difference for gypa (0.113) is exceptionally high (mean for the other genes = 0.018). Hence, the high dN/dS ratio is caused by a high rate of nonsynonymous substituion and not by a low rate of synonymous substitution (the average dS pairwise difference for gypa (0.055) is typical; mean for the other genes = 0.059). Together these data highlight the distinctive signature of diversifying selection on gypa across the evolution of higher primates. Comparable sequences are not yet available for other mammalian orders, except for the mouse homologue that is so divergent from primate sequences that a proper alignment for analysis is not possible (Matsui, Natori, and Obinata 1989 ).
Analysis of Sequence Variation in the gypa Gene Within a Human Population
To investigate whether intraspecific diversifying selection also operates within humans, the nucleotide sequence of a 3.6-kb region spanning the extracellular encoding part of the gypa gene (exons 2–4 and intron regions; fig. 1 ) was determined for one allele of each of the 33 unrelated Nigerian individuals. Among the 33 chromosomes, there were 25 different alleles of the complete sequence and three different alleles when considering only the exon coding sequence (fig. 4 ). There are five polymorphic sites in the coding sequence (all in exon 2), two of which are the nonsynonymous substitutions that determine the MN polymorphism (Huang and Blumenfeld 1995 ).
Analysis of variation among the 33 sampled chromosomes (table 2 ) showed a marked excess of nucleotide diversity in exon 2 (π = 0.024) compared with other exons (π = 0) and introns (π = 0.003). Statistical departures from neutral expectations were first tested using Tajima's (Tajima 1989 ) and Fu and Li's (Fu and Li 1993 ) methods. Tajima's D test (Tajima 1989 ) tests for a departure from neutrality as measured by the difference between π (observed average pairwise diversity) and 𝛉 (expected nucleotide diversity under neutrality derived from the number of segregating sites, S). Under balancing selection, rare alleles are selected for and maintained at intermediate frequencies, thus elevating π and, therefore, making the value of the test statistic (D) more positive. Fu and Li's test (Fu and Li 1993 ) reveals an excess or lack of singletons by comparing estimates of 𝛉 based on the number of mutations in external branches of a phylogeny versus an estimate from S (the D index) or π (the F index). Absence of singletons under balancing selection will result in a positive value for each statistic. The value of Tajima's D for exon 2 is significantly positive (D = 2.54; table 2 ), indicating that alleles tend toward intermediate frequencies in the population. Fu and Li's F value for exon 2 is also significantly positive (F = 1.86; table 2 ), reflecting the absence of singletons among the nucleotides in this exon. The positive values of both these statistics support a hypothesis of selection, maintaining diversity within humans in exon 2 and contrast with the negative values seen elsewhere in the gene (table 2 ) and at other human genetic loci (Kaessmann et al. 2001) .
Comparison of Human gypa Diversity with Divergence from Chimpanzee gypa
Alignment of the human alleles with the chimpanzee sequence for exons 2–4 (fig. 4 ) reveals that the human nucleotide polymorphisms are limited to the first half of exon 2 (up to codon 12). By marked contrast, the fixed differences between the two species all occur from codon 24 onwards (fig. 4 ). Heterogeneity between exon 2 and exons 3–4 was tested by measuring interspecific divergence (human vs. chimp) and intraspecific polymorphism (in humans) (table 3 ) using the HKA test (Hudson, Kreitman, and Aguade 1987 ). The HKA test (Hudson, Kreitman, and Aguade 1987 ) tests for heterogeneity between two genetic loci by measuring their divergence between two sister species and comparing this with polymorphism within the same loci in one of the species. Under neutrality, the degree of polymorphism and divergence for the two loci is expected to be correlated. Here, the hypothesis that exon 2 is evolving at the same rate as exons 3–4 is rejected (P = 0.02; χ2 test). This indication of non-neutrality in exon 2, in itself and in combination with Tajima's and Fu and Li's tests above, indicates that there is selective maintenance of diversity in exon 2 encoding the outermost external portion of GYPA in humans.
Discussion
Molecular diversity in cell surface receptors may be adaptive, if it provides a means to prevent the binding of specific ligands to the cell (escape variation) or a means to actively bind specific ligands (target-seeking). Given that the human malarial parasite P. falciparum uses GYPA as a receptor for erythrocyte invasion, it could be an agent of selection, but it is not likely to be the cause of the non-neutral pattern shown here, for two reasons. Firstly, the nonhuman primate malarial parasites do not use GYPA as a receptor (where studied primate malarial parasites use the Duffy antigen as a principal receptor for invasion [Miller et al. 1977] ), and therefore, could not apply selection broadly in primates. Secondly, if P. falciparum did discriminate allelic structures of GYPA in humans (evidence suggests it does not [Pasvol, Wainscoat, and Weatherall 1982 ; Binks et al. 2001] ), it is more likely that this would give an advantage of one allele over another rather than the selective maintenance of multiple alleles as seen here. An alternative hypothesis is that GYPA may be exploited by pathogens as a Trojan horse to carry them on the erythrocyte in the circulation to target cells. This would be analogous to HIV binding to dendritic cells, which transmit the virus directly to the susceptible cells in the lymph nodes, the major site of HIV replication (Pohlmann et al. 2001a, 2001b ). However, such a hypothesis is again unable to explain the selective maintenance of alleles in humans.
The joint signatures of between-species and within-species diversifying selection can, however, be explained by the ability of GYPA to bind nonerythrocytic pathogens to the erythrocyte surface (Baseman, Banai, and Kahane 1982 ; Paul and Lee 1987 ; Nishimura et al. 1988 ; Brooks et al. 1989 ; Tavakkol and Burness 1990 ; Saada et al. 1991 ; Wybenga et al. 1996 ) by acting as a decoy receptor. Surface glycoproteins containing sialic acids, are principal receptors used by pathogens to invade their target tissues (Karlsson 1995 ). Given its abundance on the erythrocyte surface and its heavy glycosylation (including sialic acid groups), GYPA could function as a decoy receptor attracting pathogens that bind cell surface sugar groups to the anucleated erythrocyte and away from more vital target tissues. Such a flypaper strategy would explain why the GYPA sequences have diverged so greatly among species, with GYPA in each species adapting specifically to its own pathogens.
This hypothesis also explains the maintenance of GYPA variation within humans by balancing selection, because heterozygotes that express two structural forms of GYPA would be potentially able to target more pathogens. In addition to the primate sequence data given here, there is also general evidence of variation in the glycosylation found on the erythrocyte surface both within humans (Gardner et al. 1989 ) and among different mammals (Gagneux and Varki 1999 ). This is supported by comparing the predicted O-linked glycosylation sites of primate GYPA sequences, which demonstrate how changes in amino acid sequence elsewhere in the protein can influence the predicted glycosylation of conserved serine or threonine residues through indirect effects such as protein folding (fig. 5 ). These differences in amino acid sequence that affect the folding and tertiary structure of surface proteins may also affect pathogen binding. Consistent with this, it has been observed that human erythrocytes with the GYPA allelic blood group M antigen are preferentially bound by certain strains of Escherichia coli, compared with cells with only N (Vaisanen et al. 1982 ; Brooks et al. 1989 ), where the difference between the two blood groups is defined by amino acid differences and not by glycosylation differences. Functional studies on the interaction between GYPA and nonerythrocytic pathogens and the effect that sequence variation has on glycosylation and pathogen binding will be important for testing the pathogen decoy hypothesis further. The strong non-neutral patterns in the gypa gene identified in this study highlight the erythrocyte surface as an important area for evolutionary studies.
Supplementary Material
New sequences described in this study: Human GYPA ex2–6 (AJ309706) Chimp GYPA ex2–6 (AJ309708). Human GYPA ex2–4 (AJ309828–45, AJ311318–32).
Howard Ochman, Reviewing Editor
Abbreviations: GYPA, Glycophorin A; dN, number of nonsynonymous substitutions per nonsynonymous site; dS, number of synonymous substitutions per synonymous site.
Keywords: natural selection erythrocyte glycophorin A infectious disease
Address for correspondence and reprints: Jake Baum, Department of Infectious and Tropical Diseases, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, United Kingdom. jacob.baum@lshtm.ac.uk
Table 1 Proportion of Nonsynonymous Differences per Nonsynonymous Site (dN) and Synonymous Differences per Synonymous Site (dS) in Exons 2–6 of the gypa Gene Between Pairs of Primate Species
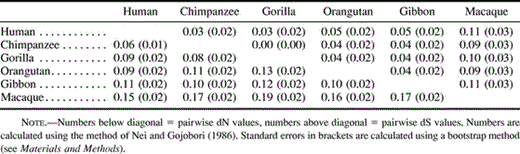
Table 1 Proportion of Nonsynonymous Differences per Nonsynonymous Site (dN) and Synonymous Differences per Synonymous Site (dS) in Exons 2–6 of the gypa Gene Between Pairs of Primate Species
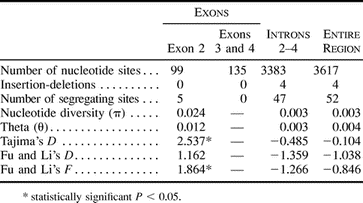
Table 3 Summary of HKA Test Comparing Human gypa Polymorphism with Human-Chimpanzee Divergence in Coding Regions

Table 3 Summary of HKA Test Comparing Human gypa Polymorphism with Human-Chimpanzee Divergence in Coding Regions
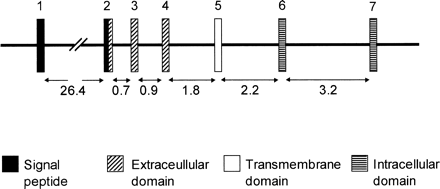
Fig. 1.—Scheme of the gypa gene. Intron sizes in kb are given below the scheme (Kudo and Fukuda 1989 ). Exons (numbered above) are not drawn to scale because of their small sizes (exons 1–7 are 37 bp, 99 bp, 96 bp, 39 bp, 86 bp, 79 bp, and 17 bp, respectively). A signal peptide (encoded by exon 1 and part of exon 2) is cleaved off to leave a 131-amino acid mature protein composed of a glycosylated extracellular domain, transmembrane, and intracellular domain
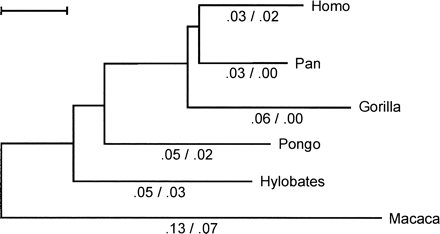
Fig. 2.—Phylogeny of the gypa gene (exons 2–6) in primates. Branches are drawn in proportion to the maximum likelihood estimates for the number of nucleotide substitutions per codon, t (scale bar t = 0.05) (Yang 1998 ). Numbers below branches indicate estimates of the proportion of nonsynonymous substitutions per nonsynonymous site versus synonymous substitutions per synonymous site (which gives the dN/dS ratio) according to the free-ratio model, where these values are independent for each branch. The tree used for the analysis is unrooted
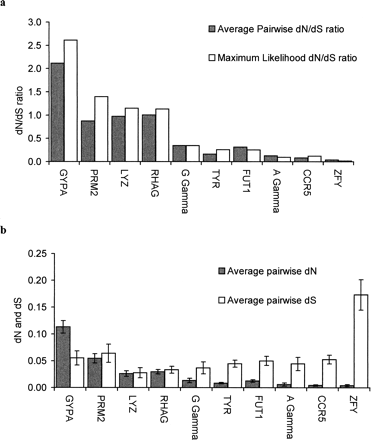
Fig. 3.—(a) Average pairwise dN/dS ratios (Nei and Gojobori 1986 ) and maximum likelihood dN/dS ratios (fixed for all branches) (Goldman and Yang 1994 ) for all six species, comparing gypa and nine other genes: protamine 2 (prm2), lysozyme (lyz), Rhesus blood group associated glycoprotein (rhag), Gγ globin (G γ), tyrosinase (tyr), α(1,2) fucosyltransferase (H) (fut1), Aγ globin (A γ), CC chemokine receptor-5 (CCR5), zinc finger protein, Y linked (zfy). Standard errors for the maximum likelihood dN/dS values calculated by CODEML (not shown) do not overlap between gypa and the other genes. No standard error can be calculated for the pairwise dN/dS ratio, but the ratio for gypa is more than twice that of any of the other genes. (b) Nei and Gojobori (Nei and Gojobori 1986 ) average pairwise dN and dS ratios show that the large dN/dS value for GYPA is the result of a large excess of dN (P = 0.001; z test) and not the absence of dS in comparison with other genes. Standard errors are calculated using a bootstrap method (see Materials and Methods section)
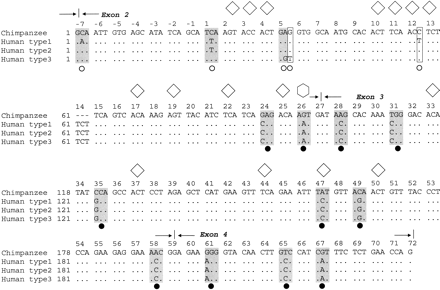
Fig. 4.—Alignment of the three human gypa coding alleles found in 33 chromosomes from Nigerian nationals (frequencies 30.3%, 24.2%, and 45.5%, respectively) with the orthologous chimpanzee gypa sequence for exons 2–4. Sites with synonymous mutations are boxed and codons with nonsynonymous mutations are shaded grey.—>|<—represents splice sites between exons. Codon numbering is according to (Kudo and Fukuda 1989 ). Codons −19 to −1 encode the signal peptide that is cleaved off the mature protein. Note that because exon 2 begins at the second position of codon −7, the first position of this codon is inferred, but this does not alter the nonsynonymous nature of the second position substitution. The MN polymorphism is determined by the nonsynonymous substitutions in codons 1 and 5. ○ is a polymorphic site within humans, is a fixed difference between species. Glycosylation sites (Pisano et al. 1993 ) are marked with ⋄ for O-linked oligosaccharides and for N-linked oligosaccharides
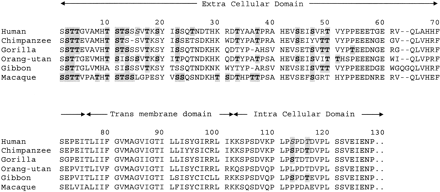
Fig. 5.—Alignment of GYPA protein sequence of six primates showing predicted glycosylation sites (shaded and bold) according to the NetOGlyc 2.0 program. Sites in italics differ in predicted O-linked glycosylation sites from published sites for human GYPA (Pisano et al. 1993 ). Signal peptide and 3′ UTA have been removed. Sites 1, 4, 11, 13, 19, 33, 44, and 50 differ in glycosylation despite identity in the amino acid at each position across primate GYPA sequences. Sites 14, 15, 22, 23, 25, and 116 differ in glycosylation despite identity in the amino acid at each position between more than one primate GYPA sequence
This work was supported by the Wellcome Trust (Prize Studentship for J.B.). We are grateful to Spencer Polley for advice on statistical analyses and Robin Weiss for helpful comments and suggestions.
References
Aminoff D.,
Baseman J. B., M. Banai, I. Kahane,
Binks R. H., J. Baum, A. M. J. Oduola, D. E. Arnot, H. A. Babiker, G. Kremsner, C. Roper, B. M. Greenwood, D. J. Conway,
Brooks D. E., J. Cavanagh, D. Jayroe, J. Janzen, R. Snoek, T. J. Trust,
Cartron J., J. London,
Gagneux P., A. Varki,
Gardner B., S. F. Parsons, A. H. Merry, D. J. Anstee,
Goldman N., Z. Yang,
Hansen J. E., O. Lund, N. Tolstrup, A. A. Gooley, K. L. Williams, S. Brunak,
Huang C. H., O. O. Blumenfeld,
Hudson R. R., M. Kreitman, M. Aguade,
Kaessmann H., V. Wiebe, G. Weiss, S. Paabo,
Karlsson K. A.,
Kudo S., M. Fukuda,
Kumar S., K. Tamura, M. Nei,
Matsui Y., S. Natori, M. Obinata,
Miller L. H., J. D. Haynes, F. M. McAuliffe, T. Shiroishi, J. R. Durocher, M. H. McGinniss,
Mourant A. E., A. C. Kopec, K. Domaniewska-Sobczak,
Nei M., T. Gojobori,
Nielsen R., Z. Yang,
Nishimura H., K. Sugawara, F. Kitame, K. Nakamura,
Pasvol G., J. S. Wainscoat, D. J. Weatherall,
Paul R. W., P. W. Lee,
Pisano A., J. W. Redmond, K. L. Williams, A. A. Gooley,
Pohlmann S., F. Baribaud, B. Lee, G. J. Leslie, M. D. Sanchez, K. Hiebenthal-Millow, J. Munch, F. Kirchhoff, R. W. Doms,
Pohlmann S., E. J. Soilleux, F. Baribaud, G. J. Leslie, L. S. Morris, J. Trowsdale, B. Lee, N. Coleman, R. W. Doms,
Rearden A., A. Magnet, S. Kudo, M. Fukuda,
Rozas J., R. Rozas,
Saada A. B., Y. Terespolski, A. Adoni, I. Kahane,
Sim B. K. L., C. E. Chitnis, K. Wasniowska, T. J. Hadley, L. H. Miller,
Tajima F.,
Tavakkol A., A. T. Burness,
Tomita M., V. T. Marchesi,
Vaisanen V., T. K. Korhonen, M. Jokinen, C. G. Gahmberg, C. Ehnholm,
Wybenga L. E., R. F. Epand, S. Nir, J. W. Chu, F. J. Sharom, T. D. Flanagan, R. M. Epand,
Yang Z.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}